ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2019; 15(7):1533-1545. doi:10.7150/ijbs.32020 This issue Cite
Research Paper
Amorphous solid dispersion of Berberine mitigates apoptosis via iPLA2β/Cardiolipin/Opa1 pathway in db/db mice and in Palmitate-treated MIN6 β-cells
Junnan Li1#, Hongwei Du2#, Meishuang Zhang1, Zhi Zhang3, Fei Teng1, Yali Zhao1, Wenyou Zhang1, Yang Yu1, Linjing Feng1, Xinming Cui4, Ming Zhang1, Tzongshi Lu5, Fengying Guan1,5 ![]() , Li Chen1
, Li Chen1 ![]()
1. Department of Pharmacology, School of Basic Medical Sciences, Jilin University, Changchun 130021, China
2. Department of Pediatric Endocrinology, The First Clinical Hospital Affiliated to Jilin University, Changchun 130021, China
3. School of Life Sciences, Jilin University, Changchun 130012, China.
4. Key Laboratory of Pathobiology, Ministry of Education, School of Basic Medical Sciences, Jilin University, Changchun 130021, China
5. Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02115, United States
#Equal contribution
Abstract

Aims: Berberine (BBR) improves beta-cell function in Type 2 diabetes (T2D) because of its anti-apoptotic activity, and our laboratory developed a new preparation named Huang-Gui Solid Dispersion (HGSD) to improve the oral bioavailability of BBR. However, the mechanism by which BBR inhibits beta-cell apoptosis is unclear. We hypothesized that the Group VIA Ca2+-Independent Phospholipase A2 (iPLA2β)/Cardiolipin(CL)/Opa1 signaling pathway could exert a protective role in T2D by regulating beta-cell apoptosis and that HGSD could inhibit β-cell apoptosis through iPLA2β/CL/Opa1 upregulation.
Methods: We examined how iPLA2β and BBR regulated apoptosis and insulin secretion through CL/Opa1 in vivo and in vitro. In in vitro studies, we developed Palmitate(PA)-induced apoptotic cell death model in mouse insulinoma cells (MIN6). iPLA2β overexpression and silencing technology were used to examine how the iPLA2β/CL/Opa1 interaction may play an important role in BBR treatment. In in vivo studies, db/db mice were used as a diabetic animal model. The pancreatic islet function and morphology, beta-cell apoptosis and mitochondrial injury were examined to explore the effects of HGSD. The expression of iPLA2β/CL/Opa1 was measured to explore whether the signaling pathway was damaged in T2D and was involved in HGSD treatment.
Results: The overexpression of iPLA2β and BBR treatment significantly attenuated Palmitate- induced mitochondrial injury and apoptotic death compared with Palmitate-treated MIN6 cell. In addition, iPLA2β silencing could simultaneously partly abolish the anti-apoptotic effect of BBR and decrease CL/Opa1 signaling in MIN6 cells. Moreover, HGSD treatment significantly decreased beta-cell apoptosis and resulted in the upregulation of iPLA2β/CL/Opa1 compared to those of the db/db mice.
Conclusion: The results indicated that the regulation of iPLA2β/CL/Opa1 by HGSD may prevent beta-cell apoptosis and may improve islet beta-cell function in Type 2 diabetic mice and in palmitate-treated MIN6 cells.
Keywords: Type 2 diabetes, Beta-cell dysfunction, Apoptosis, Dynamin-related protein(Opa1), Cardiolipin, Group VIA Ca2+-Independent Phospholipase A2 (iPLA2β), Berberine.
Introduction
Type 2 diabetes mellitus (T2D) is a growing epidemic (1), and it is characterized by insulin resistance, relative insulin deficiency, and eventual pancreatic beta-cell failure. In addition to the deficiency of insulin secretion, beta-cell apoptosis contributes to the insufficient production of insulin(2, 3). Lei et al. presented evidence that the activation of Group VIA Ca2+-Independent Phospholipase A2 (iPLA2β) in a spontaneous ER stress model can lead to the development of diabetes due to β-cell apoptosis (4). However, Zhao et al. found that iPLA2β is important in repairing oxidized mitochondrial membrane components (e.g., Cardiolipin, CL), and this may mitigate cytochrome c (cytc) release, preventing cells from undergoing apoptosis (5).
Phospholipases A2 (PLA2s) are enzymes that can release fatty acids from the second carbon group of glycerol, which can be classified into the following three groups according to their cellular location and the calcium-dependent enzymatic activity: 1) secretory PLA2; 2) cytosolic PLA2; and 3) calcium-independent PLA2 (iPLA2). iPLA2β has been investigated and cloned in both human and mouse pancreatic islet beta-cells(6-8). In a recent study, Song et al. found that oxidized CL increased in beta-cells that were incubated with Palmitate(PA), and this effect was attenuated by iPLA2β overexpression(9). These results suggest that iPLA2β/CL deficiency plays a key role in beta-cell injury. However, whether oxidized CL remodeling prevents apoptosis by iPLA2β is still unknown.
Recent studies have shown that CL is closely related to the inner mitochondrial membrane (IMM), and CL can regulate dynamin-related protein optic atrophy 1 (Opa1) (10). Both the protein expression of l-Opa1 and s-Opa1 were decreased in cells that lacked CL (11). In addition, Opa1 plays a key role in controlling cytc release (12, 13). Opa1 abnormality has been shown to be an indicator of impaired glucose tolerance and the impaired insulin response to hyperglycemia in both fed and fasted state (13, 14). Therefore, we hypothesized that the iPLA2β/CL/Opa1 pathway may play a crucial role in preventing apoptosis in β-cells. Additionally, we hypothesized that the drug that regulated iPLA2β/CL/Opa1 would reverse β-cell failure.
Berberine (BBR) is a traditional Chinese medicine that has been extensively used in Asia for the treatment of diarrhea and has also been reported to exert a hypoglycemic effect in type 2 diabetes (15). However, the poor oral bioavailability of BBR limits its clinical anti-diabetic application (16). We developed an amorphous solid dispersion of berberine with the absorption enhancer sodium caprate, referred to as Huang-Gui Solid Dispersion (HGSD) preparations (17). Our previous studies and those by Gao have found that BBR can promote insulin release and suppress β-cell apoptosis (18). In addition, we found that BBR could enhance the expression of iPLA2β in MIN6 mitochondria. Chang et al. have observed that BBR can increase CL levels in H9C2 cells (19). Given these results, we hypothesized that BBR could inhibit beta-cell apoptosis by enhancing the iPLA2β/CL/Opa1 pathway.
Materials and methods
Materials. BBR (purity quotient N 99.8%) was purchased from Northeast Pharmaceutical Group Co., Ltd., (Shenyang, China). The HGSD was prepared by solvent evaporation in the laboratory as previously described (17). Radio- immunoprecipitation assay (RIPA) buffer, phenylmethanesulfonyl fluoride (PMSF), hematoxylin, eosin and phosphatase inhibitors were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, Jiangsu, China). Pierce BCA protein assay reagents and Pierce ECL Western Blotting Substrate were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Polyvinylidene difluoride (PVDF) membranes were purchased from Bio-Rad Laboratories (Hercules, CA, USA). Anti-iPLA2β (sc-25504) were purchased from (Santa Cruz Biotechnology, CA, USA). Anti-COXIV (sc-423), and anti-Caspase-3 (9662) were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-Opa1 (ab42364), anti-cytc (ab13575), anti-GAPDH (ab181602) were purchased from Abcam (Cambridge, Cambridgeshire, UK). Peroxidase-conjugated AffiniPure goat anti-mouse(SA00001-1) and peroxidase- conjugated AffiniPure goat anti-rabbit (SA00001-2) were purchased from Proteintech Group (Rosemont, PA, USA). Chromatographic grade methanol and Chloroform were purchased from Sigma-Aldrich (St Louis, MO, USA). Internal standard tetramyristoyl-Cardiolipin [(C14:0)4-CL] sodium salt in powder form was purchased from Avanti PolarLipids Inc (Alabaster, AL, USA) and standard Cardiolipin (CL (18:2)4) sodium salt was purchased from Sigma-Aldrich (St Louis, MO, USA). All other chemicals used in this study were purchased from Sigma-Aldrich (St Louis, MO, USA).
Animal experiments. Male db/db mice and C57 mice (7 weeks old) were purchased from Model Animal Research Center of Nanjing University (Nanjing, China) and housed in constant room temperature (15-25 °C) with 12 h day-night cycle for 1 week before experiments. Both C57 control (CON) group and T2D (db/db mice) were fed with regular diet for 4 weeks. In the CON group, mice were given NS (10 ml/kg, b.w.). The db/db mice were randomly divided into four groups. In the model group (DB/DB), diabetic mice were given NS (10 ml/kg, b.w.). In the BBR group, diabetic mice were given BBR (160 mg/kg, b.w.), in the low dosage of HGSD group (HGSD-40), diabetic mice were given HGSD (40 mg/kg, b.w.) and in the high dosage of HGSD group (HGSD-160), diabetic mice were given HGSD (160 mg/kg, b.w.). Mice body weight, Fast blood glucose(FBG), Regular blood glucose (RBG), Oral Glucose Tolerance Test (OGTT), Tolerance Test (ITT), triglyceride (TG) and total cholesterol (T-CHO) were measured weekly in these four weeks. Fast blood insulin, Glucose- stimulated insulin secretion (GSIS) test and the mouse pancreases harvest were performed at the end of experiment. All animal studies were performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Jilin University (permit number, SYSK 2013-0005). Three groups CON, DB/DB and HGSD were used in the animal experiment of GSIS and western blot after the beneficial dosage of HGSD was chosen.
Biochemical analysis. The FBG and RBG were detected by glucometer (ACCU-CHEK Performa Roche, Germany) with or without fasting the mice. Fasting mice blood were drawn from orbital vein and standing for 15 minutes in room temperature, then centrifuged (3,500g) for 15 minutes at 4°C. Biochemical panel including T-CHO, TG and fasting blood insulin was examined by using Total cholesterol assay kit(A111-1), Triglyceride assay kit(A110-2) from Nanjing Jiancheng Bioengineering institute, (Nanjing, China) and ELISA kit (80-INSMSU-E01) from American Laboratory Products Company(ALPCO, American).
Oral glucose tolerance test (OGTT) and insulin tolerance test (ITT). Both C57 mice and db/db mice groups were fasted for 12 hours before OGTT test. 10 uL blood was obtained from the tail tip and was measured by test strip (1784892, Roche, America) and glucometer. Following an oral feeding of 40% glucose (2 g/kg, b.w., glucose powder from 492-62-6, Xilong Scientific Company, China.), blood glucose was tested at 0, 30, 60, 90, and 120 minutes respectively. For ITT test, both C57 and db/db mice groups were fasting for 2 hours, then injected insulin (0.5IU/kg b.w., i.p, Novolin R, Novo Nordisk A/S, Denmark). Blood glucose was tested using same protocol as OGTT test at 0, 15, 30, 60, and 120 minutes respectively.
HE staining, Immunohistochemistry (IHC) and TUNEL. Fresh pancreas tissues were harvested and soaked overnight in 4% paraformaldehyde at 4℃, then dehydrated in an ascending series of ethanol, and equilibrated with xylene, followed by embedding in paraffin and sectioning into 5-10 μm slices. Then, the samples were dewaxed with xylene and a descending series of ethanol. Continued sections were stained with both Mayer's hematoxylin and eosin (HE) and immunohistochemistry studies. Anti-Insulin antibody (1:500, Abcam) and TUNEL assay kit (C1082, Beyotime Company, China) were used to evaluate levels of insulin expression and pancreas apoptosis. Five fields from each slide from three mice at each group were chosen and analyzed. The percentage of insulin positive cells was calculated by Image Pro Plus 6.0 using mean density of Integrated optical density (IOD).
Electron microscopy. Islets isolated from mice were minced in fixative solution consisting of 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M cacodylate buffer (pH 7.4) for 2 hours at 4°C. And then, islets were post fixed in 1% osmium tetraoxide buffer for 1.5 hours. Tissues were then dehydrated with alcohol and embedded with Epon. 80 nm sections were prepared and photographed via X-650 electron microscope (Hitachi, Japan). The mitochondria and insulin granules in β-cells were observed by Images.
iPLA2β overexpression plasmid and shRNA plasmid construction. The plasmid containing pla2g6 sequence was purchased from GenePharma (China). The targeting sequence for iPLA2β is (5'-AACAGCACAGAGAAUGAGGAG-3'). The shRNA was inserted into pGPU6-GFP-NEO (GenePharma, China).
Cell culture. Mouse insulinoma cells (MIN6) were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% FBS and 50 μM β-mercaptoethanol at 37 °C and 5% CO2. Firstly, MIN6 cells were stably transfected with plasmid to over-express iPLA2β while control MIN6 cells stably transfected with empty vector only. After transfected with different plasmids for 24h, we treated cells with or without the palmitate (PA, 0.4mM) for 24h to observe the injury induced by PA and the effect of over-express iPLA2β. Secondly, MIN6 cells were stably transfected with plasmid to silence iPLA2β, and control MIN6 cells stably transfected with empty vector only. After transfected with different plasmids for 24h, BBR (10uM) and/or PA was added simultaneously to observe the role of iPLA2β in BBR's effect.
Detection of beta cell viability and apoptosis by MTT assay and Hoechst staining. The MTT assay was performed to evaluate the viability of MIN6 cells. Briefly, MIN6 were cultured in a 96-well microplate. After treatment, 20 μL of MTT solution was added to each well, and the cells were incubated for 2-4 hours. Then 200 μL of DMSO was added to dissolve the formazan crystals. Plates were read at 562 nm using Thermo Scientific Microplate Reader (Multiskan Spectrum). MIN6 were cultured on cover slips in a 24-well microplate. After treatment, Hoechst (Beyotime Biotechnology 33258) staining were used to detect cell apoptosis according to the manufacturer's instructions, cellular fluorescence was observed by OLYMPUS IX71 fluorescence microscope.
The insulin secretion. MIN6 cells were cultured on glass coverslips in 24-well plates and after treatment, the supernatant buffer of different groups were collected for basal insulin secretion measurement. The insulin content was detected by ELISA (EZRMI-13K, Merck Millipore Corporation, China) and normalized via total protein content. Cells were washed with PBS and lysed in the Radio Immuno Precipitation Assay (RIPA) buffer to isolate total protein. Protein concentration was determined by the Pierce BCA protein assay reagents.
Immunocytochemistry of Mitochondrial tracker and iPLA2β. MIN6 cells were cultured on glass cover slips in 24-well plates, and stained with MitoTracker Red (invitorgen detection technologies) according to the manufacturer's protocol. Then, cover slips were removed from the plate and fixed with 4% paraformaldehyde. After fixation, cells were incubated with 2% BSA for 30min and primary antibody mixture of iPLA2β (1:150, Santa Cruz Biotechnology) at 4 °C overnight. Lastly, cover slips were hybridized with secondary antibody mixture anti-rabbit IgG FITC (1:100, Santa Cruz Biotechnology) for 30 minutes at room temperature in dark chamber, then sealed with a drop of Fluoromount-G on glass slides. The results were observed by OLYMPUS IX71 fluorescence microscope.
Assessment of Mitochondrial Membrane Potential. Mitochondrial membrane potential (MMP) in MIN6 cell was measured using Mitochondrial membrane potential assay kit (JC-1) according to the manufacturer's instructions (C2006, Beyotime Biotechnology, China). The cells were washed twice with PBS, then incubated with JC-1 at 37℃ for 30 minutes. After incubation, cells were washed twice with PBS. Cellular fluorescence was observed by OLYMPUS IX71 fluorescence microscope.
Protein expression analyses. Mitochondrial and cytosolic fractions were isolated from islets and cells with a Mitochondria/Cytosol Fractionation kit (K256-100, BioVsion Research Products, USA) according to the manufacturer's instructions. The mitochondrial proteins were analyzed by immunoblotted with antibodies to iPLA2β, Opa1 and the mitochondrial marker COXIV. The cytosolic proteins extracted from tissue and cell lysates were analyzed by western blotting to detect expressions of caspase-3, cytc and GAPDH. Secondary antibodies goat-anti-rabbit or goat-anti-mouse (Proteintech) were diluted in 1:5000. The membrane was incubated in western ECL substrate (32106, Thermo fisher, USA) and exposed to Tanon imager, with Quantity One software for results analyses.
Lipid Extraction. Detached mitochondria from islets or MIN6 cells were homogenized in 100μl methanol, then centrifuged in 2800g for 5 minutes to remove tissue debris. Supernatants were transferred to 300μl of chloroform. Samples were Vortex-mixed and centrifuged in 900g for 5 min. Lipid phosphorus content in the supernatants was collected.
Cardiolipin analysis by HPLC/MS. Internal standard [tetramyristoyl-Cardiolipin (C14:0)4-CL] was added to extracted lipid solution, and mixture was concentrated, reconstituted. Then, it was analyzed by HPLC/MS on a Survey or HPLC on a C8 column (15 cm x2.1 mm, 581421-U, Sigma, USA) interfaced with the ion source of a AB Sciex triplequadruple mass spectrometer (AB SCIEX Triple Quad™ 5500 LC/MS/MS system) with extended mass range operated in negative ion mode. The lower limit of quantitation (LLOQ) and the calibration standard curve were obtained to calculated the CL concentration of each sample.
Statistical analyses. All the data were presented as means ± SEM. One-way ANOVA on GraphPad Prism5 showed statistical significance between multiple groups. P<0.05 and P<0.01 were considered statistically different and significantly different respectively.
Results
Palmitate-induced cell apoptosis in MIN6 Cells could be reversed by the overexpression of iPLA2β. To investigate the role of iPLA2β in apoptosis and in mitochondrial dysfunction, iPLA2β-overexpressing MIN6 cells were used in a Palmitate (PA)-induced cell injury model, and cell viability and apoptosis were evaluated. After incubation with PA, there was a remarkable decrease in the viability of MIN6 cells that were stably transfected with vector only, while those that overexpressed iPLA2β in MIN6 cells exhibited a reduction of the PA-induced injury (Figure 1A). However, when iPLA2β-silenced cells were established and were treated or not treated with PA, the results showed that silencing iPLA2β can slightly aggravate the injury induced by PA compared to that of the PA-treated MIN6 cells (as shown in the supplementary Figure). In PA-treated MIN6 cells that were transfected with vector only, the insulin content in the supernatant was lower than that in nontreated cells. However, the PA-treated cells that overexpressed iPLA2β secreted more insulin than the normal cells that were treated with PA (Figure 1B). These results indicated that the function of insulin secretion was impaired after PA treatment, while overexpressed iPLA2β could mitigate this impairment. Moreover, Hoechst staining was used to evaluate the cell apoptosis level. More apoptotic cells were observed in the PA-treated group than in the nontreated group, and this damage was decreased in iPLA2β-overexpressing cells compared to that of the PA-treated MIN6 cells (Figure 1C). In addition, cytosolic cytc and caspase3, which are key proteins in the execution of apoptosis via the mitochondrial pathway, were evaluated by Western blotting. Compared to those of the controls, both cytc and caspase3 were significantly increased in the PA-treated group and this increase was partly blocked by iPLA2β overexpression (Figure 1D). Overall, these results demonstrated that iPLA2β might serve as a key target to inhibit beta-cell apoptosis.
Palmitate(PA) induce the apoptosis in MIN6 cells and over-expression iPLA2β can mitigate this injury. The influence of PA-induced cytotoxicity and the mediation of over-expression iPLA2β were assessed by MTT assay(A). Data was expressed as means ± SEM (n=6). Compared with control group, *P<0.05, **P<0.01; compared with PA group, #P<0.05, ##P<0.01. Basal insulin release and content were measured in MIN6(B). Data was expressed as means ± SEM (n=6). Compared with control group, **P<0.01; compared with PA group, #P<0.05. After treated with PA for 24h in normal cells and iPLA2β over-expressed cells, the apoptosis were observed by Hoechst staining, bright blue means the apoptotic cells and marked with arrows(C). The protein was extracted from cells and cytosolic cytc and caspase3 were measured and analyzed by Western Blot(D). Data was expressed as means ± SEM (n=4). Compared with control group, *P<0.05, **P<0.01; compared with PA group, #P<0.05, ##P<0.01. Mitochondrial membrane potential in MIN6 were observed by JC-1 fluorescent dying and the intensity has been analyzed, ratio of green fluorescence to red fluorescence can represent the loss of mitochondrial membrane potential(E) Data was expressed as means ± SEM (n=4).Compared with control group,**P<0.01; compared with PA group, ##P<0.01.
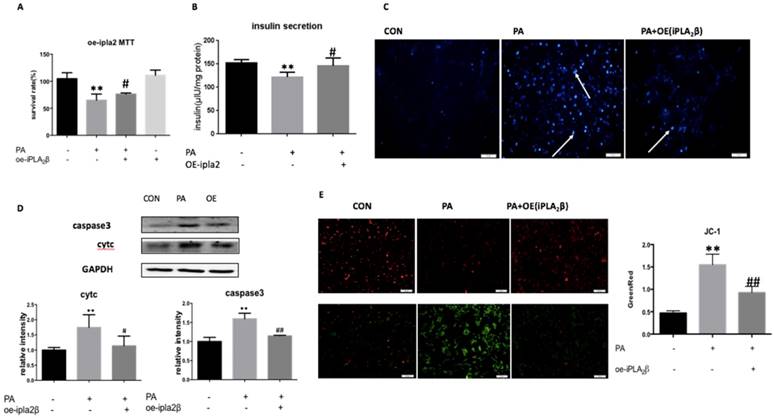
Mitochondrial membrane potential collapse in Palmitate-treated cells could be reversed by the overexpression of iPLA2β. JC-1 is a potential-sensitive MMP indicator. With high MMP, JC-1 accumulates in the mitochondria matrix (matrix) and produces more red fluorescence than green fluorescence, which can only be detected from the JC-1 monomer in the low MMP condition. Our data showed that PA-treated MIN6 cells expressed higher levels of green fluorescence compared with that in nontreated cells, and that the overexpression of iPLA2β could partly inhibit PA-induced mitochondrial injury and the loss of MMP compared to those of PA-treated MIN6 cells (Figure 1E).
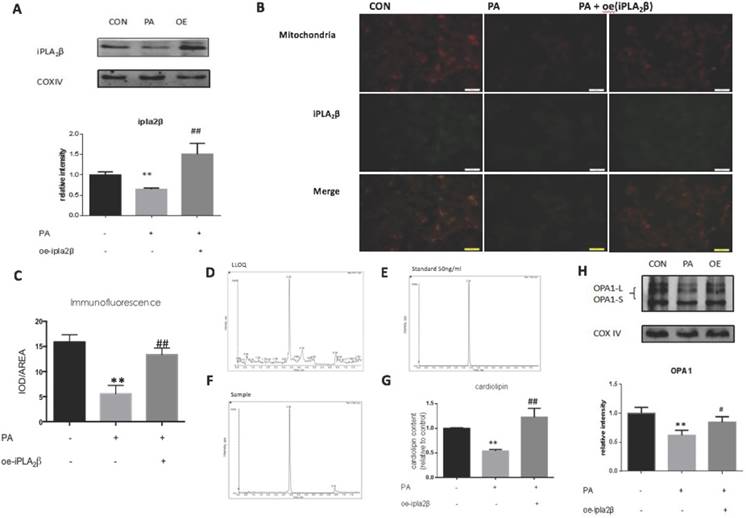
Overexpression of iPLA2β in mitochondria might enhance Cardiolipin and Opa1 expression to alleviate cell apoptosis. To further explore the mechanism of iPLA2β in the inhibition of apoptosis, mitochondrial proteins were extracted from MIN6 cells and analyzed by Western blotting. Our data showed that compared those of the controls, iPLA2β in mitochondria was increased significantly in iPLA2β-overexpressing MIN6 cells after PA treatment and was decreased in PA-treated MIN6 cells that were transfected with vector only (Figure 2A). Furthermore, immunofluorescence analysis using MitoTracker Red (labelled mitochondria) and GFP (green fluorescent protein that labelled iPLA2β) was used to examine the distribution of iPLA2β in MIN6 cells. Yellow spots represent the merged areas of iPLA2β and mitochondria labelling. The intensity of the yellow area was analyzed with IOD/AREA. The results showed that iPLA2β-GFP was uniformly distributed in the cytoplasm without PA treatment, as illustrated in the first column, and that PA reduced the iPLA2β distribution in the mitochondria, as shown in the second column; in contrast, the over-expressed iPLA2β was more concentrated in mitochondria than that in the PA treated MIN6 cells, as shown in the third column (Figure 2B-C). In addition to evaluating the mitochondrial protective function of iPLA2β, we further investigated its effect on CL with HPLC/MS. The detection method was established, and to ensure the accuracy of the method, the lower limit of quantitation (LLOQ) and the standard sample were obtained (Figure 2D-E). Our data showed that compared to those of the controls, CL expression was also significantly decreased in cells that were undergoing PA-induced apoptosis and was increased in iPLA2β-overexpressing cells after PA treatment (Figure 2F-G). Additionally, Opa1 was examined by Western blotting, which showed a similar trend (Figure 2H). Overall, these results demonstrated that iPLA2β could act as an anti-apoptotic target via the CL/Opa1 pathway.
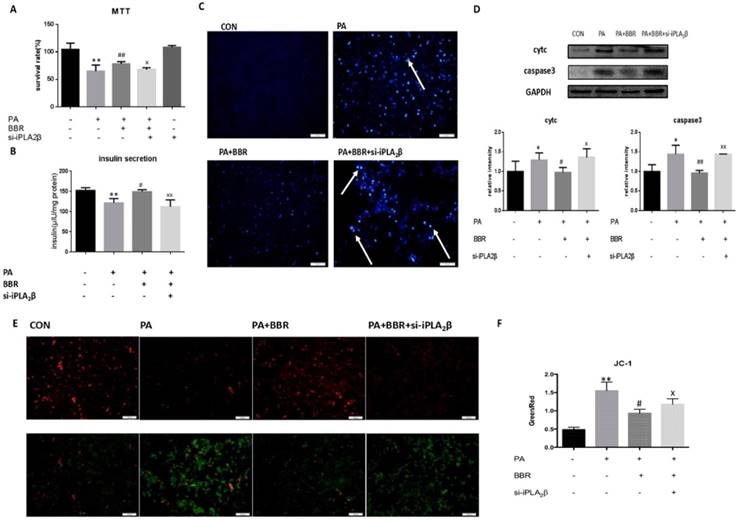
Anti-apoptotic activity, MMP maintenance and insulinotropic effect of BBR is diminished by iPLA2β silencing. To further investigate the role of iPLA2β in the hypoglycemic effect of BBR, iPLA2β-silenced cells were established and were treated with PA together with BBR. All results showed that compared to those of the PA-treated MIN6 cells, BBR could increase cell viability (Figure 3A), insulin secretion (Figure 3B) and reduce the apoptosis induced by PA in MIN6 cells by reducing the expression of caspase3 and cytc (Figure 3C-D). In addition, BBR significantly increased the red fluorescence intensity compared with that of cells treated with PA only, mitigating the loss of the MMP (Figure 3E-F). However, these effects were reduced in the iPLA2β-silenced cells (Figure 3A-F). Overall, these results demonstrated that BBR might have an anti-apoptotic effect on beta-cells via iPLA2β.
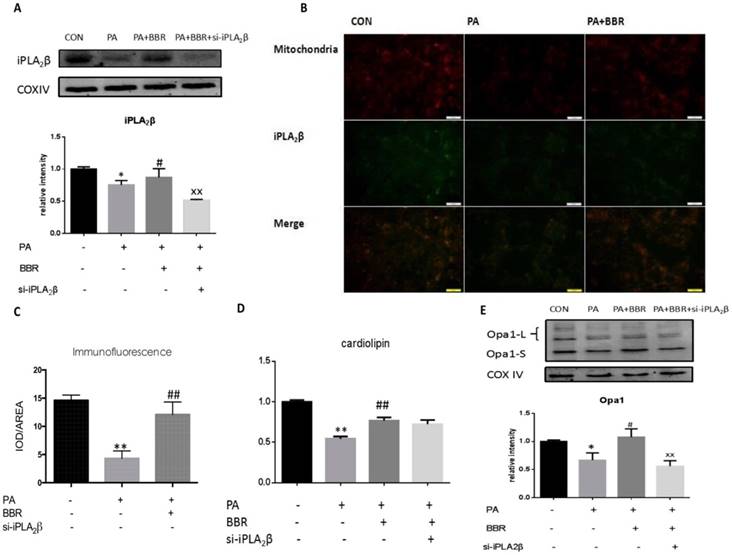
BBR may relieve PA impairment via the iPLA2β/CL/Opa1 pathway. Furthermore, we examined iPLA2β, CL and Opa1 in mitochondria after the BBR treatment of PA-induced injury. Our results showed that iPLA2β/CL/Opa1 expression in the PA group was reduced compared with that of the control group. However, iPLA2β/CL/Opa1 expression was increased compared with that of the PA group (Figure 4A-E) after treatment with BBR. However, the effect of BBR on iPLA2β/CL/Opa1 was inhibited by the silencing of iPLA2β. These data suggest that BBR inhibited apoptotic death in MIN6 cells that were treated with a high level of PA partly by activating the iPLA2β/CL/Opa1 pathway (Figure 4A-E).
iPLA2β/CL/Opa1 play an important role in protecting cells from injury induced by PA. After treated with PA in normal cells and iPLA2β over-expressed cells, mitochondria were isolated from cells and the protein was extracted from mitochondria. iPLA2β was analyzed by Western blot and analyzed(A). Distribution of iPLA2β was detected by fluorescence microscopy. Mitochondria was marked by Mito-Tracker Red and iPLA2β was marked by Green fluorescence respectively, then the two colors were merged to observe the iPLA2β's distribution and the intensity of yellow area has been analyzed via IOD/AREA (B-C) Data was expressed as means ± SEM (n=4). Compared with control group,**P<0.01; compared with PA group, ##P<0.01. Lower limit of quantitation(LLOQ) and standard sample were detected to ensure the accuracy of measurement (D,E). The Cardiolipin content of mitochondria isolated from cells was observed by HPLC/MS(F). The content of CL in samples were detected by high resolution HPLC/MS and analyzed later(G). Data was expressed as means ± SEM (n=4). Compared with control group, **P<0.01; Compared with PA group, ##P<0.01. Opa1 was detected by Western blot and analyzed(H). Data was expressed as means ± SEM (n=4). Compared with control group, **P<0.01; Compared with PA group, #P<0.05.
Anti-apoptotic activity, MMP maintenance and insulinotropic effect of BBR partly diminished by iPLA2β silence. The influence of BBR's anti-apoptosis function and the block of silence iPLA2β were assessed by MTT assay(A). Data was expressed as means ± SEM (n=6). Compared with control group, **P<0.01; compared with PA group, ##P<0.01; compared with BBR treated group in vector cells, xP<0.05. Basal insulin release and content were measured in MIN6(B). Compared with control group, **P<0.01; compared with PA group, #P<0.05; compared with BBR treated group in vector cells, xxP<0.01. After using BBR to treat PA-induced apoptosis for 24h in normal cells and iPLA2β silenced cells, the apoptosis were observed by Hoechst staining, bright blue means the apoptotic cells and marked with arrows(C). The protein was extracted from cells and cytosolic cytc and caspase3 were measured and analyzed by Western Blot(D). Data was expressed as means ± SEM (n=4). Compared with control group, *P<0.05; compared with PA group, #P<0.05, ##P<0.01; compared with BBR treated group in vector cells, xP<0.05, xxP<0.01. Mitochondrial membrane potential in MIN6 were observed by JC-1 fluorescent dying and the intensity has been analyzed, ratio of green fluorescence to red fluorescence can represent the loss of mitochondrial membrane potential(E,F) Data was expressed as means ± SEM (n=4). Compared with control group,**P<0.01; compared with PA group, #P<0.01, compared with BBR treated group in vector cells, xP<0.05.
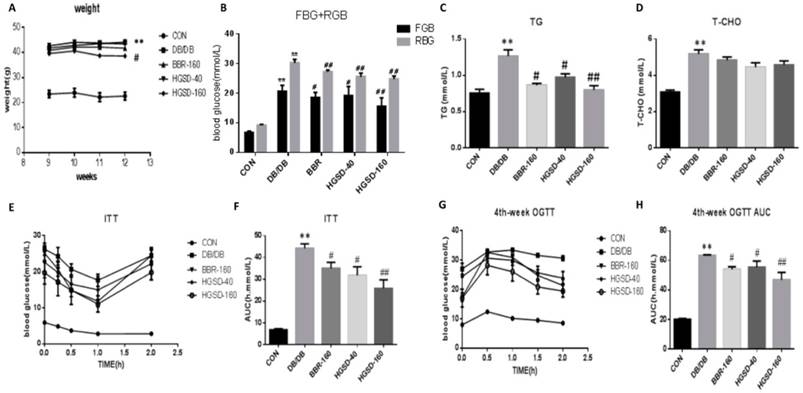
Effects of BBR and HGSD on Body Weight, blood glucose, and blood lipids in db/db mice. To test our hypothesis and to observe the effect of BBR in vivo, we conducted experiments on db/db mice, and the physiological and biochemical characteristics were examined. As we wanted to determine whether HGSD improves the antidiabetic effect of BBR and which dosage is best, we used a lower dose of HGSD and a higher dose of HGSD together with BBR. Our data show that mouse body weight, blood glucose, triglyceride levels and total cholesterol levels were significantly increased in db/db mice compared to that in the C57 mice. The HGSD‐treated db/db mice showed a significant decrease in body weight (Figure 5A) compared to that of the db/db mice. Furthermore, the FBG, RBG and TG in BBR- and HGSD-treated mice were significantly lower than those in db/db mice (Figure 5B-D), and the OGTT and ITT were significantly improved compared with those in the db/db group (Figure 5E-H). BBR- and HGSD‐treated db/db mice showed significant hypoglycemic and lipid-lowering effects and an improvement in both the OGTT and ITT compared to those in the db/db group. Additionally, the effect of the treatment with HGSD (160 mg) was greater than that of treatment with BBR (160 mg) and HGSD (40 mg).
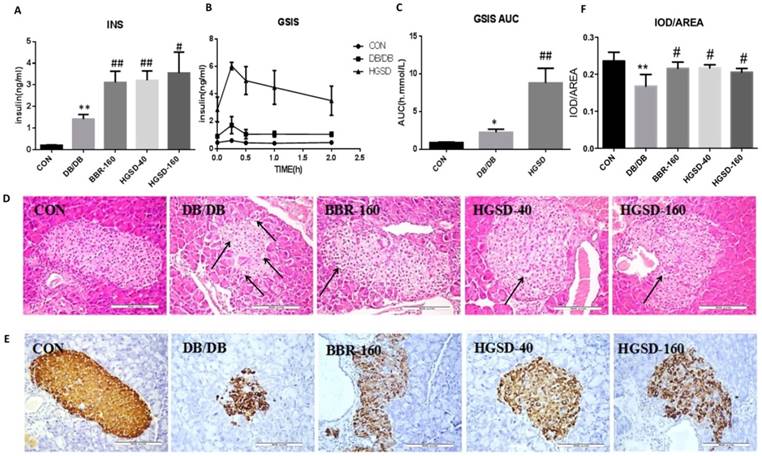
The effect of BBR and HGSD on insulin secretion and islet morphology in db/db mice. The fasting blood insulin in db/db mice was significantly higher than that in C57 mice (Figure 6A), and the GSIS and the AUC of GSIS in db/db mice was also significantly higher than that in C57 mice (Figure 6B-C). Although db/db mice had higher insulin levels than that of C57 mice, the islet cells of db/db mice showed damage in the histology study. The pancreas of C57 mice were normal and nucleus were clear, while the islet cells in db/db mice presented a disrupted distribution with the characteristic vacuolation and a large quantity of inflammatory cells had infiltrated (Figure 6D). The average insulin per unit area in the islets of db/db mice was lower compared with that of C57 mice (Figure 6E-F). However, db/db mice had smaller and more islets than those of C57 mice. Therefore, the total insulin was higher in db/db mice compared with that in C57 mice. Meanwhile, the results also showed that BBR and HGSD could effectively stimulate insulin secretion compared to that of the controls. The fasting blood insulin levels in the medicine-treated mice were higher than those in the db/db mice (Figure 6A), and the GSIS results and insulin immunohistochemical results were in accordance with these results (Figure 6B-C, 6E-F). In addition, BBR and HGSD improved the islet cell morphology by increasing the islet area, reducing inflammation and vacuolation compared to those of the db/db controls (Figure 6D). Overall, these results demonstrated that BBR and HGSD could prevent db/db islet damage and enhance the insulin secretion, especially in the mice treated with HGSD (160 mg).
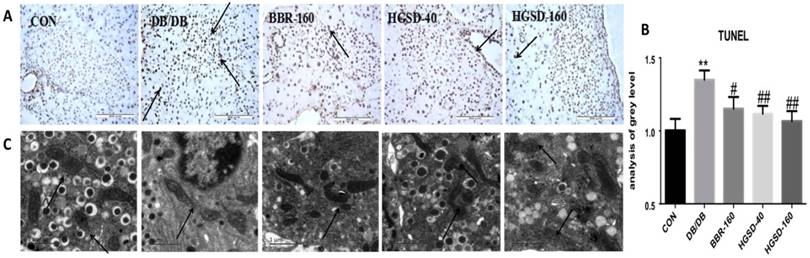
BBR and HGSD treatment Mitigated apoptosis and mitochondrial injury in the islet cells of db/db mice. TUNEL staining was used to evaluate apoptosis in mouse pancreas islets. Our data showed that the number of apoptotic cells was significantly decreased in BBR- and HGSD-treated mice compared to that in db/db mice, based on a TUNEL assay (Figure 7A-B). To investigate the injury level of the mitochondria in mouse islets, the ultrastructure of the mouse islets was examined using transmission electron microscopy. Compared with the control group, in the db/db mice, the mitochondria cristae was laxer and the shape of the mitochondria in was abnormal. Moreover, the insulin vesicle in the db/db group was more exhausted than that in the control group. After BBR and HGSD treatment, the mitochondrial cristae, the shape of mitochondria and the insulin vesicle were improved compared to those in db/db mice (Figure 7C). Additionally, the effects of the treatment with HGSD (160 mg) was greater than those of treatment with BBR(160 mg) and HGSD(40 mg), especially in regard to the anti-apoptotic effect. Therefore, a higher dose of HGSD (160 mg) was chosen for further experiments in vivo.
BBR may relieve the PA impairment via iPLA2β/CL/Opa1 pathway. After using BBR to treat PA-induced apoptosis in vector cells and iPLA2β silenced cells, mitochondria were isolated from cells and the protein was extracted from mitochondria. iPLA2β was analyzed by Western blot and analyzed(A). Compared with control group, Data was expressed as means ± SEM (n=6), *P<0.05; compared with PA group, #P<0.05; compared with BBR treated group in vector cells, xxP<0.01. Distribution of iPLA2β was detected by fluorescence microscopy. Mitochondria was marked by Mito-Tracker Red and iPLA2β was marked by Green fluorescence respectively, then the two colors were merged to observe the iPLA2β's distribution and the intensity of yellow area has been analyzed via IOD/AREA (B,C) Data was expressed as means ± SEM (n=4). Compared with control group,**P<0.01; compared with PA group, ##P<0.01. The Cardiolipin content of mitochondria isolated from cells was observed by HPLC/MS analyzed(D). Data was expressed as means ± SEM (n=4). Opa1 was extracted from mitochondria detected by Western blot and analyzed(E). Data was expressed as means ± SEM (n=4). Compared with control group, *P<0.05; **P<0.05; compared with PA group, #P<0.05, ##P<0.01; compared with BBR treated group in vector cells, xxP<0.01.
The effect of BBR and HGSD on higher weight, blood glucose, blood lipid and insulin resistance in db/db mice. Body weight was tested from 10-week to 13-week (A). At the end of 12 week, fasting blood glucose and random blood glucose were measured(B). Blood lipid included triglyceride(TG), total cholesterol(T-CHO) were measured by biochemical kits at 12-week old (C, D). Plasma glucose concentrations in different phases were measured in Oral Glucose Tolerance Test(OGTT) and Insulin tolerance test (ITT) at 12-week old(E-H). Data was expressed as means ± SEM (n=6). Compared with CON group, **P<0.01. Compared with DB/DB group, #P<0.05, ##P<0.01.
The effect of BBR and HGSD on insulin secretion and islet morphology in db/db mice at the end of 12 week. Fasting blood insulin was measured in mice serum(A). After stimulation of glucose, plasma insulin concentrations were measured in different phases during glucose stimulated insulin secretion (GSIS) (B, C). Histology were used to observe islet morphology. Islet was severely damaged in T2D mice as the images marked with arrow. (D) (200×). Pancreatic insulin level in db/db mice are measured using Immunohistochemistry (E) (200×) and insulin was analyzed. IOD/AREA means the average insulin of islet cells (F). Data was expressed as means ± SEM (n=3). Compared with CON group, **P<0.01. Compared with DB/DB group, #P<0.05, ##P<0.01.
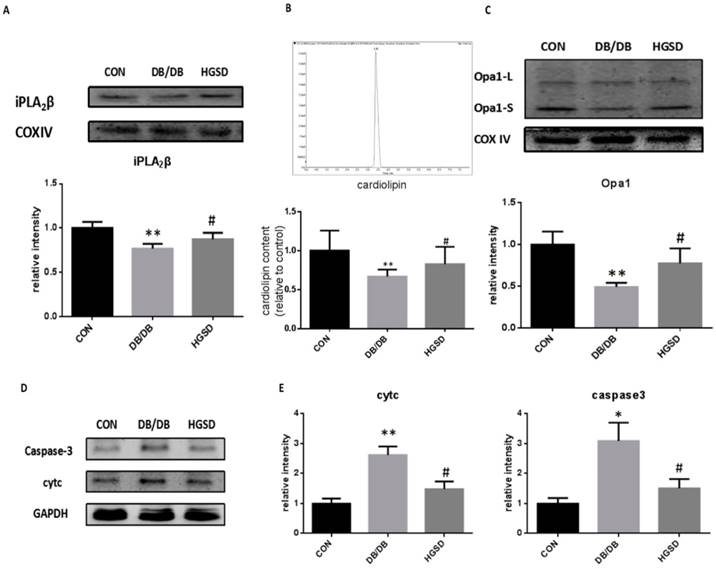
HGSD treatment increased iPLA2β, Cardiolipin and Opa1 expression in the mitochondria of islets and mitigated apoptosis by regulating cytc and caspase3 in type 2 diabetes. The iPLA2β and Opa1 expression in the mitochondria of pancreatic islets was detected by Western blotting. The content of CL was measured in mitochondria by detection with HPLC/MS. Our results showed that iPLA2β/CL/Opa1 expression in the db/db group was reduced compared with that in the control group. After treatment with HGSD, iPLA2β/CL/Opa1 expression was increased compared with that in untreated db/db mice (Figure 8A-C). In addition, both cytc and caspase3 were also significantly decreased in HGSD-treated mice compared to the untreated db/db mice. (Figure 8D-E). The results regarding the iPLA2β/CL/Opa1 levels in response to HGSD treatment were in accordance with the effect of BBR in PA-damaged MIN6 cells that occurred in response to the upregulation of iPLA2β/CL/Opa1 expression. Therefore, the anti-apoptotic and the insulinotropic effect of HGSD is related to iPLA2β/CL/Opa1.
The effect of BBR and HGSD on apoptosis of beta cells and the injury of mitochondria in db/db mice at the end of 12 week. Apoptotic cells were detected by TUNEL using brown staining(200×) and IOD/AREA has been calculated (A,B). Injury of mitochondria and insulin vesicle was showed by electron microscope images and marked with arrows.(C) Data was expressed as means ± SEM (n=3). Compared with CON group, **P<0.01. Compared with DB/DB group, #P<0.05, ##P<0.01.
HGSD improved the deterioration of iPLA2β/CL/Opa1 and mitigate apoptosis via regulating cytc and caspase3 in db/db mice islets. Mitochondria was isolated from mice pancreas and the protein was extracted from mitochondria. iPLA2β was analyzed by Western blot and analyzed (A). The Cardiolipin content of mitochondria isolated from mice pancreas was observed by HPLC/MS and the content of CL in con and db/db mice were analyzed (B). Opa1 was analyzed by Western blot and analyzed(C). Data was expressed as means ± SEM (n=4). Compared with CON group, *P<0.05, **P<0.01. Compared with DB/DB group, #P<0.05. Protein was exacted from islets. Cytosolic cytc and caspase-3 were measured by Western blot and analyzed. (D-E). Data was expressed as means ± SEM (n=4). Compared with CON group, *P<0.05, **P<0.01. Compared with DB/DB group, #P<0.05.
Discussion
Berberine (BBR), which is extracted from a traditional Chinese herb, has been shown to be effective in lowering blood sugar, alleviating insulin resistance and reducing the severity of type 2 diabetes mellitus and the diabetes-related complications. Many studies have attempted to demonstrate the potential mechanism by which BBR mitigates diabetes. For example, BBR can activate the AMPK pathway and then inhibit the synthesis of lipids (20). Furthermore, BBR may play an important role in promoting the uptake and usage of glucose (21). Ko's studies suggest that BBR can activate the IRS-1-PI3K-Akt-GLUT4 pathway and then increase glucose uptake in adipocytes, mitigating insulin resistance (22). However, how BBR affects insulin secretion remains controversial. It has been reported that BBR can protect islet cells by inhibiting apoptosis or by increasing HNF4α (18, 23). Other studies have illustrated that BBR could inhibit insulin secretion through the cAMP pathway (15, 24). Since accumulating evidence has indicated that the decrease of the pancreatic β-cell mass is a major factor contributing to the pathogenesis of diabetes and apoptosis is now considered an important contributor to the β-cell mass reduction in T2D(25), we paid special attention to the anti-apoptotic effect of BBR on pancreatic β-cells and its mechanism in vivo and in vitro. In the current study, we demonstrated that the regulation of HGSD in iPLA2β/CL/Opa1 pathway which might contribute to the inhibition of beta-cell apoptosis and might promote islet insulin release in Type 2 diabetic mice and in PA-treated MIN6 cells. The principal findings of our study include the following: 1. the overexpression of iPLA2β not only inhibited PA-induced β-cell apoptosis but also caused CL/Opa1 upregulation in MIN6 beta-cells compared to those of the PA-treated MIN6 cells; 2. iPLA2β silencing could partly weaken the anti-apoptotic effect of BBR and the BBR-induced upregulation of iPLA2β/CL/Opa1 compared to those of treated cells; and 3. the new preparation of BBR-HGSD (160 mg) has a stronger protective effect on beta-cells against apoptosis, which is related to the enhancement of the iPLA2β/CL/Opa1 pathway in islet cells.
There is increasing evidence that iPLA2β plays a key role in β-cell apoptosis and in insulin secretion. Studies have indicated that the activation of iPLA2β is a requisite for optimal glucose-stimulated insulin secretion from islet β-cells (26, 27). In a recent study, Song et al. showed that the overexpression of iPLA2β reduced the sensitivity of INS-1 cells to PA-induced apoptosis and mitochondrial injury (11). Previous studies showed that iPLA2β is involved in the remodeling of CL through its role in the repair of oxidized cardiolipin (ox-CL)(28-30), and CL is critical for mitochondrial function and the retention of cytochrome c(31, 32). Song examined the effects of iPLA2β on CL by using global iPLA2β knock-out (KO) mice and transgenic (TG) mice that overexpressed iPLA2β in pancreatic islet β-cells(9). It has been proven that the β-cell monolysocardiolipin (MLCL) content increased with increasing iPLA2β expression, and iPLA2β contributes to CL remodeling by excising oxidized residues from oxidized CL to yield MLCL species for reacylation with unoxidized C18:2-CoA to regenerate the native CL structure and function. In our study, we developed a Palmitate(PA)- induced apoptotic death model in mouse insulinoma cells (MIN6), and we treated cells that were overexpressing iPLA2β to explore the anti-apoptotic mechanism of iPLA2β. Our results further demonstrated that the overexpression of iPLA2β not only inhibited beta-cell apoptosis but also alleviated mitochondrial injury via CL upregulation. However, the mechanism by which iPLA2β/CL regulates cytc to exert its anti-apoptotic effect in diabetes beta-cells is still unknown.
Recent studies showed that CL could regulate mitochondrial dynamics by promoting the functional dimer formation of Opa1 in a CL-dependent manner (11, 33, 34). Electron tomography showed that Opa1 regulates the shape and length of mitochondrial cristae and keeps the cristae junctions tight, which is very important during apoptosis for the regulation of the mobilization of cytc to the IMS following BID treatment (35). Moreover, a previous study showed that CL oxidation destabilizes its interaction with cytc, which enables cytc to detach from the membrane and to be released into the cytoplasm through pores in the outer membrane (36, 37). Thus, cytc can be regulated by Opa1, which is involved in the development of diabetes and other metabolic diseases (5, 38, 39). Opa1, in turn, is regulated by CL. In our study, we showed that iPLA2β overexpression can reverse the decrease of Opa1 and cytc in PA-treated MIN6 cells, along with the increase of CL, for the first time, and that silencing iPLA2β can slightly aggravate the injury induced by PA (shown in the supplementary data). These data indicate that Opa1 regulation is involved in the interaction between iPLA2β and CL, and iPLA2β/CL/Opa1 degradation plays a key role in beta-cell dysfunction. The regulation of the iPLA2β/CL/Opa1 pathway provides a new therapeutic target for the treatment of T2D.
To further elucidate the specific mechanism of iPLA2β in the anti-apoptotic effect of BBR, we used iPLA2β silencing technology to investigate the relationship of iPLA2β with CL and Opa1 in a PA-induced apoptotic model. We generated iPLA2β-silenced cells and treated the cells with PA and BBR. The results showed that compared to non-transfected cells, the anti-apoptotic effect of BBR was weakened and the insulinotropic effect was also reduced, which indicated that BBR could inhibit apoptosis by regulating iPLA2β. We also found that when iPLA2β was silenced, CL/Opa1 upregulation with BBR was decreased at the same time, further indicating that BBR attenuates beta-cell apoptosis by enhancing the iPLA2β/CL/Opa1 signaling pathway.
To confirm the mechanism in vivo, a mouse with a functional defect in the long-form leptin receptor named db/db mice, which developed obesity and hyperglycemia, was used as a T2D animal model to validate the above mentioned hypothesis. Our data showed that the diabetic model was successfully developed. The beta-cells were damaged in db/db mice, and mitochondrial cristae remodeling was involved in β-cell apoptosis. Moreover, our data showed that both an abnormality of iPLA2β/CL/Opa1 and mitochondrial-triggered apoptosis in beta-cells were involved in T2D mice that were not observed in the control group. These data further demonstrated that iPLA2β/CL/Opa1 damage contributed to beta-cell apoptosis in T2D mice. After 4 weeks of treatment, compared to that of the other treatment groups, HGSD (160 mg) exhibited greater hypoglycemic properties and beta-cell protection in db/db mice, and the expression of iPLA2β, CL and Opa1 were all increased in the mitochondria of islet cells. Therefore, the effect of HGSD on beta-cell anti-apoptosis in T2D is related to iPLA2β/CL/Opa1 upregulation.
In summary, our results showed that the iPLA2β/CL/Opa1 signaling pathway exerted a protective role in beta-cell apoptosis, which could provide a novel therapeutic option for type 2 diabetes. The HGSD regulation of iPLA2β/CL/Opa1 contributes to the inhibition of beta-cell apoptosis and the improvement of islet insulin release in T2D mice. Given these findings, our studies have important implications for the use of HGSD as a therapeutic agent in T2D.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81503122), Science and technology projects of the Education Department of Jilin Province (JJKH20180248KJ) , Preclinical Pharmacology R&D Center of Jilin Province, Science and technology development projects of Jilin Province (20170623062TC, 20180201025YY) and Norman Bethune Program of Jilin University (2015224). Thanks are given to Tapas Ranjan Behera for assistance with literary revisions (Internal Medicine, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, USA).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Smyth S, Heron A. Diabetes and obesity: the twin epidemics. Nat Med. 2006;12(1):75-80
2. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102-10
3. Rhodes CJ. Type 2 diabetes-a matter of beta-cell life and death?. Science. 2005;307(5708):380-4
4. Lei X, Zhang S, Barbour SE, Bohrer A, Ford EL, Koizumi A. et al. Spontaneous development of endoplasmic reticulum stress that can lead to diabetes mellitus is associated with higher calcium-independent phospholipase A2 expression: a role for regulation by SREBP-1. J Biol Chem. 2010;285(9):6693-705
5. Zhao Z, Zhang X, Zhao C, Choi J, Shi J, Song K. et al. Protection of pancreatic beta-cells by group VIA phospholipase A(2)-mediated repair of mitochondrial membrane peroxidation. Endocrinology. 2010;151(7):3038-48
6. Lei X, Bone RN, Ali T, Wohltmann M, Gai Y, Goodwin KJ. et al. Genetic modulation of islet beta-cell iPLA(2)beta expression provides evidence for its impact on beta-cell apoptosis and autophagy. Islets. 2013;5(1):29-44
7. Lei X, Zhang S, Bohrer A, Barbour SE, Ramanadham S. Role of calcium-independent phospholipase A(2)beta in human pancreatic islet beta-cell apoptosis. Am J Physiol Endocrinol Metab. 2012;303(11):E1386-95
8. Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, Jones SS. A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J Biol Chem. 1997;272(13):8567-75
9. Song H, Wohltmann M, Tan M, Ladenson JH, Turk J. Group VIA phospholipase A2 mitigates palmitate-induced beta-cell mitochondrial injury and apoptosis. J Biol Chem. 2014;289(20):14194-210
10. Unsay JD, Cosentino K, Subburaj Y, Garcia-Saez AJ. Cardiolipin effects on membrane structure and dynamics. Langmuir. 2013;29(51):15878-87
11. Joshi AS, Thompson MN, Fei N, Huttemann M, Greenberg ML. Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J Biol Chem. 2012;287(21):17589-97
12. Molina AJ, Wikstrom JD, Stiles L, Las G, Mohamed H, Elorza A. et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes. 2009;58(10):2303-15
13. Stiles L, Shirihai OS. Mitochondrial dynamics and morphology in beta-cells. Best Pract Res Clin Endocrinol Metab. 2012;26(6):725-38
14. Zhang Z, Wakabayashi N, Wakabayashi J, Tamura Y, Song WJ, Sereda S. et al. The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol Biol Cell. 2011;22(13):2235-45
15. Perez-Rubio KG, Gonzalez-Ortiz M, Martinez-Abundis E, Robles-Cervantes JA, Espinel-Bermudez MC. Effect of berberine administration on metabolic syndrome, insulin sensitivity, and insulin secretion. Metab Syndr Relat Disord. 2013;11(5):366-9
16. Hua W, Ding L, Chen Y, Gong B, He J, Xu G. Determination of berberine in human plasma by liquid chromatography-electrospray ionization-mass spectrometry. J Pharm Biomed Anal. 2007;44(4):931-7
17. Zhaojie M, Ming Z, Shengnan W, Xiaojia B, Hatch GM, Jingkai G. et al. Amorphous solid dispersion of berberine with absorption enhancer demonstrates a remarkable hypoglycemic effect via improving its bioavailability. Int J Pharm. 2014;467(1-2):50-9
18. Gao N, Zhao TY, Li XJ. The protective effect of berberine on beta-cell lipoapoptosis. J Endocrinol Invest. 2011;34(2):124-30
19. Chang W, Zhang M, Chen L, Hatch GM. Berberine Inhibits Oxygen Consumption Rate Independent of Alteration in Cardiolipin Levels in H9c2 Cells. Lipids. 2017;52(11):961-7
20. Zhou L, Yang Y, Wang X, Liu S, Shang W, Yuan G. et al. Berberine stimulates glucose transport through a mechanism distinct from insulin. Metabolism. 2007;56(3):405-12
21. Brusq JM, Ancellin N, Grondin P, Guillard R, Martin S, Saintillan Y. et al. Inhibition of lipid synthesis through activation of AMP kinase: an additional mechanism for the hypolipidemic effects of berberine. J Lipid Res. 2006;47(6):1281-8
22. Ko BS, Choi SB, Park SK, Jang JS, Kim YE, Park S. Insulin sensitizing and insulinotropic action of berberine from Cortidis rhizoma. Biol Pharm Bull. 2005;28(8):1431-7
23. Wang ZQ, Lu FE, Leng SH, Fang XS, Chen G, Wang ZS. et al. Facilitating effects of berberine on rat pancreatic islets through modulating hepatic nuclear factor 4 alpha expression and glucokinase activity. World J Gastroenterol. 2008;14(39):6004-11
24. Zhou L, Wang X, Shao L, Yang Y, Shang W, Yuan G. et al. Berberine acutely inhibits insulin secretion from beta-cells through 3',5'-cyclic adenosine 5'-monophosphate signaling pathway. Endocrinology. 2008;149(9):4510-8
25. Thomas HE, McKenzie MD, Angstetra E, Campbell PD, Kay TW. Beta cell apoptosis in diabetes. Apoptosis. 2009;14(12):1389-404
26. Ramanadham S, Gross R, Turk J. Arachidonic acid induces an increase in the cytosolic calcium concentration in single pancreatic islet beta cells. Biochem Biophys Res Commun. 1992;184(2):647-53
27. Ramanadham S, Gross RW, Han X, Turk J. Inhibition of arachidonate release by secretagogue-stimulated pancreatic islets suppresses both insulin secretion and the rise in beta-cell cytosolic calcium ion concentration. Biochemistry. 1993;32(1):337-46
28. Nigam S, Schewe T. Phospholipase A(2)s and lipid peroxidation. Biochim Biophys Acta. 2000;1488(1-2):167-81
29. Wiswedel I, Gardemann A, Storch A, Peter D, Schild L. Degradation of phospholipids by oxidative stress-exceptional significance of cardiolipin. Free Radic Res. 2010;44(2):135-45
30. Zachman DK, Chicco AJ, McCune SA, Murphy RC, Moore RL, Sparagna GC. The role of calcium-independent phospholipase A2 in cardiolipin remodeling in the spontaneously hypertensive heart failure rat heart. J Lipid Res. 2010;51(3):525-34
31. Salamon Z, Tollin G. Interaction of horse heart cytochrome c with lipid bilayer membranes: effects on redox potentials. J Bioenerg Biomembr. 1997;29(3):211-21
32. Shidoji Y, Hayashi K, Komura S, Ohishi N, Yagi K. Loss of molecular interaction between cytochrome c and cardiolipin due to lipid peroxidation. Biochem Biophys Res Commun. 1999;264(2):343-7
33. Ban T, Heymann JA, Song Z, Hinshaw JE, Chan DC. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum Mol Genet. 2010;19(11):2113-22
34. DeVay RM, Dominguez-Ramirez L, Lackner LL, Hoppins S, Stahlberg H, Nunnari J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J Cell Biol. 2009;186(6):793-803
35. Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T. et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126(1):177-89
36. Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA. et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1(4):223-32
37. Orrenius S, Zhivotovsky B. Cardiolipin oxidation sets cytochrome c free. Nat Chem Biol. 2005;1(4):188-9
38. Li J, Romestaing C, Han X, Li Y, Hao X, Wu Y. et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010;12(2):154-65
39. Han X, Yang J, Cheng H, Yang K, Abendschein DR, Gross RW. Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry. 2005;44(50):16684-94
Author contact
![]() Corresponding authors: Li Chen, Tel: +86-431-85619366; E-mail: submitchencom or Fengying Guan, Tel: +86-431-85619799; E-mail: guanfyedu.cn
Corresponding authors: Li Chen, Tel: +86-431-85619366; E-mail: submitchencom or Fengying Guan, Tel: +86-431-85619799; E-mail: guanfyedu.cn
Received 2018-12-5
Accepted 2019-4-12
Published 2019-6-2