ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2019; 15(8):1618-1629. doi:10.7150/ijbs.33323 This issue Cite
Research Paper
Tripartite Motif 8 Deficiency Relieves Hepatic Ischaemia/reperfusion Injury via TAK1-dependent Signalling Pathways
Qiu Tao*, Wang Tianyu*, Zhou Jiangqiao ![]() , Chen Zhongbao, Ma Xiaoxiong, Zhang Long, Zou Jilin
, Chen Zhongbao, Ma Xiaoxiong, Zhang Long, Zou Jilin
Department of Organ Transplantation, Renmin Hospital of Wuhan University, Wuhan University, Wuhan, 430060, China.
*These authors contributed equally to this work as co-first authors in this paper.
Abstract

Tripartite motif (Trim) 8 is an E3 ubiquitin ligase, interacting with and ubiquitinating diverse substrates, and is closely involved in innate immunity. However, the function of Trim8 in hepatic ischaemia/reperfusion (I/R) injury remains largely unknown. The aim of this study is to explore the role of Trim8 in hepatic I/R injury. Trim8 gene knockout mice and primary hepatocytes were used to construct hepatic I/R models. The effect of Trim8 on hepatic I/R injury was analysed via pathological and molecular analyses. The results indicated that Trim8 was significantly upregulated in liver of mice subjected to hepatic I/R injury. Trim8 knockout relieved hepatocyte injury triggered by I/R. Silencing of Trim8 expression alleviated hepatic inflammation responses and inhibited apoptosis in vitro and in vivo. Mechanistically, our study suggests that Trim8 deficiency may elicit hepatic protective effects by inhibiting the activation of transforming growth factor β-activated kinase 1 (TAK1)-p38/JNK signalling pathways. TAK1 was required for Trim8 function in hepatic I/R injury as TAK1 activation abolished Trim8 function in vitro. In conclusion, our study demonstrates that Trim8 deficiency plays a protective role in hepatic I/R injury by inhibiting the activation of TAK1-dependent signalling pathways.
Keywords: Trim8, Ischaemia/reperfusion, Inflammation, Apoptosis, Hepatology, Ubiquitination
Introduction
Ischaemic reperfusion (I/R) occurs during surgical procedures, including hepatic resection and liver transplantation (LT) [1]. Hepatic I/R is an important cause of liver damage and is also the underlying cause of graft dysfunction in marginal organs. Hepatic I/R injury can result in loss of viability of a liver graft, which is directly correlated with the recipient's survival [2].
I/R is a common pathological disorder leading to acute liver injury, a potentially life-threatening clinical condition with high morbidity, mortality, and prolonged patient hospitalization. Unfortunately, there are no good treatments to improve hepatic I/R injury. Hepatic I/R injury is characterized by endothelial and Kupffer cell swelling, vasoconstriction, leukocyte infiltration, and platelet aggregation in sinusoids [3]. The end result is microcirculatory failure. Activation of Kupffer cells and neutrophils leads to release of inflammatory cytokines and free radicals, which further aggravate liver injury [4, 5]. This concept has led to intense efforts to develop agents with direct anti-inflammatory and anti-apoptotic properties; however, to date, there is still no effective drug for the treatment of I/R in clinical practice [6].
Tripartite motif (Trim) 8, a member of the Trim family, is defined by the presence of a common domain structure composed of a tripartite motif that includes a RING-finger domain, one or two B-box domains, and a coiled-coil motif. The Trim8 gene maps to chromosome 10 within a region which is frequently deleted or rearranged in tumours and transcribes a 3.0-kB mRNA transcript [7]. Trim8 expression is mostly ubiquitous in murine and human tissues, and in epithelial and lymphoid cells, which can be induced by IFN-γ. Trim8 functions as an E3 ubiquitin ligase that interacts with and ubiquitinates diverse substrates. Previous study on Trim8 showed it is implicated in various pathological processes, including inflammation, autophagy and cancer [8-10]. Trim8 promotes K63-linked polyubiquitination of transforming growth factor β-activated kinase 1 (TAK1), leading to the activation of TAK1 and enhanced inflammatory responses in Pseudomonas aeruginosa induced keratitis [9]. In nonalcoholic steatohepatitis, Trim8 directly binds to and ubiquitinates TAK1, thus promoting its phosphorylation and the activation of downstream c-Jun N-terminal kinase/p38 and nuclear factor κB signalling [11]. Trim8 also contributes to pathological cardiac hypertrophy through enhancing TAK1-dependent signalling [12]. Previous research also showed that TAK1 is closely related to liver ischaemia and reperfusion [13]. Therefore, we speculated that Trim8 could regulate hepatic ischemia reperfusion injury by regulating TAK1 function.
In the present study, we found that Trim8 protein expression was significantly increased in livers after I/R injury, indicating that Trim8 may play an important role in the pathogenesis of hepatic I/R injury. Using Trim8 knockout mice, we demonstrated that Trim8 deficiency alleviated hepatic I/R injury by suppressing inflammation and apoptosis. Mechanistically, we found that Trim8 deficiency blocked activation of the TAK1-JNK/p38 signalling axis. Collectively, this work reveals a novel function of Trim8 in hepatic I/R injury and suggests that Trim8 may serve as a drug target for hepatic I/R injury.
Materials and methods
Reagents
Antibody against Trim8 was purchased from ProSci Incorporated (25-217). Antibodies against p-IkBα (9246), IkBα (4814), p65 (4764), p-p65 (3033) p-Ikkβ (2078), Ikkβ (8943), Bax (2772), Bcl2 (3498), C-Caspase3 (9664), p-ERK (4370), ERK (4695), p-JNK (4668), JNK (9252), p-p38 (4511), p38 (9212), p-ASK1 (3765), p-TAK1 (4531), TAK1 (4505) and GAPDH (2118) were purchased from Cell Signalling Technology. Antibody against ASK1 was purchased from Abclonal (A6274). Goat anti-mouse (115-035-003) and goat anti-rabbit (111-035-003) secondary antibodies were purchased from Jackson Laboratory. Foetal calf serum was purchased from HyClone. Cell culture reagents and all other reagents were purchased from Sigma.
Animal models and procedures
All the animal experimental protocols were approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University. Animal maintenance was in accordance with Animal Experiment Center of Wuhan University standard guidelines. The experiments were conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Mice were kept in an air-filtered, temperature-controlled (22-24°C), light-controlled room with humidity between 40 and 70% and were permitted free access to a standard diet.
Trim8 gene knockout mice were generated according to a previously described protocol [12]. The mouse hepatic I/R injury model was constructed according to the classical method with little modification [14]. A ventral midline incision was made into the abdomen to expose the hepatic pedicles of the left and middle lobe of the liver. The portal vein and hepatic artery of the middle and left lobes were blocked with non-invasive vascular clamps, resulting in approximately 70% hepatic ischaemia to prevent severe mesenteric vein congestion. After 0.5 min, compared with the non-blocked right lobe, the blocked lobes turned white, indicating successful blood flow blockage. No hepatic blood flow blockage was performed in Sham group mice.
After 60 min of ischaemia, the vascular clamps were removed, and the hepatic blood flow was restored. Sampling: Mice in the sham operation group (sham operation group) and ischemia reperfusion group were anesthetized with 3% pentobarbital sodium at 6 h and 24 h after surgery.1ml of orbital venous plexus blood was taken and serum was separated. Meanwhile, the left liver lobe of the ischemic area was collected, frozen in liquid nitrogen or fixed in 10% formalin medium for 24 hours, and paraffin sections were embedded after dehydration.
Isolation, culture and in vitro hypoxia/reoxygenation (H/R) of primary hepatocytes
Primary hepatocytes were isolated from 6-8-week-old male mice via type IV collagenase digestion as described previously [14, 15]. After anaesthesia, the mouse abdominal cavity was opened, and 0.5% type IV collagenase (catalogue no. 17104-019, Thermo Fisher Scientific) was injected into the portal vein. When the liver was completely digested, the liver was then excised, minced, filtered, and centrifuged to obtain primary hepatocytes. The cells were cultured in DMEM (catalogue no. 11965092; Invitrogen) supplemented with 10% FBS in plates coated with rat tail collagen at 37 ℃ with 5% CO2.
In order to establish an in vitro ischemia reperfusion model, the primary hepatocytes were divided into Control group and H/R group. The hepatocytes in the Control group were cultured in complete medium at 37℃ and 5% CO2, while the hepatocytes in the H/R group were cultured with sugar-free and serum-free DMEM in the O2 / CO2 cell culture system in (37℃ and 5% CO2, 1% O2) for hypoxia training for 6 hours. After completion of the hypoxia treatment, the H/R group was restored under normal conditions for the indicated time, similar to the Control group.
Recombinant adenovirus production and transfection
Recombined adenoviruses mediating gene expression were constructed as previously described [16]. Briefly, AdCaTAK1 was generated to overexpress the constitutively active form of TAK1 (CaTAK1), and AdGFP was used as control. Primary hepatocytes were infected with the recombinant adenoviruses at a multiplicity of infection (MOI) of 100 for 24 h.
Serum biochemical analysis
Serum alanine aminotransferase (ALT), serum aspartate aminotransferase (AST) and alkaline phosphatase (ALP) levels in the serum were detected with an ADVIA 2400 biochemical analyser (Siemens, Tarrytown, NY, USA).
The inflammation markers tumour necrosis factor TNF-α and Ccl2 in the serum were measured according to the standard procedure recommended by the kit manufacturers (Murine TNF-α Standard ABTS ELISA Development Kit, Peprotech, 900-K54; Mouse CCL2 Quantikine ELISA Kit, R&D Systems, MJE00).
Pathological analysis
HE staining
The liver was excised, fixed with 10% phosphate-buffered formalin, embedded in paraffin, and sectioned to a 5-μm thickness according to the standard procedure. We deparaffinized and gradually hydrated the sections before examining them with HE staining. A pathologist who was blinded to the experimental protocol provided the morphological assessments.
TUNEL staining
In order to detect apoptosis induced by I/R, we used an in situ apoptosis detection kit (ApopTag® Plus In Situ Apoptosis Fluorescein Detection Kit, 11684817910; Roche). TUNEL analysis was performed according to the kit's instructions. After observing the TUNEL-positive cells under a fluorescence microscope (version 2.2, Olympus), we evaluated the number of TUNEL-positive cells per high field of the tissue section using a quantitative digital analysis imaging system, Image-Pro Plus (version 6.0).
Immunofluorescence
The infiltration of inflammatory cells into the liver sections was detected via immunofluorescence staining by applying primary anti-CD11b antibody (ab75476, Abcam), anti-Ly6g antibody (ab25024, Abcam), anti-F4/80 antibody (MCA497, Serotec). After incubation with primary antibody overnight at 4°C, liver sections were incubated with fluorescence-labelled secondary antibodies (Goat anti-rabbit-IgG, A-11011, Thermo Fisher Scientific; Conjugate Anti-rat IgG, 4417, Cell Signalling Technology) for 1 hour. Liver slices were washed and then mounted with DAPI-Fluoromount-G (Thermo Fisher Scientific). Immunofluorescence images were obtained using a fluorescence microscope with DP2-BSW software (version 2.2, Olympus). Positive-staining cells in the images were quantified using Image-Pro Plus software (version 6.0).
Western blotting
Protein was extracted from the liver tissues and primary hepatocytes according to standard operating procedures. Protein concentrations were detected using a BCA Protein Assay kit (Thermo Fisher Scientific, 23225). In short, the protein samples were separated on 12.5% a sodium dodecyl sulphate-polyacrylamide gel and then transferred them to a nitrocellulose membrane. The membrane were blocked using 5% non-fat dry milk in a TBS-T buffer and incubated it overnight at 4°C with primary antibody. After the blots were rinsed extensively with TBS-T buffer, they were incubated with HRP-conjugated secondary antibody and developed using an enhanced chemiluminescence system, and images were captured on a light-sensitive imaging film.
qPCR
Total RNA was extracted from cultured hepatocytes and hepatic tissues using TRIzol reagent (T9424, Sigma) according to the manufacturer's instructions. After RNA was reverse transcribed into cDNA using a Transcriptor First Strand cDNA Synthesis Kit (R312-02, vazyme), quantitative real-time PCR amplification was performed using SYBR Green PCR Master Mix (R323-01, vazyme). Each experiment was performed in triplicate, and the results were calculated by normalizing the expression of β-actin in the mean gene expression. The PCR reactions were performed using the following cycling conditions: incubation at 95℃ for 10 min, followed by 40 cycles of 95℃ for 10 s and 60℃ for 1 min. The relative mRNA expression levels were calculated using the 2-ΔΔCt method and were normalized against β-actin. All the primers information are listed in Table 1. These data represent the means of three experiments.
Primer information
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Trim8 | GTGGAGATACGGAGGAATGAGA | TGGTGCAGCTTTTCGTACTGC |
| IL1β | CCGTGGACCTTCCAGGATGA | GGGAACGTCACACACCAGCA |
| IL6 | TAGTCCTTCCTACCCCAATTTCCTACA | TTGGTCCTTAGCCACTCCTTC |
| Ccl2 | AGAGGATCACCAGCAGC | ACCTTAGGGCAGATGCAGTT |
| Ccl5 | TGCTGCTTTGCCTACCTCTC | TCTTCTCTGGGTTGGCACAC |
| Cxcl2 | GCGCCCAGACAGAAGTCATA | CAGTTAGCCTTGCCTTTGTTCA |
| Bad | CCAGAGTTTGAGCCGAGTGAGCA | ATAGCCCCTGCGCCTCCATGAT |
| Bax | TGAGCGAGTGTCTCCGGCGAAT | GCACTTTAGTGCACAGGGCCTTG |
| Bcl2 | TGGTGGACAACATCGCCCTGTG | GGTCGCATGCTGGGGCCATATA |
| β-actin | GTGACGTTGACATCCGTAAAGA | GCCGGACTCATCGTACTCC |
Statistical analysis
All data are presented as the mean ± standard error. The SPSS 19.0 software was applied for all statistical analyses. Statistical differences among more than 2 groups were examined using one-way ANOVA, followed by Bonferroni analysis (for data meeting homogeneity of variance) or Tamhane's T2 analysis (for data demonstrating heteroscedasticity). Statistical differences between two groups were examined with a two-tailed Student's t-test. Differences were considered statistically significant when P < 0.05.
Results
Hepatic ischaemia and reperfusion induces upregulation of Trim8 protein expression in vivo and in vitro
To explore whether Trim8 was associated with hepatic I/R, we constructed the hepatic I/R model in mouse and in primary hepatocytes. In a mouse model of hepatic I/R injury, Trim8 protein expression was upregulated in the liver samples at 6 hours after reperfusion (Fig. 1A). However, the mRNA expression of Trim8 was not affected whether 3 or 6 hours after reperfusion (Fig. 1B), indicating that the regulation of Trim8 protein expression after hepatic I/R injury may be dependent on posttranslational modification. The protein expression of Trim8 was also consistently significantly increased in primary hepatocytes at 6 hours after H/R (Fig. 1C). These results suggested that Trim8 is involved in the pathogenesis of hepatic I/R injury.
Trim8 deficiency relieves hepatic I/R injury
To explore the function of Trim8 on I/R induced liver damage, the Trim8 knockout mice were generated (Fig. 2A). Trim8 knockout (KO) mice and wild-type (WT) mice were used to construct a hepatic I/R model. Compared with WT mice, the serum levels of ALT, AST and ALP in the Trim8 KO mice were significantly decreased at 6 hours and 24 hours after reperfusion (Fig. 2 B-D). According to the result of HE staining, compared with the WT group with I/R, the necrotic area in livers was significantly reduced in the Trim8 KO group with I/R (Fig. 2E). These results demonstrate that Trim8 deficiency relieves hepatic I/R injury.
Trim8 expression is upregulated in hepatic I/R models in vivo and vitro. (A) Western blot analysis (Left) and quantification (right) of TRIM8 protein levels in livers from wild type mice with sham treatment or ischemia for 1h followed by reperfusion for 6h (n=4 per group). (B) qPCR analysis of Trim8 mRNA levels in livers from wild type mice with sham treatment or ischemia for 1h followed by reperfusion for 3h and 6h (n=4 per group). (C) Western blot analysis (Left) and quantification (right) of Trim8 expression in cultured primary hepatocytes after H/R stimuli for 6 h, n=3 independent experiments. All data are shown as the mean ± SD. n.s., not significant; **P < 0.01.
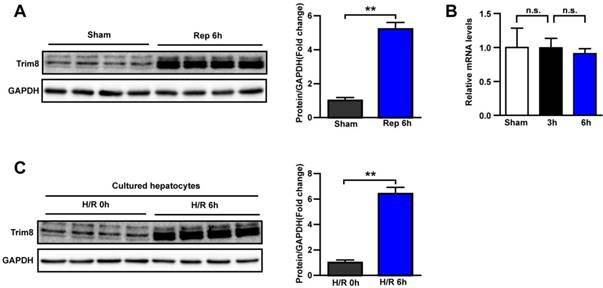
Trim8 deficiency relieves liver injury in an ischaemia/reperfusion model. (A) Trim8 protein levels in livers from wild type (WT) and Trim8-knockout (KO) mice (n=4 per group). (B-D) The levels of ALT, AST and ALP in the serum of WT and Trim8 KO mice at 6 h and 24 h after I/R (n=8 per group). (E) Representative H&E staining images (Left) and quantification of the necrotic areas in ischaemic liver lobes of the indicated mice at 6 or 24h after I/R (Scale bar:100μm, n=6 per group). *P < 0.05; **P < 0.01. All data are shown as the mean ± SD.
Trim8 deficiency inhibits liver inflammation during hepatic I/R
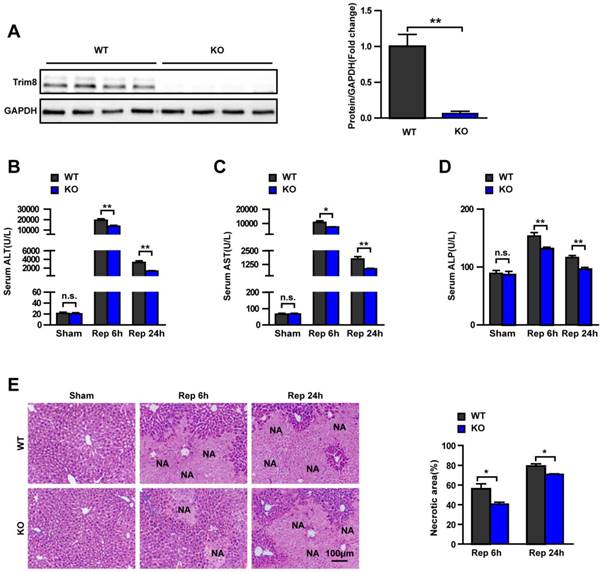
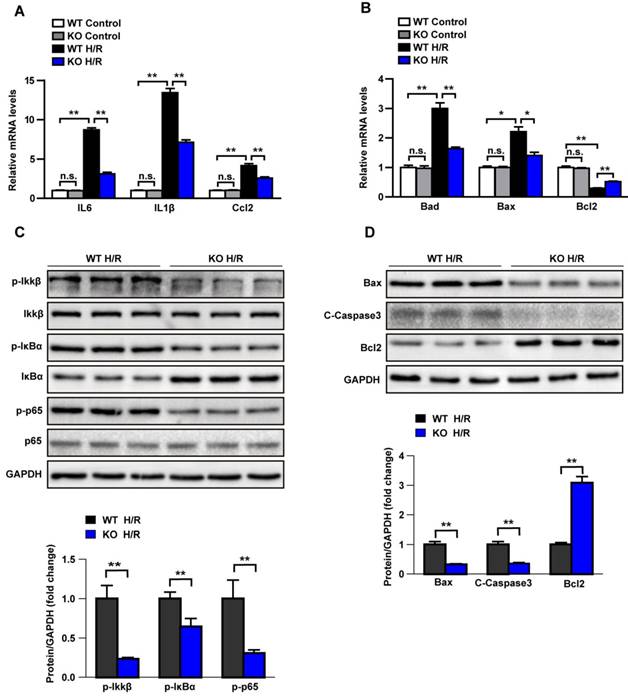
During the development of I/R, multiple inflammatory cytokines are secreted and the NF-κB pathway is activated [4]. Moreover, Previous investigations have revealed that Trim8 plays an important role in regulating inflammatory responses and NF-κB signaling pathways[8].To further evaluate the effect of Trim8 on inflammation responses during hepatic I/R injury, the relative mRNA levels of IL6, IL1β, Ccl2, Ccl5 and Cxcl2 were examined in the livers from WT and KO mice at 6h post reperfusion and the result showed that the mRNA levels of these cytokines significantly decreased in KO mice (Fig. 3A). Immunofluorescence staining showed that CD11b+ cells infiltration in the liver tissue of KO group was significantly less than that of the WT group at 6h post-reperfusion (Fig. 3B). Compared with WT group at 24h post-reperfusion, the TNF-α and Ccl2 concentration in the serum was significantly reduced in KO group (Fig. 3C), the mRNA expression of inflammatory factors, such as IL1β, IL6, Ccl2, Ccl5 and Cxcl2, was also dramatically reduced (Fig. 3D). In addition, the infiltration of CD11b, Ly6g and F4/80 positive cells in liver tissue was decreased by Trim8 deficiency (Fig. 3E). Western blotting showed that the classic NF-κB pro-inflammatory signalling pathway was dramatically inhibited by Trim8 deficiency, which was demonstrated by the significant reduction in activation of p-IκBα, p-p65 and p-IKKβ proteins (Fig. 3F). The above results indicated that the Trim8 deficiency inhibits inflammatory response during hepatic I/R injury.
Trim8 deficiency alleviates apoptosis after hepatic I/R in vivo
Apoptosis also plays an important role in hepatic I/R injury. Apoptosis features in liver were measured in WT and KO mice after hepatic I/R. TUNEL staining showed that Trim8 deficiency dramatically alleviated hepatocyte apoptosis (Fig. 4A). Western blot results validated the up regulation of anti-apoptotic protein Bcl2 and the down regulation of pro-apoptotic protein Bax and C-Caspase3 in the livers of Trim8 KO mice compared with WT mice at 24 hours after I/R (Fig. 4B). Moreover, RT-PCR was applied into detect the mRNA expression of apoptosis signalling pathway factors. The results showed that the mRNA expression of the pro-apoptotic Bax and Bad was decreased and the expression of the anti-apoptotic Bcl2 was significantly increased in the KO group at 24 h after I/R (Fig. 4C).
Trim8 deficiency relieves the inflammation response after hepatic I/R. (A) mRNA levels of IL6, IL1β, Ccl2, Ccl5 and Cxcl2 in WT and KO group at 6h after I/R (n=4 per group). (B) Immunofluorescence staining (20×) of CD11b-positive inflammatory cells (red) in ischaemic liver sections from mice in the indicated groups (n=6 per group). (C) Expression levels of TNF-α and Ccl2 in the serum of WT and KO mice 24 h after I/R treatment (n=8 per group). (D) mRNA levels of cytokines and chemokines were determined by qPCR in liver tissues from mice in the KO and WT groups at 24 h after hepatic I/R (n=4 per group). (E) Representative immunofluorescence staining (20×) of CD11b, Ly6g and F4/80-positive inflammatory cells (red) in ischaemic liver sections from mice in the indicated groups. The nuclei were labelled with DAPI (blue) (n=6 per group). (F) Western blot analysis of the activation of NF-κB signalling in livers from Trim8-KO or WT mice, which were challenged by I/R stimulation for 24 h (n=4 per group). GAPDH served as the loading control. All data are shown as the mean ± SD. n.s., not significant; *P < 0.05; **P < 0.01.
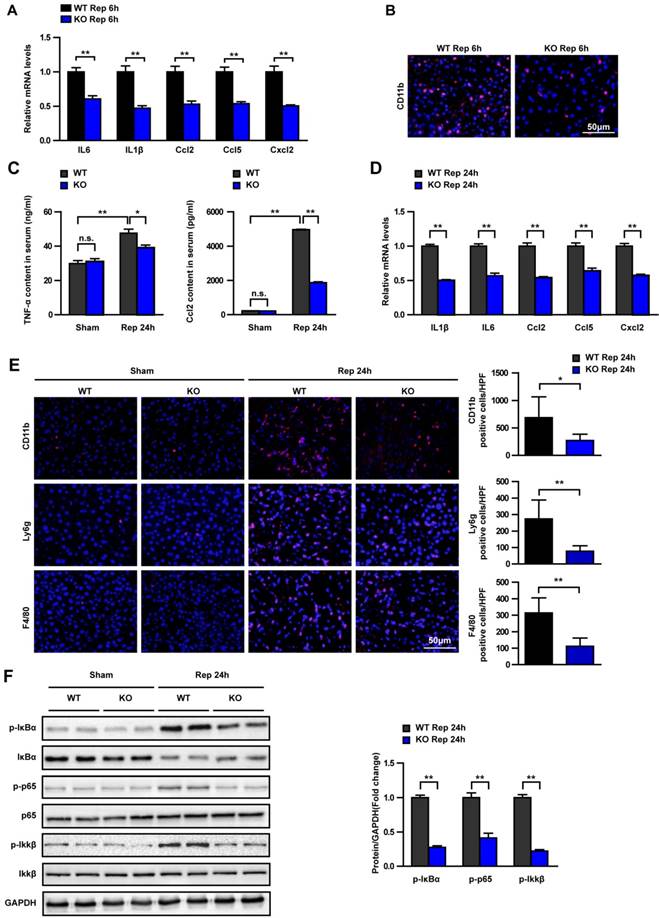
Trim8 deficiency relieves apoptosis in hepatic I/R. (A) TUNEL staining (20×) of liver sections at 24 h after reperfusion (n=6 per group). (B) Western blot analysis of Bax, Bcl2 and cleaved-caspase-3 expression in livers from WT and KO mice at 24 h after I/R or a sham operation (n=4 per group). GAPDH served as the loading control. (C) The mRNA levels of Bad, Bax and Bcl2 in livers from WT and KO mice at 24 h after I/R (n=4 per group). All data are shown as the mean ± SD. **P < 0.01.
Trim8 deficiency alleviates inflammation and apoptosis in primary hepatocyte
To further clarify whether the pro-inflammatory effect of Trim8 is mediated by hepatocytes, the primary hepatocytes isolated from the Trim8 KO mice and wild type mice were treated with H/R or Control treatment. The results showed that the Trim8 deficiency in hepatocytes inhibits the expression of inflammatory cytokines, chemokines and pro-apoptotic associated molecules, promotes the expression of anti-apoptotic molecule (Fig.5A-5B). Western blot showed that the classic pro-inflammatory NF-κB signalling pathway was dramatically inhibited by Trim8 deficiency after H/R treatment, as demonstrated by the significant decrease in p-IKKβ, p-IκBα and p-p65 (Fig.5C). Additionally, Western blot results further validated the up regulation of Bcl-2 and down regulation of Bax and C-Caspase3 in Trim8 deficiency hepatocytes (Fig.5D). Taken together, these results demonstrated that Trim8 deficiency inhibits inflammation and apoptosis in hepatocytes after H/R treatment.
TAK1-JNK/p38 signalling is involved in Trim8-regulated hepatic I/R injury
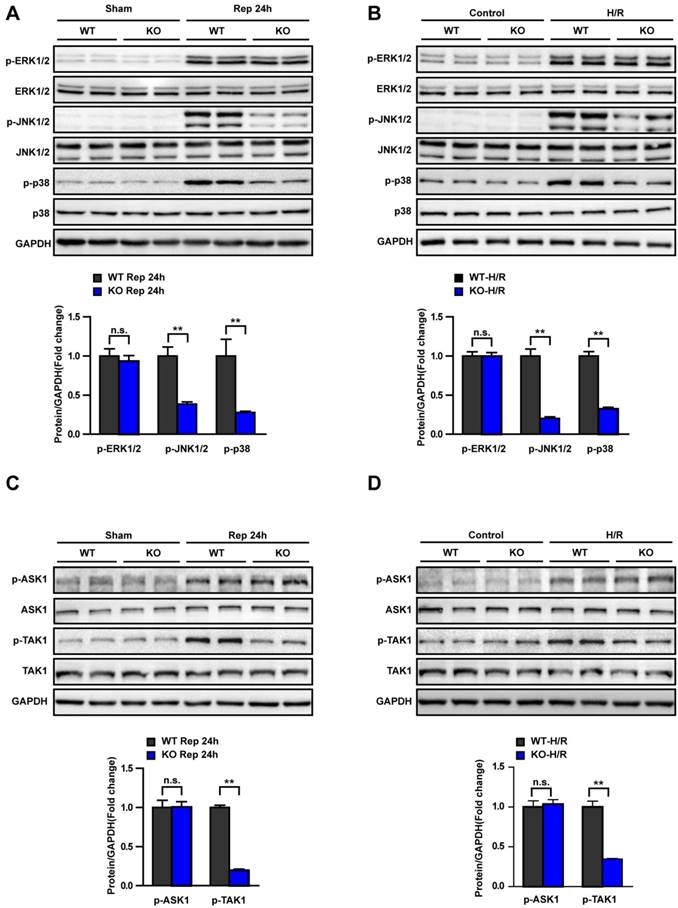
The underlying mechanisms of Trim8 in hepatic I/R injury were explored in vitro and in vivo. We detected the level of MAPK activation in the livers of WT and KO mice at 24 h after I/R. The results showed that Trim8 deficiency blocked activation of p-JNK and p-p38 but had no effect on p-ERK (Fig. 6A). Consistent with the vivo results, phosphorylation level of JNK and p38 was significantly decreased in primary hepatocytes from KO mice at 6h after H/R (Fig. 6B). Furthermore, we examined the activation of p-ASK1 and p-TAK1, which are the classical upstream mediators of the JNK and p38 signalling pathway. We found that only TAK1 activation was responsive to Trim8 expression changes in vivo and in vitro (Fig. 6C and D). These results demonstrate that Trim8 deficiency protects against hepatic I/R injury by inhibiting activation of the ASK1/JNK/p38 axis.
The effect of Trim8 on hepatic I/R is mediated by TAK1 activity
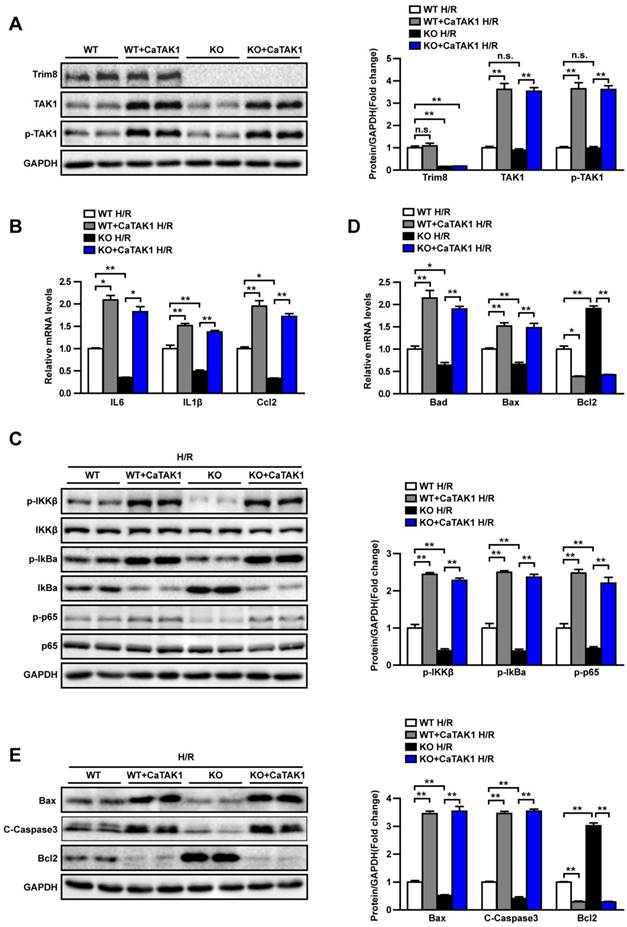
To further elucidate the molecular mechanism, we isolated primary hepatocytes from WT and KO mice, overexpressing CaTAK1, and exploring the phenotype of hepatocytes after H/R treatment. Western blot was used to detect the expression of Trim8, TAK1 and the phosphorylation level of TAK1. The expression of Trim8 in KO group is extremely low, the protein expression of TAK1 and the phosphorylation level of TAK1 were significantly increased after overexpressing the CaTAK1 (Fig. 7A). RT-PCR (Fig. 7B, 7D) and Western blotting (Fig. 7C, 7E) revealed that H/R treatment induced the activation of NF-κB signaling pathway, the secretion of inflammatory factors and the up-regulation of apoptosis-related molecules are significantly aggravated by CaTAK1. CaTAK1 abolished the difference between WT H/R and KO H/R hepatocytes. Taken together, the effect of Trim8 on hepatic I/R is mediated by TAK1 activity.
Trim8 deficiency inhibits inflammation and apoptosis in H/R-treated hepatocytes. (A-B) Relative mRNA levels of cytokines and chemokines (A) and apoptosis associated gene (B) in control or H/R-treated primary hepatocytes. (C) Protein levels of NF-κB signalling pathway in H/R-treated hepatocytes. (D) Protein levels of apoptosis-related molecules (Bax, C-Caspase3 and Bcl2) in H/R-treated hepatocytes. All data are shown as the mean ± SD. n=3 independent experiments. n.s., not significant; *P < 0.05; **P < 0.01.
Trim8 deficiency inhibits TAK1-JNK/p38 signalling. (A) The protein levels of total and phosphorylated ERK, JNK and p38 in liver tissues from the indicated groups (n=4 per group). (B) Total and phosphorylated ERK, JNK and p38 protein levels in Control or H/R-treated hepatocytes. n=3 independent experiments. (C) The protein expression of total and phosphorylated ASK1 and TAK1 in liver tissues from the indicated groups (n=4 per group). (D) Total and phosphorylated ASK1 and TAK1 protein levels in Control or H/R-treated hepatocytes. n=3 independent experiments. All data are shown as the mean ± SD. n.s., not significant; **P < 0.01.
Discussion
I/R injury is a complex disorder with multiple causative factors in a variety of settings and with varied clinical manifestations, especially in the field of liver transplantation. The molecular mechanisms of I/R are likely associated with apoptosis, oxidative stress, Ca2+ overload, and inflammation. A large number of factors and mediators play an important role in hepatic I/R injury. The involved signalling pathways are highly complex and not yet clearly elucidated.
The effect of Trim8 on hepatic I/R is mediated by TAK1 activity. (A) The expression levels of Trim8, TAK1 and the phosphorylation level of TAK1 (left) and quantification results (right) in the indicated groups. (B, D) Relative mRNA levels of cytokines and chemokines (B) and apoptosis associated gene (D) in H/R-treated indicated groups. (C) The levels of total and phosphorylated Ikkβ, IkBα and p65 (left) and quantification results (right) in indicated groups. (E) The expression levels of apoptosis-related molecules (Bax, C-Caspase3 and Bcl2) (left) and quantification results (right) in indicated groups. All data are shown as the mean ± SD. n=3 independent experiments. *P < 0.05; **P < 0.01.
Hepatic I/R activates the innate immune system to drive full development of inflammatory hepatocellular injury. Damage-associated molecular patterns (DAMPs) stimulate myeloid and dendritic cells via pattern recognition receptors (PRRs) to initiate the immune response [17, 18]. Then, a complex intracellular signalling network transduces inflammatory signalling to regulate both innate immune cell activation and parenchymal cell death [19].
Trim8 is a ubiquitously expressed factor in the Trim family that orchestrates multiple biological processes, including cell survival, differentiation, apoptosis, and in particular, the innate immune response [11]. The role of Trim8 in hepatic I/R injury has not yet been reported. To explore Trim8 role in hepatic I/R, we examined Trim8 expression in vitro and in vivo. Our results demonstrated that the protein expression of Trim8 is upregulated in hepatic I/R mice and cultured primary hepatocytes (Fig. 1). To further explore Trim8 function in hepatic I/R, we verified Trim8 effect in Trim8 knockout mice. The results preliminarily demonstrated that Trim8 deficiency alleviates the hepatic I/R injury (Fig. 2).
Many studies have shown that Trim8 plays an important role in innate immunity. According to one recent study, Trim8 can negatively regulate TLR3 and TLR4 mediated innate immune and inflammatory responses. Trim8 deficiency leads to increased polyinosinic-polycytidylic acid- and LPS-triggered induction of downstream antimicrobial genes, including TNF, IL-6, and Rantes [8]. Trim8 also contributes to pathological cardiac hypertrophy by enhancing transforming growth factor β-activated kinase 1-dependent signalling pathways and regulates the TNF-induced NF-κB pathway [12]. Moreover, different domains of Trim8 show discrete functions at different steps in regulation of the TNF-induced NF-κB pathway. The ubiquitin ligase activity of Trim8 is essential for regulation of NF-κB activation in both the cytoplasm and nucleus [20]. Therefore, the main mechanism by which Trim8 inhibits inflammation depends on the NF-κB pathway. In our study, we further demonstrated that Trim8 deficiency inhibits the production of inflammatory cytokines and the infiltration of inflammatory cells in liver tissue. The main mechanism also relies on inhibition of the NF-κB signalling pathway (Fig. 3).
Excerpt the inflammatory response, cell death is another mechanism involved with liver damage during hepatic I/R [18]. During I/R process, initial parenchymal damage is caused by glycogen depletion, lack of oxygen supply and ATP depletion, which lead to later activation of Kupffer cells, recruitment of inflammatory cells, and secretion of cytokines and chemokines produced by different cells [21]. The inflammatory response further promotes liver cell damage [22, 23]. Therefore, the apoptosis mechanism was investigated in our experiments. The results showed that Trim8 deficiency inhibited apoptosis by down regulating Bad, Bax and C-Caspase3 expression and upregulating Bcl-2 expression (Fig. 4). The Trim8 effect in hepatic I/R was validated by detecting inflammation and apoptosis in primary hepatocytes (Fig. 5).
Regulation of Trim8 expression involves multiple mechanisms. In cancer research, Trim8 is down regulated in glioma affects cell proliferation and is associated with patient survival [24]. Trim8 also regulates stemness in glioblastoma through PIAS3-STAT3[25] and inhibits tumour growth by restoring p53 tumour suppressor function through blunting of N-MYC activity in chemoresistant tumours [26]. In innate immunity under human non-alcoholic fatty liver disease and non-alcoholic steatohepatitis conditions, the function of Trim8 involves targeting of TAK1 to promote insulin resistance and steatohepatitis. Trim8 directly ubiquitinates transforming growth factor-beta-activated kinase 1, thus promoting its phosphorylation and activation of downstream c-Jun N-terminal kinase/p38 and NF-κB signalling [11]. The MAP3-kinase TGF-beta-activated kinase 1 (TAK1) critically modulates innate and adaptive immune responses and connects cytokine stimulation with activation of inflammatory signalling pathways, including cell survival, apoptosis, autophagy, oxidative processes, cancer and the immune response [27-30]. TAK1 is a MAP3K protein and is considered a common upstream molecule in the MKK-JNK/p38 and IKK-NF-κB cascade, which induces activation of downstream JNK and NF-κB signalling [31-33]. Previous research also showed that TAK1 is closely related to liver ischaemia and reperfusion [34-37]. In our study, the mechanism by which Trim8 regulates hepatic I/R was found to also depend on the TAK1-JNK1/2/p38 axis (Fig. 6), and the effect of Trim8 on hepatic I/R is mediated by TAK1 activity (Fig.7). We have proved that Trim8 in hepatocytes played an important role in hepatic I/R process in vitro and in vivo. However, since Trim 8 completely knockout mice were applied in this study, the knockout of Trim8 in non-parenchymal cells of liver may also play a role in regulating I/R injury. It needs further study to verify it.
In conclusion, we provide evidence that hepatocyte endogenous Trim8 is an essential molecule for development of hepatic I/R and that its role is dependent on TAK1-JNK1/2/p38 signalling. Thus, targeting Trim8 in hepatocytes may represent a promising strategy to alleviate hepatic ischaemia and reperfusion.
Abbreviations
ALT: alanine aminotransferase; AST: aspartate aminotransferase; ALP: alkaline phosphatase; H&E: haematoxylin and eosin; I/R: ischaemic reperfusion; KO: knockout; LT: liver transplantation; MAPK: mitogen-activated protein kinase; mRNA: messenger RNA; NF-κB: nuclear factor kappa B; PCR: polymerase chain reaction; Trim8: tripartite motif 8; TNF-α: tumour necrosis factor α; TAK1: transforming growth factor-beta-activated kinase 1; WT: wild type.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (no. 81870067 and no.81400753).
Author Contributions
Qiu Tao and Zhou Jiangqiao designed the research study and performed the research; Chen Zhongbao and Ma Xiaoxiong contributed essential reagents or tools; Zhang Long and Zou Jilin analysed the data; Qiu Tao and Wang Tianyu wrote the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Saidi RF, Kenari SK. Liver ischemia/reperfusion injury: an overview. J Invest Surg. 2014;27(6):366-79
2. Quesnelle KM, Bystrom PV, Toledo-Pereyra LH. Molecular responses to ischemia and reperfusion in the liver. Arch Toxicol. 2015;89(5):651-7
3. Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. Am J Transplant. 2011;11(8):1563-9
4. Eltzschig HK, Eckle T. Ischemia and reperfusion-from mechanism to translation. Nat Med. 2011;17(11):1391-401
5. Peralta C, Jiménez-Castro MB, Gracia-Sancho J. Hepatic ischemia and reperfusion injury: effects on the liver sinusoidal milieu. J Hepatol. 2013;59(5):1094-106
6. Zhang XJ, Cheng X, Yan ZZ. et al. An ALOX12-12-HETE-GPR31 signaling axis is a key mediator of hepatic ischemia-reperfusion injury. Nat Med. 2018;24(1):73-83
7. Caratozzolo MF, Marzano F, Mastropasqua F, Sbisà E, Tullo A. TRIM8: Making the Right Decision between the Oncogene and Tumour Suppressor Role. Genes (Basel). 2017:8 (12). doi: 10.3390/genes8120354
8. Ye W, Hu MM, Lei CQ. et al. TRIM8 Negatively Regulates TLR3/4-Mediated Innate Immune Response by Blocking TRIF-TBK1 Interaction. J Immunol. 2017;199(5):1856-1864
9. Guo L, Dong W, Fu X. et al. Tripartite Motif 8 (TRIM8) Positively Regulates Pro-inflammatory Responses in Pseudomonas aeruginosa-Induced Keratitis Through Promoting K63-Linked Polyubiquitination of TAK1 Protein. Inflammation. 2017;40(2):454-463
10. Roy M, Tomar D, Singh K. et al. TRIM8 regulated autophagy modulates the level of cleaved Caspase-3 subunit to inhibit genotoxic stress induced cell death. Cell Signal. 2018;48:1-12
11. Yan FJ, Zhang XJ, Wang WX. et al. The E3 ligase tripartite motif 8 targets TAK1 to promote insulin resistance and steatohepatitis. Hepatology. 2017;65(5):1492-1511
12. Chen L, Huang J, Ji YX. et al. Tripartite Motif 8 Contributes to Pathological Cardiac Hypertrophy Through Enhancing Transforming Growth Factor β-Activated Kinase 1-Dependent Signaling Pathways. Hypertension. 2017;69(2):249-258
13. Wu H, Zhou J, Ou W, Li Y, Liu M, Yang C. TAK1 as the mediator in the protective effect of propofol on renal interstitial fibrosis induced by ischemia/reperfusion injury. Eur J Pharmacol. 2017;811:134-140
14. Zhou J, Qiu T, Wang T. et al. USP4 deficiency exacerbates hepatic ischaemia/reperfusion injury via TAK1 signalling. Clin Sci (Lond). 2019;133:335-49
15. Gan LT, Van Rooyen DM, Koina ME, McCuskey RS, Teoh NC, Farrell GC. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J Hepatol. 2014;61(6):1376-84
16. Hu J, Zhu XH, Zhang XJ. et al. Targeting TRAF3 signaling protects against hepatic ischemia/reperfusions injury. J Hepatol. 2016;64(1):146-59
17. van Golen RF, van Gulik TM, Heger M. The sterile immune response during hepatic ischemia/reperfusion. Cytokine Growth Factor Rev. 2012;23(3):69-84
18. Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59(3):583-94
19. Lu L, Zhou H, Ni M. et al. Innate Immune Regulations and Liver Ischemia-Reperfusion Injury. Transplantation. 2016;100(12):2601-2610
20. Tomar D, Sripada L, Prajapati P, Singh R, Singh AK, Singh R. Nucleo-cytoplasmic trafficking of TRIM8, a novel oncogene, is involved in positive regulation of TNF induced NF-κB pathway. PLoS One. 2012;7(11):e48662
21. van Golen RF, Reiniers MJ, Olthof PB, van Gulik TM, Heger M. Sterile inflammation in hepatic ischemia/reperfusion injury: present concepts and potential therapeutics. J Gastroenterol Hepatol. 2013;28(3):394-400
22. Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deaths. Hepatology. 2006;43(2 Suppl 1):S31-44
23. Sun P, Zhang P, Wang PX. et al. Mindin deficiency protects the liver against ischemia/reperfusion injury. J Hepatol. 2015;63(5):1198-211
24. Micale L, Fusco C, Fontana A. et al. TRIM8 downregulation in glioma affects cell proliferation and it is associated with patients survival. BMC Cancer. 2015;15:470
25. Zhang C, Mukherjee S, Tucker-Burden C. et al. TRIM8 regulates stemness in glioblastoma through PIAS3-STAT3. Mol Oncol. 2017;11(3):280-294
26. Mastropasqua F, Marzano F, Valletti A. et al. TRIM8 restores p53 tumour suppressor function by blunting N-MYC activity in chemo-resistant tumours. Mol Cancer. 2017;16(1):67
27. Sato S, Sanjo H, Takeda K. et al. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol. 2005;6(11):1087-95
28. Greten FR. TAK1: Another mesh in the NF-κB - JNK controlled network causing hepatocellular carcinoma. J Hepatol. 2011;55(3):721-723
29. Kobayashi A, Kobayashi S, Miyai K. et al. TAK1 inhibition ameliorates survival from graft-versus-host disease in an allogeneic murine marrow transplantation model. Int J Hematol. 2018;107(2):222-229
30. Iriondo O, Liu Y, Lee G. et al. TAK1 mediates microenvironment-triggered autocrine signals and promotes triple-negative breast cancer lung metastasis. Nat Commun. 2018;9(1):1994
31. Liu L, Liu Y, Liu X. et al. Resibufogenin suppresses TAK1-mediated NF-κB activity via protein kinase C-dependent inhibition of glycogen synthase kinase 3. Cancer Sci. 2018;109:3611-3622
32. Zhou J, Fan Y, Zhong J. et al. TAK1 mediates excessive autophagy via p38 and ERK in cisplatin-induced acute kidney injury. J Cell Mol Med. 2018;22(5):2908-2921
33. Mangan MS, Latz E. TAK1ng control: TAK1 restrains NLRP3 activation. J Exp Med. 2018;215(4):1007-1008
34. Nace GW, Huang H, Klune JR. et al. Cellular-specific role of toll-like receptor 4 in hepatic ischemia-reperfusion injury in mice. Hepatology. 2013;58(1):374-87
35. Wang X, Mao W, Fang C. et al. Dusp14 protects against hepatic ischaemia-reperfusion injury via Tak1 suppression. J Hepatol. 2017 [Epub ahead of print]
36. Zhang M, Yang D, Gong X. et al. Protective benefits of AMP-activated protein kinase in hepatic ischemia-reperfusion injury. Am J Transl Res. 2017;9(3):823-829
37. Yang L, Wang W, Wang X. et al. Creg in Hepatocytes Ameliorates Liver Ischemia/Reperfusion Injury in a TAK1-Dependent Manner in Mice. Hepatology. 2019;69(1):294-313
Author contact
![]() Corresponding author: Zhou Jiangqiao, Department of Organ Transplantation, Renmin Hospital of Wuhan University, Wuhan University, ZiYang Road 99#, Wuhan 430060, China. Tel.: 86-27-88041911-81849, Fax: 86-27-88072292, Email: zhoujqedu.cn
Corresponding author: Zhou Jiangqiao, Department of Organ Transplantation, Renmin Hospital of Wuhan University, Wuhan University, ZiYang Road 99#, Wuhan 430060, China. Tel.: 86-27-88041911-81849, Fax: 86-27-88072292, Email: zhoujqedu.cn
Received 2019-1-19
Accepted 2019-5-21
Published 2019-6-4