ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion and Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2019; 15(8):1664-1675. doi:10.7150/ijbs.35216 This issue Cite
Research Paper
CD44 3'-Untranslated Region Functions as a Competing Endogenous RNA to Enhance NK Sensitivity of Liver Cancer Stem Cell by Regulating ULBP2 Expression
Jun Weng1*, Xu Han1*, Kaiyu Liu1, Jiong Yang2, Shiruo Wei3, Yue Zhang1, Fanhong Zeng1, Yang Li1, Li Shen3 ![]() , Yi Gao1
, Yi Gao1 ![]()
1. Second Department of Hepatobiliary Surgery, Zhujiang Hospital, State Key Laboratory of Organ Failure Research, Co-Innovation Center for Organ Failure Research, Southern Medical University, Guangzhou, China
2. Department of Geriatrics, The Affiliated Hospital of Qingdao University, Qingdao, China
3. Department of Cell Biology, School of Basic Medical Sciences, Peking University Health Science Center, Beijing, China
* contributed equally to this work.
Abstract

Liver CSCs are a rare subpopulation of heterogenous liver cancer cells with self-renewal and differentiation properties, which has emerged as a promising therapeutic target. Compelling data shows that NK cells selectively eliminate human cancer derived CSCs like colorectal carcinoma, melanoma, and glioblastoma. But the effect of NK cells on liver CSCs still remains unknown. To study the cytotoxic effect of NK cells on liver CSCs and the mechanism, we performed cytotoxicity assay, ELISA assays, CRISPRi, qRT-PCR, immunoblotting, RNA immunoprecipitation, and luciferase reporter using two types of CSCs reprogrammed from HCC. CSCs derived from liver cancer were susceptible to NK cell mediated cytotoxicity. The susceptibility of liver CSCs to NK cell-mediated cytotoxicity declined significantly after silencing CD44 by CRISPRi-mediated gene knockdown. CD44 3ʹ UTR functioned as a ceRNA to regulate the expression of ULBP2 mainly by competing miR-34a. CD44 3ʹ UTR functioned as a ceRNA to enhance NK sensitivity of liver cancer stem cell by regulating ULBP2 expression.
Keywords: liver Cancer Stem Cell • Natural Killer • Post-translational regulation • ceRNA • miR-34a-5p
Introduction
Liver cancer is the second leading cancer type worldwide with high mortality rate. Hepatocellular carcinoma (HCC) is the main histopathology type of primary liver cancers[1]. In the past 10 years, although therapeutic improvement has been positively made, the prognosis of HCC still remains poor. Recent studies indicate HCC progression are driven by cancer stem cells (CSC), a stem-cell like population, which possess self-renewing and pluripotency properties through an asymmetric proliferating pattern[2]. Occupying a minor subpopulation of malignant tumor, CSCs, which present in various human cancers including liver cancer, have been postulated as the key for chemotherapeutic resistance, tumor relapse, and seeding metastasis by mounting studies. In order to eradicate malignant tumor, CSC is a promising target, thus, anti-CSC strategy has been an urgent task in HCC treatment.
Increasing evidence supports that in addition to their remarkable role played in hematological malignancies, activated natural killer (NK) cells preferentially kill CSCs derived from a variety of human solid tumors[3]. Being classified as a large granular member of innate lymphoid cells (ILCs), NK cells are phenotypically characterized by the absence of CD3 and the expression of surface molecules like CD56 and CD16[4]. They exhibit powerful protective and cytotoxic function in recognizing and eliminating both infected cells and tumor cells by producing proinflammatory and lymphocytotoxicity cytokines. Tallerico et al. demonstrated that NK cells show a significant cytotoxic effect on CSCs derived from colorectal carcinoma cells (CRC)[5]. Pietra et al. found that IL-2-activated NK cells could efficiently recognize and lysis CSCs derived from melanoma through activating a different combination of NK receptors[6]. Castriconi et al. reported that CSCs isolated from glioblastoma could be killed by IL-2 or IL-15 activated allogeneic and autologous NK cells[7]. But the effect of NK cells on liver CSCs still remains unknown.
CSCs express high levels of surface CD44 and M to NK cell mediated cytotoxicity, while differentiated tumor cells express lower levels of surface CD44 and are resistant to NK cell mediated cytotoxicity. The increase of surface receptor CD44 expression is identified in nearly all types of CSCs which have been reported previously[8]. Stated thus, two types of CSCs reprogrammed from HCC by combining different reprogramming factors were used in our research which verified that CSCs derived from liver cancer were susceptible to NK cell mediated cytotoxicity. We then detected that the expression level of CD44 corresponded with the level of ULBP2, an activating NK ligand, which then further influenced the susceptibility of CSCs to NK cell mediated cytotoxicity. Our present work also suggested that CD44 may function as a ceRNA (Competing endogenous RNA) to regulate the expression of ULBP2 mainly by competing miR-34a.
Materials and Methods
Cell culture
Transcription factors Oct4, Sox2, Klf4 and c-Myc (OSKM), with or without shMBD3, were ectopically expressed in C3A cells to generate CD44highiCSC (also named as shMBD3-iCSCs) and CD44intiCSC (also named as C3A-iCSCs). All cells were cultured in a humidified atmosphere (37°C, 5% CO2). Liver cancer stem cells were cultured in DMEM/F-12 (11320; Thermo Fisher Scientific, Waltham, MA, USA) containing 20% knockout serum replacement (10828028; Thermo Fisher Scientific, Waltham, MA, USA), 1 mM L-glutamine, 0.1 mM 2-mercaptoethanol, 0.1 mM nonessential amino acids, and 10 ng/ ml recombinant human basic fibroblast growth factor (13256029; Thermo Fisher Scientific, Waltham, MA, USA)[9]. Both cells were passaged with 0.5 mM EDTA. In all experiments, CSCs were in the state between P10 to P20. NK-92 cells were cultured in NK Cell Culture Medium (CL-0530; Procell, Wuhan, China). HepG2 cells were cultured in DMEM (11965; Thermo Fisher Scientific, Waltham, MA, USA) containing 10% Fetal Bovine Serum (FBS) (SH30084; GE Healthcare Life Sciences, Chicago, IL, USA). Hep3B cells were cultured in MEM (11095; Thermo Fisher Scientific, Waltham, MA, USA) containing 10% FBS.
Cytotoxicity Assay and ELISA
CytoTox 96 ® Non-Radioactive Cytotoxicity Assay (G1780; Promega, Madison, WI, USA) was preformed to measure NK cells cytotoxicity. %Cytotoxicity = (Experimental - Effector Spontaneous - Target Spontaneous)/(Target Maximum - Target Spontaneous) × 100. NK-92 cells were incubated with the respective target cells in 96 well plates for 4 hours at 37°C. The E:T ratios were indicated in the text. Antibodies used for masking experiments were against ULBP2 (M311; Amgen, Seattle, WA, USA).
Concentrations of secreted IFN-γ were determined using Human Interferon gamma ELISA Kit (ab46048; Abcam, Cambridge, MA, USA).
Plasmid constructs and reagents
Guide sequences (5'-TCCATGGTGTCCGGAGCGAA) against CD44 1st exon was inserted into pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-Puro (Addgene plasmid #71236) to create CRISPRi mediated CD44 knockdown vector. 3'UTR expressing lentivirus plasmid (pITA-CD44 3'UTR) was created as follow: CD44 3'UTR was amplified from cDNA prepared from CD44highiCSC by PCR; The PCR product of 3'UTR was subcloned between NotI and BamHI sites of pITA vector. CD44 CDS expressing plasmid used here was previously constructed in our laboratory[9]. CD44 3'UTR was subcloned into XbaI site of pGL3-Promoter to create CD44 3'UTR luciferase report plasmid. ULBP2 promoter was amplified from genome prepared from CD44highiCSC by PCR and further subcloned into XbaI site of pGL3-Promoter to create ULBP2 3'UTR luciferase report plasmid. ULBP2 3'UTR was amplified from cDNA prepared from CD44highiCSC by PCR and further subcloned into XbaI site of pGL3-Promoter to create ULBP2 3'UTR luciferase report plasmid. Mutant constructs were generated by using Fast Mutagenesis System (FM111-01; TransGen Biotech, Beijing, China). pCDNA3.1 (-) + FLAG-NLS-MS2-eGFP was a gift from Carl Novina (Addgene plasmid # 86827). MS2 tagged 3'UTR expressing vector was built by fusing MS2 stem-loop (MS2 tag) repeats and 3'UTR into pCDNA3.1(+).
The miRNA mimics for miR-16-5p, miR-34a-5p, miR-373-3p, miR-520c-3p and miRNA inhibitor for inhibition of endogenous miR-34a activity were purchased from GeneCopoeia (I-270 Hi-Tech corridor, MD, USA). Transfection was carried out with the Lipofectamine 2000 (11668; Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions.
Viral transduction
Lentivirus was produced by transiently transfecting lentivector, pspAX2, and pMD2.G into 293T cells followed by ultracentrifugation to concentrate viral supernatants. Concentrated viral supernatants were then supplemented with 8 μg/mL of polybrene (TR-1003-G; Merck Millipore, Burlington, MA, USA), and incubated with target iCSCs at 37 ℃ for 22 hours. Those iCSCs were subsequently drug-selected (2 μg/mL puromycin) for successful proviral integration[10].
RNA isolation and Quantitative RT-PCR
Total RNA was extracted using TRIzol reagent (15596; Thermo Fisher Scientific, Waltham, MA, USA). RNA was reverse transcribed into cDNA using the ReverTra Ace® qPCR RT Master Mix (FSQ201; Toyobo, Osaka, Japan). Real-time PCR was performed on the CFX Connect™ Real-Time System (Bio-Rad Laboratories, Hercules, CA, USA) using SYBR ® Green Realtime PCR Master Mix (QPK201; Toyobo, Osaka, Japan) according to the manufacturer's instructions. Primer sets used were as follows: CD44 primer set 1: 5'- CATCAGTCACAGACCTGCCCAATGC and 5'- ATGTAACCTCCTGAAGTGCTGCTCC; CD44 primer set 2: 5'-AGAGCTGGCCAAGTCTTCAC and 5'-GCTTCCAGAGTTACGCCCTT; MICA 5'-ACTTGACAGGGAACGGAAAGGA and 5'-CCATCGTAGTAGAAATGCTGGGA; MICB 5'-ATCTGTGCAGTCAGGGTTTCTC and 5'-TGAGGTCTTGCCCATTCTCTGT; ULBP1 5'-TGGGTATCATGCTTACTGTCTGGG and 5'-GGGTTTGGGTTCATAGTGCAGAGTT; ULBP2 5'-CTTTGCTGCCTCCTCATCATCC and 5'-GCCAGACAGAAGGGCGAGTTT; ULBP3 5'-AGTTCAGCTTCGATGGACGGAAGT and 5'-AGCCAGCTCCTTGCAGTCTCTCATT; CD48 5'-GCCTGAGAACTACAAACAACTAACC and 5'-GCAGCTTGATCTTCCATTCTTGCTC; CD112 5'-TGGACTGGGAAGCCAAAGAGA and 5'-TACAGAGAGGGTCACAGGTATCAGG; CD155 5'-GCTCTGCTGTTTGTTCTGCTTTCC and 5'-TTTCTGCTGCTGGATGCGGTTT; ICAM1 5'- GACTAAGCCAAGAGGAAGGAGCAA and 5'- TCAGCATACCCAATAGGCAGCAAG.
miRNA expression analyses were carried out on the CFX Connect™ Real-Time System (Bio-Rad Laboratories, Hercules, CA, USA) using All-in-One miRNA qRT-PCR Detection Kit (QP015; GeneCopoeia, I-270 Hi-Tech corridor, MD, USA) according to the manufacturer's instructions.
Immunoblotting
Cells were rinsed with PBS. Total cellular protein was extracted with RIPA lysis buffer (C1051; APPLYGEN, Beijing, China) containing protease inhibitor cocktail (P1265; APPLYGEN, Beijing, China). The lysates were clarified. Protein amount in the lysate was measured using Quick Start™ Bradford 1x Dye Reagent (500-0205; Bio-Rad Laboratories, Hercules, CA, USA). After being heated at 95℃ for 3 min, protein samples were subjected to SDS-PAGE and electrophoretically transferred to nitrocellulose membranes (PALL, Port Washington, NY, USA) under proper conditions. The membranes were blocked with 5% skim milk dissolved in TBS-Tween 20 for 2-4 hours. Primary antibodies were incubated with corresponding membranes at 4 ℃ overnight, and then washed with TBS-Tween 20. The blots were incubated with secondary antibodies at room temperature for 2 hours. The protein bands were visualized by Plus ECL Plus (P1010; APPLYGEN, Beijing, China).
Antibodies used for western blotting were against β-actin (1:10000; PM053; MBL, Nagoya, Japan), CD44 (1:1000; 15675-1-AP; proteintech, Rosemont, IL, USA;) and ULBP2 (1:800; 13133-1s-AP; proteintech, Rosemont, IL, USA;). The densitometry data were analyzed by ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Luciferase assay
For luciferase activity assay, cells were seeded in 24 well plates and transfected with plasmids and mimics described in the text. 48 hours after transfection, luciferase activity was measured using the Dual-Glo luciferase assay system (E2920; Promega, Madison, WI, USA) with a luminometer (Centro LB 960; Berthold Technologies, Bad Wildbad, Germany). Luciferase activity was normalized to the renilla control.
RNA immunoprecipitation
RNA immunoprecipitation was performed using the EZ-Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (17-701; Merck Millipore, Burlington, MA, USA) according to the manufacturer's instructions. Antibodies used for immunoprecipitations were against GFP (ab290; Abcam, Cambridge, MA, USA) and Ago2 (ab32381; Abcam, Cambridge, MA, USA). The precipitated RNAs were purified using the TRIzol reagent (15596; Thermo Fisher Scientific, Waltham, MA, USA) and detected by All-in-One miRNA qRT-PCR Detection Kit (QP015; GeneCopoeia, I-270 Hi-Tech corridor, MD, USA).
Statistical analysis
Data were presented as mean ± SD from at least three independent experiments. The Student's t-test was employed to evaluate the significance. All statistical analyses were performed using Prism 8.0 (GraphPad, La Jolla, CA, USA).
Results
CSCs derived from liver cancer were susceptible to NK cell mediated cytotoxicity in correlation with CD44 expression
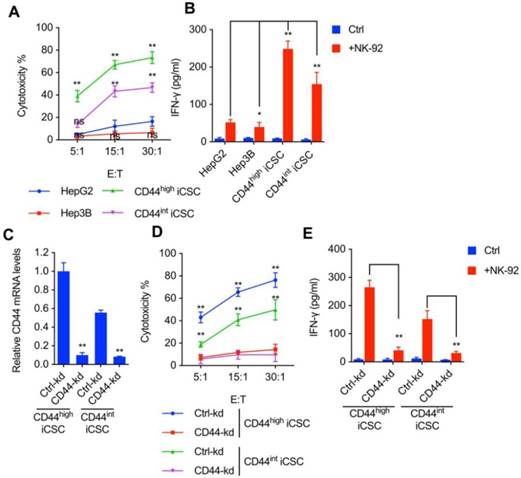
It has been previously demonstrated that NK cells selectively eliminate CSCs derived from colorectal carcinoma[5], melanoma[6], and glioblastoma[7]. To explore the effect of NK cells on liver CSCs, we performed lactate dehydrogenase (LDH) cytotoxic assay with NK-92 cells and challenged them in vitro with two types of CSCs reprogrammed from HCC by combining different reprogramming factors. CD44s expression was dominant in Liver CSCs. shMBD3-iCSCs with high level of CD44 expression was named CD44highiCSC while C3A-iCSC with intermediate level of CD44 expression was named CD44intiCSC. Moreover, we examined the IFN-γ release in supernatants of cytotoxic assay described previously. As shown in Fig. 1A and Fig. 1B, both CSCs are more sensitive to NK Cells than conventional hepatocellular carcinoma cell lines (HepG2 and Hep3B).
In agreement with previous reports that CSCs express high levels of surface CD44 and are susceptible to NK cell-mediated cytotoxicity, we observed NK cells are more toxic to Liver CSC with higher CD44 expression (Fig. 1A, B). Hence, we speculated that CD44 might play a role in NK cell-mediated cytotoxicity in liver CSCs. To verify this hypothesis, CRISPRi was used to deplete endogenous CD44 expression in CD44highiCSC, and the CD44 knockdown effect was measured by qPCR (Fig. 1C)[11]. As an evidence for our theory, loss of CD44 significantly impaired NK cell-mediated lysis and IFN-γ release in CD44highiCSC (Fig. 1D, E). Similar results were obtained from the CD44intiCSC (Fig. 1D, E).
CSCs derived from liver cancer were susceptible to NK cell mediated cytotoxicity in correlation with CD44 expression. (A) CD44highiCSC, CD44intiCSC, HepG2, and Hep3B were used in the 4 hours NK cell cytotoxicity assay with NK-92 cells at different E:T ratios. Data are showed in means ± SD from three independent experiments which are performed in triplicate (ns: not significant, **: p < 0.01). (B) IFN-γ release in the supernatants of cytotoxic assay (Fig. 1A, E:T ratio = 15:1) was determined by ELISA. Data were presented as mean ± SD (n=3) (*: p<0.05, **: p<0.01). (C) CD44 transcript level of CD44highiCSC/CD44intiCSC stably expressing dCas9-KRAB and sgRNA against CD44 (CD44-knockdown, CD44-kd) was analyzed by qRT-PCR. Levels are represented relative to those found in control-infected cells as means ± SD (n=3) (**: p < 0.01). (D) Identical cells (Fig. 1C) were used in the 4 hours NK cell cytotoxicity assay with NK-92 cells at different E:T ratios. Data are showed in means ± SD from three independent experiments which are performed in triplicate (**: p < 0.01). (E) IFN-γ release in the supernatants of cytotoxic assay (Fig. 1D, E:T ratio = 15:1) was determined by ELISA. Data were presented as mean ± SD (n=3) (**: p<0.01).
CD44 regulated ULBP2 expression, which then further influenced the susceptibility of CSCs to NK cell mediated cytotoxicity. (A) MICA/B, ULBP1-3, CD48, CD112, CD155, and ICAM1 transcript levels of CD44highiCSC/CD44intiCSC stably expressing dCas9-KRAB and sgRNA against CD44 (CD44-knockdown, CD44-kd) were analyzed by qRT-PCR. Levels are represented relative to those found in control-infected cells as means ± SD (n=3) (**: p < 0.01). (B) CD44 and ULBP2 protein levels of identical cells (Fig. 2A) were analyzed by Western blotting. β-actin served as a loading control. (C) CD44highiCSC and CD44intiCSC were used in the 4 hours NK cell cytotoxicity assay with NK-92 cells at different E:T ratios. Assays were performed either in the presence of M311 mAb (anti-ULBP2) or control IgG. Data are showed in means ± SD from three independent experiments which are performed in triplicate (**: p < 0.01). (D) IFN-γ release in the supernatants of cytotoxic assay (Fig. 2C, E:T ratio = 15:1) was determined by ELISA. Data were presented as mean ± SD (n=3) (**: p<0.01).
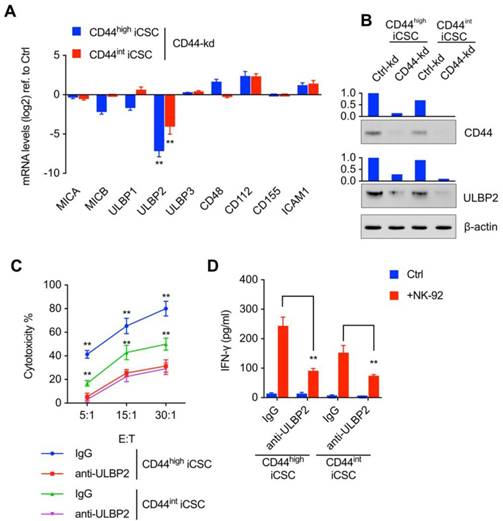
CD44 regulated ULBP2 expression, which then further influenced the susceptibility of CSCs to NK cell mediated cytotoxicity
To further explore the mechanism of CD44 in NK cell-mediated cytotoxicity, we went on to detect the ligands which were critical for NK cell receptors to bind in this process. We then analyzed the ligands of NK receptors in mRNA level and found that knockdown of CD44 resulted in a vast decrease in ULBP2 expression (Fig. 2A). Western blot analysis showed notable decrease of ULBP2 expression in CD44 depletion Liver CSCs (Fig. 2B). Thus, these data suggested that CD44 might influence the susceptibility of CSCs to NK cell-mediated cytotoxicity by regulating ULBP2 expression in both mRNA and protein level. What's more, a marked decrease in NK cell-mediated cytotoxicity (Fig. 2C) and IFN-γ secretion (Fig. 2D) was observed after antibody-mediated masking of ULBP2.
The regulation of ULBP2 was performed by CD44 3'-untranslated region
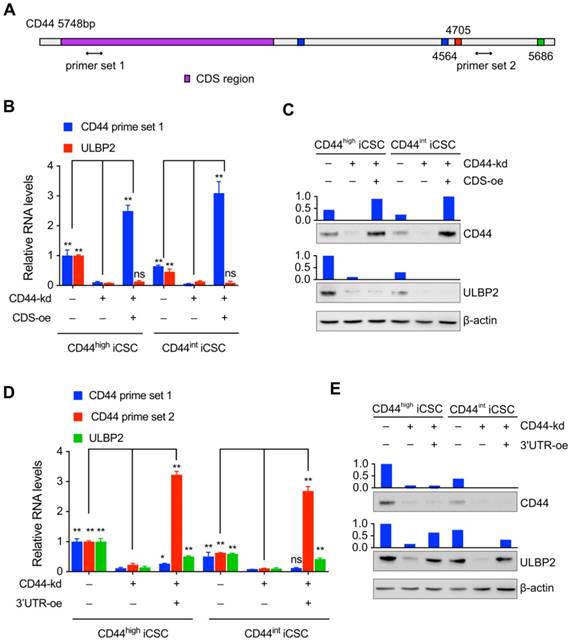
Only a handful of protein-coding mRNAs has been validated as ceRNAs and CD44 is one of them[12]. It has been reported that CD44 may regulate downstream genes by a mechanism independent of its protein[13,14]. Hence, we further explored which part of CD44 was involved in the regulation of ULBP2 expression. Different primers set targeting the 3'UTR or CDS of CD44 mRNA was designed to monitor the expression level of different CD44 parts (Fig. 3A). Our results showed that ectopic expressing CD44 CDS didn't affect ULBP2 mRNA and protein expression level in endogenous CD44 depletion CD44highiCSC (Fig. 3B, C).
In contrast, introducing CD44 3'UTR in endogenous CD44 knockdown CD44highiCSC rescued ULBP2 expression in mRNA and protein level compared with control group. Similar results were obtained in CD44intiCSC (Fig. 3D, E). All of these results indicate that CD44 3'UTR plays a vital role in regulating ULBP2 expression.
The regulation of ULBP2 was performed by CD44 3'-untranslated region. (A) Schematic diagram of CD44 genes and the primer sets used in the qPCR assays. (B) CD44highiCSC/CD44intiCSC stably expressing dCas9-KRAB and sgRNA against CD44 (CD44-knockdown, CD44-kd) were transfected with blank expression vector (Ctrl) or CD44 coding sequence expression vector (CD44 CDS-overexpression, CDS-oe). CD44 and ULBP2 transcript levels were determined by qRT-PCR. Levels are represented relative to those found in control-transfected cells as means mean ± SD (n=3) (ns: not significant, **: p < 0.01). (C) CD44 and ULBP2 protein levels of identical cells (Fig. 3B) were analyzed by Western blotting. β-actin served as a loading control. (D) CD44highiCSC/CD44intiCSC stably expressing dCas9-KRAB and sgRNA against CD44 (CD44-knockdown, CD44-kd) were transfected with blank expression vector (Ctrl) or CD44 3'UTR expression vector (CD44 3'UTR -overexpression, 3'UTR-oe). CD44 CDS, CD44 3'UTR, and ULBP2 transcript levels were determined by qRT-PCR. Levels are represented relative to those found in control-transfected cells as means mean ± SD (n=3) (ns: not significant, *:p<0.05, **:p < 0.01). (E) CD44 and ULBP2 protein levels of identical cells (Fig. 3D) were analyzed by Western blotting. β-actin served as a loading control.
Loss of CD44 downregulated ULBP2 mRNA stability while upregulated miR-34a-5p, miR-373-3p and miR-520c-3p expression
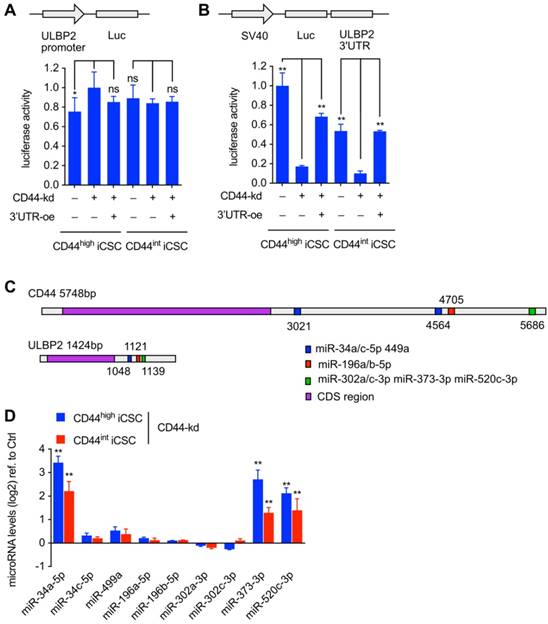
After the observation of changed ULBP2 mRNA expression level under CD44 knockdown and CD44 3'UTR ectopic expressing, we performed luciferase activity assay to determine the basal activity of ULBP2 promoter.
About 1.5 kb ULBP2 promoter was subcloned into pGL3-Basic vector and transfected into CD44highiCSC and CD44intiCSC with pRL-CMV. We found that CD44 knockdown and CD44 3'UTR ectopic expressing had no effect on both ULBP2 promoter-driven luciferase activity (Fig. 4A) and transcription initiation rate of ULBP2 promoter (Fig. S1B). These results suggested that CD44 3'UTR regulated ULBP2 in post-transcriptional steps. 3' UTR luciferase activity assay and mRNA stability assay were performed to determine whether CD44 3'UTR could stabilize ULBP2 mRNA. ULBP2 3'UTR was subcloned into pGL3-Promoter vector and transfected into CD44highiCSC and CD44intiCSC with pRL-CMV. Notably, depletion of CD44 significantly attenuated the luciferase activity of ULBP2 3'UTR and introducing CD44 3'UTR could rescue it (Fig. 4B), which worked in concert with the data of ULBP2 mRNA half-life. Together, these data indicated that CD44 3'UTR regulated ULBP2 in post-transcriptional steps by targeting its 3'UTR. According to ceRNA theory, CD44 may function as a decoy to sponge miRNA[15]. We speculated that CD44 might regulate the expression of ULBP2 through ceRNA mechanism. miRNAs targeting both CD44 and ULBP2 were predicted by StarBase software (Fig. 4C)[16]. As ceRNA has the ability to influence miRNA expression, those candidates were further screened by detecting their variation before and after CRISPRi-mediated depletion of CD44. As shown in Fig. 4D, miR-34a-5p, miR-373-3p, and miR-520c-3p were upregulated in CD44 knockdown cells.
Bond both CD44 and ULBP2
MS2 tagging based RNA immunoprecipitation (RIP) analysis was performed to detect miRNA-target interactions between miRNAs (miR-34a-5p, miR-373-3p, and miR-520c-3p) and CD44/ULBP2. If the endogenous RNA contains MS2 stem-loop (MS2 tag) repeats, it could be specifically bound by MS2 coat protein (MCP), so MS2 tagging based RIP could be used to detect RNA-RNA or RNA-protein interaction[17]. The vector expressing MS2 tagged CD44 3'UTR or ULBP2 3'UTR was transfected into 293T cells together with a vector expressing MCP-GFP fusion protein and a miRNA mixture (miR-16-5p, miR-34a-5p, miR-373-3p, and miR-520c-3p). The lysates were incubated with IgG or GFP-antibody (Fig. 5A). Western blot analysis showed that Ago2 was immunoprecipitated from both cells expressing MS2 tagged CD44 or ULBP2 3'UTR (Fig. 5B). qPCR assay was used to measure the enrichment of miRNA after immunoprecipitate. As shown in Fig. 5C, compared with the control group (miR-16-5p), miR-34a-5p, miR-373-3p, and miR-520c-3p were directly bound by CD44 and ULBP2 3'UTR.
Loss of CD44 downregulated ULBP2 mRNA stability while upregulated miR-34a-5p, miR-373-3p and miR-520c-3p expression. (A) CD44highiCSC/CD44intiCSC stably expressing dCas9-KRAB and sgRNA against CD44 (CD44-knockdown, CD44-kd) were transfected with pGL3-Basic containing ULBP2 promoter, pRL-CMV, and CD44 3'UTR sequence expression vector (CD44 3'UTR -overexpression, 3'UTR -oe). Blank expression vector served as the control. Luciferase activity of ULBP2 promoter was determined by luciferase reporter assay. Data were presented as mean ± SD (n=3) (ns: not significant, *:p<0.05). (B) CD44highiCSC/CD44intiCSC stably expressing dCas9-KRAB and sgRNA against CD44 (CD44-knockdown, CD44-kd) were transfected with pGL3-Promoter containing ULBP2 3'UTR, pRL-CMV, and CD44 3'UTR sequence expression vector (CD44 3'UTR -overexpression, 3'UTR-oe). Blank expression vector served as the control. Luciferase activity of ULBP2 3'UTR was determined by luciferase reporter assay. Data were presented as mean ± SD (n=3) (**: p < 0.01). (C) Schematic diagram of miRNA binding sites on CD44 and ULBP2 transcripts. (D) miRNA expression levels of CD44highiCSC/CD44intiCSC stably expressing dCas9-KRAB and sgRNA against CD44 (CD44-knockdown, CD44-kd) were analyzed by qRT-PCR and normalized to U6. Levels are represented relative to those found in control-infected cells as means ± SD (n=3) (**: p < 0.01).
miR-34a-5p, miR-373-3p, and miR-520c-3p bond both CD44 and ULBP2. (A) Schematic diagram of MS2 tagging based RNA immunoprecipitation assay. (B) MS2 tagging based RIP was performed to examine in vivo binding of Ago2 to CD44 3'UTR or ULBP2 3'UTR. A Vector expressing MS2 tagged luciferase (MS2-Ctrl) served as the control. (C) The enrichment of miR-34a-5p, miR-373-3p, and miR-520c-3p after immunoprecipitate (Fig. 5B) were analyzed by qRT-PCR and normalized to U6. miR-16-5p served as the control. The enrichment of miRNAs are represented as means ± SD (n=3) (**: p < 0.01). (D) pGL3-Promoter vector containing CD44 3'UTR or ULBP2 3'UTR was transfected into 293T cells together with miRNA mimics and pRL-CMV. Luciferase activity of CD44 3'UTR and ULBP2 3'UTR were determined by luciferase reporter assay. pGL3-Promoter (luc-Ctrl) served as the control. Data were presented as mean ± SD (n=3) (ns: not significant, **:p<0.01). (E) Schematic of mutations in miR145 target sites. The red nucleotides were removed in the report vectors. (F) pGL3-Promoter vector containing wild-type (WT) or mutant (mut) 3'UTR was transfected into 293T cells together with miR-34a mimics and pRL-CMV. Scramble negative control RNA (NC RNA) served as the control. Luciferase activity of mutant 3'UTR were determined by luciferase reporter assay. Data were presented as mean ± SD (n=3) (**: p<0.01).
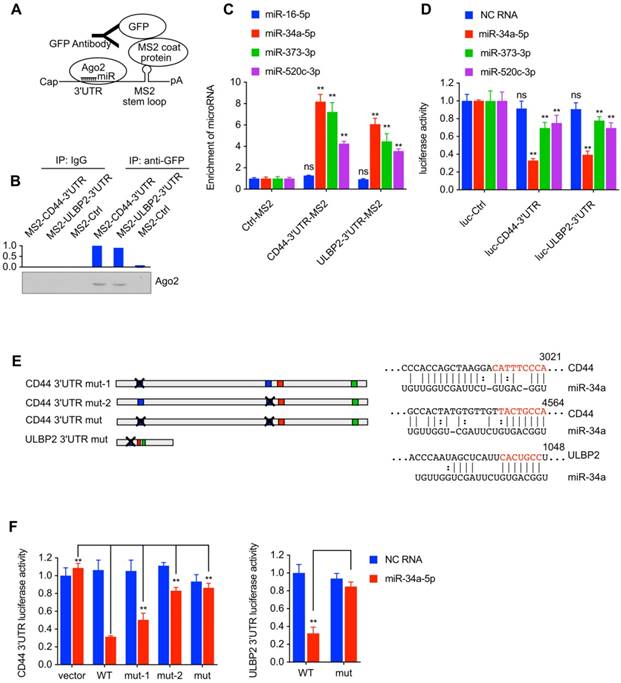
miR-34a-5p, miR-373-3p, and miR-520c-3p
pGL3-Promoter vector containing CD44 3'UTR or ULBP2 3'UTR was transfected into 293T cells together with miRNA mimics and pRL-CMV to find out whether these miRNAs could decrease the activity of luciferase activity by directly interacting with the 3'UTR of CD44 and ULBP2. Compared with miR-373-3p and miR-520c-3p mimics, miR-34a-5p mimics significantly reduced the luciferase activity in both cells (Fig. 5D), indicating that miR-34a-5p could downregulate both CD44 and ULBP2 by directly binding their 3'UTR. Notably, the 3'UTR of CD44 and ULBP2 contains serval miR-34a response element, the mutation (Fig. 5E) of which could rescue the luciferase activity performed previously in 293T cells (Fig. 5F).
CD44 functioned as a ceRNA to protect ULBP2 in liver CSCs by competitively binding miR-34a
Wang et al. reported that the expression level of miRNA could not only be influenced by its targeting ceRNA but also reach a saturated state in high concentrations[15]. miR-34a and miR-16 mimics were transfected into 293T cells to form different concentrations (from 20 nM to 80 nM) together with CD44 3'UTR WT or CD44 3'UTR mut expression vector. Result showed that CD44 3'UTR WT significantly decreased the level of miR-34a in 20 nM concentration while a moderate decrease of miR-34a was detected in 40 nM concentration and the decrease was nearly diminished in 80nM concentration (Fig. 6A). in supporting to Wang's report, the result present here indicated that CD44 3'UTR WT performed as a ceRNA to rescue miR-34a in an appropriate concentration but if the concentration of miR-34a got high enough to saturate CD44 3'UTR WT, this reducing effect could be diluted.
We then examined whether miR-34a, ULBP2 3'UTR, and CD44 3'UTR could establish a regulatory ceRNA network in 293T cells. CD44 3'UTR overexpressing vector and ULBP2 3'UTR containing pGL3-Promoter vector was co-transfected in 293T cells together with miR-34a-5p mimics and pRL-CMV. As expected, loss of miR-34a-5p expression or mutation of its response element diminished the interaction between CD44 and ULBP2 expression (Fig. 6B).
Because CD44 3'UTR and ULBP2 3'UTR could interact with Ago2 and miR-34a, we further researched whether CD44 and ULBP2 could be suppressed by endogenous miR-34a in liver CSCs[18]. Our results showed that transfection of miR-34a inhibitor in CD44 knockdown CD44highiCSC or CD44intCSC partly rescued ULBP2 and CD44 expression (Fig. 6C, D).
CD44 functioned as a ceRNA to protect ULBP2 in liver CSCs by competitively binding miR-34a. (A) miR-16 and miR-34a concentrations in saturation assays were determined by qRT-PCR. microRNA levels were represented as means ± SD (n=3) (**: p < 0.01). (B) CD44 3'UTR overexpressing vector and ULBP2 3'UTR containing pGL3-Promoter vector were transfected in 293T cells together with miR-34a-5p mimics and pRL-CMV. Luciferase activity of ULBP2 3'UTR were determined by luciferase reporter assay. (C) CD44highiCSC/CD44intiCSC stably expressing dCas9-KRAB and sgRNA against CD44 (CD44-knockdown, CD44-kd) were transfected with negative control or miR-34a inhibitor. CD44 and ULBP2 transcript levels were determined by qRT-PCR. Levels are represented relative to those found in control- transfected cells as means mean ± SD (n=3) (ns: not significant, **: p < 0.01). (D) CD44 and ULBP2 protein levels of identical cells (Fig. 6C) were analyzed by Western blotting. β-actin served as a loading control. (E) CD44highiCSC/CD44intiCSC stably expressing dCas9-KRAB and sgRNA against CD44 (CD44-knockdown, CD44-kd) were transfected with CD44 3'UTR WT expression vector or CD44 3'UTR mut expression vector. CD44 3'UTR, miR-34a, pri-miR-34a, and pre-miR-34a transcript levels were determined by qRT-PCR and normalized to Ctrl (blank expression vector). Levels are represented relative to those found in control- infected cells as means mean ± SD (n=3) (ns: not significant, **: p < 0.01).
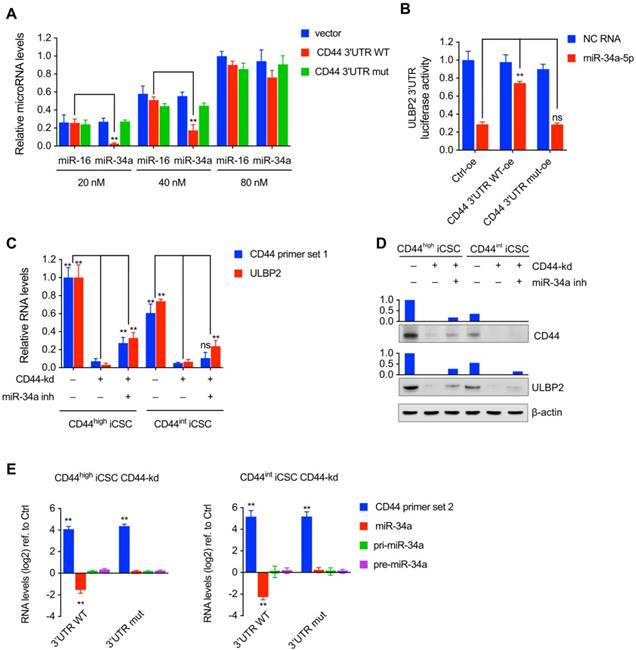
Since miR-34a was a critical component to regulate ULBP2 expression, we analyzed whether the expression level of miR-34a could be influenced by CD44 3'UTR in liver CSCs. Ectopic overexpressing CD44 3'UTR WT decreased mature miR-34a expression compared with CD44 3'UTR mut. We also investigated whether CD44 3'UTR could affect miR-34 biogenesis in liver CSCs. As shown in Fig. 6E, CD44 3'UTR did not change pri-miR-34a and pre-miR-34a expression. These results suggested that CD44 3'UTR regulated miR-34a mainly in post-transcriptional steps, which was aligned with ceRNA theory.
Taken together, all of those data described above strongly suggested that CD44 protected ULBP2 in a ceRNA manner mainly by specifically binding miR-34a.
Discussion and Conclusion
Liver CSCs are a rare subpopulation of heterogenous liver cancer cells with self-renewal and differentiation properties, which has emerged as a promising therapeutic target. So far, different strategies have been used to isolate or induce CSCs based on classical surface stem cell markers, side population cells, activity of intracellular enzymes, promoter-driven fluorescent protein expression, suspension culture, cytotoxic and hypoxic resistance, and transfection with defined factors[19]. All of those methods mentioned above except transfection with defied factors require fluorescence activated cell sorting (FACS), serum deprivation, cytotoxic drugs, hypoxia, lacking cell adhesion or other stress-induced environments[20]. The activity of NK cells relies on a series of germ-line encoded activating receptors including CD16, NKG2D, DNAM-1, and NKp46, which possess corresponding ligands[21]. Ligation of NKG2D is critical to trigger NK-mediated cytotoxicity. Grouped into 2 families termed MICs (MICA, MICB) and ULBPs (ULBP1, ULBP2, ULBP3, ULBP4, RAET1G, RAET1L)[22,23], NKG2D ligands are completely or nearly completely absent on the surface of normal cells while overexpressed on the surface of infected and stressed cells[24]. Thus, stress-induced environments could not only promote the sensitivity to apoptosis-inducing pathways but also artificially enhance NK sensitivity to CSCs through activating NKG2D. Therefore, reprogramming with defined factors was used in our research to avoid unwanted additional stress, making our experiment status more analogous to natural state.
Cancer stem cells have been postulated to be responsible for sustaining tumor progression though asymmetrical growth and low proliferation rate which made them resistant to clinical chemotherapy and radiotherapy[25,26]. To eliminate tumor stem compartment, several studies evaluated the killing effect of immune cytotoxic cells (NK cells, CD8 T cells, and γδ T cells). Unfortunately, CSCs are poorly targeted by T-lymphocytes, but compelling data shows that NK cells selectively eliminate human cancer derived CSCs like colorectal carcinoma[5], melanoma[6], and glioblastoma[7]. The effect of NK cells on CSCs derived from breast cancer is controversial: Yin et al. reported that CSCs derived from breast cancer are sensitive to NK mediated cytotoxicity through upregulating the expression of NKG2D ligands ULBP1, ULBP2, and MICA[27]; while Wang et al. observed that breast cancer CSCs could reduce NK killing by shedding MICA and MICB[28]. Conclusion drawn from our research is aligned with colorectal carcinoma, melanoma, and glioblastoma that NK cells selectively kill CSCs reprogrammed from liver cancer cells (Fig. 1A, B).
As a famous multi-structural transmembrane glycoprotein, CD44 exhibits a variety of cellular functions including adhesive cell-cell and cell-matrix interactions, lymphocyte activation and homing, cell migration, cell proliferation, angiogenesis, and tumor metastasis[29]. CD44 is also a well-known assistant CSC marker in liver cancer, gastric cancer, breast cancer, and acute myeloid leukemia[30].
This research was initiated by our original observation that the susceptibility of liver CSCs to NK cell-mediated cytotoxicity declined significantly after silencing CD44 by CRISPRi-mediated gene knockdown (Fig. 1C-E). We then detected that the expression level of CD44 corresponded with the level of ULBP2 (Fig. 2A, B), an activating NK ligand, which then further influenced the susceptibility of CSCs to NK cell mediated cytotoxicity (Fig. 2C, D). By ectopic expressing CD44 3'UTR or CD44 CDS, we discovered that overexpress CD44 3'UTR (Fig. 3D, E), rather than CD44 CDS (Fig. 3B, C), could rescue the expression of ULBP2 in CD44 silencing liver CSCs.
The non-coding 3'-untranslated region (3'UTR) of CD44 has been shown to inhibit cell proliferation and colony formation, while enhance cell adhesion, motility, and invasion. Through binding and sequestering miR-216a, miR-330 and miR-608, CD44 regulates the level of CDC42, a Rho-GTPase which plays important role in cell migration, morphology, and cell-cycle progression[14]. Similar to CDC42, Col1α1 and FN1 could also be modulated by CD44 through miRNA binding[13], which link CD44 to a broader miRNA-ceRNA interaction network revolving around PTEN and VCAN[31,32].
Anja Heinemann et al. reported that miR-34a and miR-34c inversely corelated with ULBP2 surface molecules and control ULBP2 expression[33]. Can Liu et al. validated that miR-34a inhibited prostate CSCs by directly and functionally repressing CD44[34]. According with those former findings, our results elucidated that CD44 protected ULBP2 in a ceRNA manner mainly by specifically binding miR-34a to alleviate the degradation of ULBP2. As a miRNA with tumor-suppressive activity, miR-34a possess a relatively low concentration which is just right to form the ceRNA network.
Our results suggested that CD44 may function as a ceRNA to regulate the expression of ULBP2 by competing miR-34a, miR-373, and miR-520c, which broadened the ceRNA function of CD44 3ʹ UTR in ULBP2 regulation. Hence, the NK cell mediated cytotoxicity in liver CSCs reported here is unaffected by anti-CD44 antibody mediated CD44 signaling blockage in liver CSCs (Fig. S1E, F), which is also interpreted as a potential strategy to eradicate liver CSCs.
Abbreviations
3'UTR: 3'-untranslated region; Ago2: Argonaute; CD112: NECTIN-2; CD155: poliovirus receptor; CD16: Fc fragment of IgG, low affinity IIIa receptor; CD3: cluster of differentiation 3; CD44: homing cell adhesion molecule; CD48: signaling lymphocytic activation molecule 2; CD56: Neural cell adhesion molecule; CDC42: Cell division control protein 42 homolog; ceRNA: competing endogenous RNA; CRC: colorectal carcinoma cells; CRISPR: clustered regularly interspaced short palindromic repeats; CSC: cancer stem cells; CMV: Cytomegalovirus; Col1α1: Collagen Type Alpha 1; dCas9: Cas9 Endonuclease Dead; FACS: fluorescence activated cell sorting; FBS: Fetal Bovine Serum; FN1: Fibronectin; GFP: Green fluorescent protein; HCC: Hepatocellular carcinoma; ICAM-1: Intercellular Adhesion Molecule 1; IL-2: Interleukin 2; IL-15: Interleukin 15; ILCs: innate lymphoid cells; KRAB: krüppel-associated box; LDH: lactate dehydrogenase; MBD3: Methyl-CpG-binding domain protein 3; MICA: MHC class I polypeptide-related sequence A; MICB: MHC class I polypeptide-related sequence B; MCP: MS2 coat protein; NK: natural killer; NKG2D: killer cell lectin like receptor K1; OSKM: Oct4, Sox2, Klf4 and c-Myc; PTEN: Phosphatase and tensin homolog; sgRNA: single guide RNA; ULBP1: UL16 binding protein 1; ULBP2: UL16 binding protein 2; ULBP3: UL16 binding protein 3; VCAN: Versican; RIP: RNA immunoprecipitation.
Supplementary Material
Supplementary figures.
Acknowledgements
This project was funded by National Natural Science Foundation of China (81572313), Science and Technology Planning Project of Guangdong Province, China (2015B020229002), Science and Technology Planning Project of Guangdong Province (2014B020227002), and Science and Technology Program of Guangzhou (201604020002).
Author Contributions
J.Weng, K. Liu, Y. Gao designed research; K. Liu, X. Han, L. Shen and Yang Li analyzed data; K. Liu, X. Han, S. Wei, J. Weng, L. Shen, Yue Zhang and Y. Gao performed research; K. Liu, J. Yang, Fanhong Zeng and Y. Gao wrote the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Bray F, Center MM. et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
2. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755-68
3. Tallerico R, Garofalo C, Carbone E. A New Biological Feature of Natural Killer Cells: The Recognition of Solid Tumor-Derived Cancer Stem Cells. Front Immunol. 2016;7:179
4. Vivier E, Tomasello E, Baratin M. et al. Functions of natural killer cells. Nat Immunol. 2008;9:503-10
5. Tallerico R, Todaro M, Di Franco S. et al. Human NK cells selective targeting of colon cancer-initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules. J Immunol Baltim Md 1950. 2013;190:2381-90
6. Pietra G, Manzini C, Vitale M. et al. Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. Int Immunol. 2009;21:793-801
7. Castriconi R, Daga A, Dondero A. et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol Baltim Md 1950. 2009;182:3530-9
8. Kaur K, Nanut MP, Ko M-W. et al. Natural killer cells target and differentiate cancer stem-like cells/undifferentiated tumors: strategies to optimize their growth and expansion for effective cancer immunotherapy. Curr Opin Immunol. 2018;51:170-80
9. Li R, He Q, Han S. et al. MBD3 inhibits formation of liver cancer stem cells. Oncotarget. 2017;8:6067-78
10. Kutner RH, Zhang X-Y, Reiser J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc. 2009;4:495-505
11. Thakore PI, D'Ippolito AM, Song L. et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods. 2015;12:1143-9
12. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344-52
13. Rutnam ZJ, Yang BB. The non-coding 3' UTR of CD44 induces metastasis by regulating extracellular matrix functions. J Cell Sci. 2012;125:2075-85
14. Jeyapalan Z, Deng Z, Shatseva T. et al. Expression of CD44 3'-untranslated region regulates endogenous microRNA functions in tumorigenesis and angiogenesis. Nucleic Acids Res. 2011;39:3026-41
15. Wang Y, Xu Z, Jiang J. et al. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev Cell. 2013;25:69-80
16. Li J-H, Liu S, Zhou H. et al. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92-97
17. Schmidt K, Joyce CE, Buquicchio F. et al. The lncRNA SLNCR1 Mediates Melanoma Invasion through a Conserved SRA1-like Region. Cell Rep. 2016;15:2025-37
18. Chi SW, Zang JB, Mele A. et al. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479-86
19. Duan J-J, Qiu W, Xu S-L. et al. Strategies for isolating and enriching cancer stem cells: well begun is half done. Stem Cells Dev. 2013;22:2221-39
20. Wuputra K, Lin C-S, Tsai M-H. et al. Cancer cell reprogramming to identify the genes competent for generating liver cancer stem cells. Inflamm Regen. 2017;37:15
21. Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225-74
22. Bauer S, Groh V, Wu J. et al. Pillars Article: Activation of NK Cells and T Cells by NKG2D, a Receptor for Stress-Inducible MICA. Science. 1999. 285: 727-729. J Immunol Baltim Md 1950. 2018;200:2231-3
23. Cosman D, Müllberg J, Sutherland CL. et al. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 2001;14:123-33
24. Chan CJ, Smyth MJ, Martinet L. Molecular mechanisms of natural killer cell activation in response to cellular stress. Cell Death Differ. 2014;21:5-14
25. Reya T, Morrison SJ, Clarke MF. et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11
26. Lobo NA, Shimono Y, Qian D. et al. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675-99
27. Yin T, Wang G, He S. et al. Human cancer cells with stem cell-like phenotype exhibit enhanced sensitivity to the cytotoxicity of IL-2 and IL-15 activated natural killer cells. Cell Immunol. 2016;300:41-5
28. Wang B, Wang Q, Wang Z. et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res. 2014;74:5746-57
29. Misra S, Hascall VC, Markwald RR. et al. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front Immunol. 2015;6:201
30. Yan Y, Zuo X, Wei D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl Med. 2015;4:1033-43
31. Tay Y, Kats L, Salmena L. et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147:344-57
32. Lee DY, Jeyapalan Z, Fang L. et al. Expression of versican 3'-untranslated region modulates endogenous microRNA functions. PloS One. 2010;5:e13599
33. Heinemann A, Zhao F, Pechlivanis S. et al. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res. 2012;72:460-71
34. Liu C, Kelnar K, Liu B. et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211-5
Author contact
![]() Corresponding author: Li Shen, Department of Cell Biology, University Health Science Center, 38 Xueyuan Rd, Beijing, 100191, China. Yi Gao, Second Department of Hepatobiliary Surgery, Zhujiang Hospital. 253 GONGYE DADAO, Guangzhou, 510280, China. Tel: +86-20-6278256 Fax: +86-20-61643207 E-mail: drgaoycom
Corresponding author: Li Shen, Department of Cell Biology, University Health Science Center, 38 Xueyuan Rd, Beijing, 100191, China. Yi Gao, Second Department of Hepatobiliary Surgery, Zhujiang Hospital. 253 GONGYE DADAO, Guangzhou, 510280, China. Tel: +86-20-6278256 Fax: +86-20-61643207 E-mail: drgaoycom
Received 2019-3-25
Accepted 2019-5-15
Published 2019-6-4