ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Overview of CAR-T techniques
Obstacles in treating solid...
Companion diagnostics for CAR-T...
Conclusions
References

Int J Biol Sci 2019; 15(12):2548-2560. doi:10.7150/ijbs.34213 This issue Cite
Review
Current Progress in CAR-T Cell Therapy for Solid Tumors
Shuo Ma1,*, Xinchun Li1,*, Xinyue Wang1,*, Liang Cheng1,2, Zhong Li1, Changzheng Zhang1, Zhenlong Ye1,3,4, ![]() , Qijun Qian1,3,4
, Qijun Qian1,3,4 ![]()
1. Shanghai Baize Medical Laboratory, Shanghai, China
2. Department of Pathology and Laboratory Medicine, Indiana University School of Medicine, Indianapolis, Indiana, USA
3. Shanghai Cell Therapy Research Institute, Shanghai, China
4. Shanghai Engineering Research Center for Cell Therapy, Shanghai, China
* These authors contributed equally to this work.
Abstract

Cancer immunotherapy by chimeric antigen receptor-modified T (CAR-T) cells has shown exhilarative clinical efficacy for hematological malignancies. Recently two CAR-T cell based therapeutics, Kymriah (Tisagenlecleucel) and Yescarta (Axicabtagene ciloleucel) approved by US FDA (US Food and Drug Administration) are now used for treatment of B cell acute lymphoblastic leukemia (B-ALL) and diffuse large B-cell lymphoma (DLBCL) respectively in the US. Despite the progresses made in treating hematological malignancies, challenges still remain for use of CAR-T cell therapy to treat solid tumors. In this landscape, most studies have primarily focused on improving CAR-T cells and overcoming the unfavorable effects of tumor microenvironment on solid tumors. To further understand the current status and trend for developing CAR-T cell based therapies for various solid tumors, this review emphasizes on CAR-T techniques, current obstacles, and strategies for application, as well as necessary companion diagnostics for treatment of solid tumors with CAR-T cells.
Keywords: CAR-T cells, chimeric antigen receptor, solid tumors, companion diagnostics, CTC
Introduction
Immunotherapy with CAR-T cells has achieved tremendous successes in treatment of hematological malignancies. Two CD19-targeting CAR-T cell products, Kymriah from the Novartis (East Hanover, NJ USA) and Yescarta from the Kite Pharma (Santa Monica, CA USA), have been approved by the US FDA for treating B cell acute lymphoblastic leukemia (B-ALL) and diffuse large B-cell lymphoma (DLBCL), respectively [1]. However, due to intricacies of solid tumors and their locations in the human body, treatment of solid tumors with CAR-T cells is facing multiple obstacles, such as the hostile tumor microenvironment, on-tumor/off-tumor toxicities, and undesired antigen specificity [2]. Many strategies and approaches have been tried to overcome these obstacles, including arming CAR-T cells with knock-out of PD-1 expression or secretion of cytokines/chemokines and using CAR-T cells in combination with other treatments [3-5]. Despite these efforts, there are still no CAR-T cells clinically approved for solid tumor treatment so far. Encouragingly and optimistically, in this landscape, more than forty clinical trials in treatment of solid tumors by CAR-T cells have been registered in China alone (Table 1) [6].
Beside the focuses on the aspects of treatment, companion diagnostics are increasingly recognized as playing important roles in patient screening, treatment regimen, efficacy evaluation, and real-time monitoring of CAR-T cell therapies. Therefore, in this review, we focus on current CAR-T techniques, obstacles, strategies for overcoming these obstacles, as well as necessary companion diagnostics in treatment of solid tumors with CAR-T cells.
Clinical trials in treatment of solid tumors in China by CAR-T cells
| Antigen | Tumor target | Sponsor | Phase | NCT number | Study start |
|---|---|---|---|---|---|
| CD133 | Liver cancer; pancreatic cancer; brain tumor; breast cancer; ovarian tumor; colorectal cancer, acute myeloid lymphoid leukemias | Chinese PLA General Hospital | Phase 1 | NCT02541370 | 2015 |
| CD138 | Multiple myeloma | Chinese PLA General Hospital | Phase 1/Phase 2 | NCT01886976 | 2013 |
| CD19 | Recurrent or refractory B-cell tumor | Second Military Medical University | Phase 1/Phase 2 | NCT02644655 | 2015 |
| CEA | Lung cancer; colorectal cancer; gastriccancer; breast cancer; pancreatic cancer | Southwest Hospital | Phase 1 | NCT02349724 | 2014 |
| Claudin 18.2 | Advanced gastric adenocarcinoma, pancreatic adenocarcinoma | Changhai Hospital | NA | NCT03159819 | 2017 |
| EGFR | Advanced EGFR-positive solid tumors | Chinese PLA General Hospital | Phase 1/Phase 2 | NCT01869166 | 2013 |
| EGFR | Advanced glioma | RenJi Hospital | Phase 1 | NCT02331693 | 2014 |
| EGFR | Advanced solid tumor | Shanghai Cell Therapy Research Institute | Phase 1/Phase 2 | NCT03182816 | 2017 |
| EGFR | EGFR-positive colorectal cancer | Shenzheng Second People's Hospital | Phase 1/Phase 2 | NCT03152435 | 2017 |
| EGFRvIII | Recurrent glioblastoma multiforme | Beijing Sanbo Brain Hospital | Phase 1 | NCT02844062 | 2016 |
| EGFRvIII | Glioblastoma multiforme | Shenzhen Geno-Immune Medical Institute | Phase 1/Phase 2 | NCT03170141 | 2017 |
| EphA2 | EphA2-positive malignant glioma | Fuda Cancer Hospital | Phase 1/Phase 2 | NCT02575261 | 2015 |
| EpCAM | Liver neoplasms | Sinobioway Cell Therapy Co., Ltd. | NA | NCT02729493 | 2015 |
| EpCAM | Stomach neoplasms | Sinobioway Cell Therapy Co., Ltd. | NA | NCT02725125 | 2015 |
| EpCAM | Malignant neoplasms of the nasopharynx, TNM staging, distant metastasis and breast cancer recurrence | Sichuan University | Phase 1 | NCT02915445 | 2016 |
| EpCAM | Colon cancer, esophageal carcinoma, pancreatic cancer, prostate cancer, gastric cancer, hepatic carcinoma | First Affiliated Hospital of Chengdu Medical College | Phase 1/Phase 2 | NCT03013712 | 2017 |
| GD2 | Neuroblastoma | Zhujiang Hospital | Phase 2 | NCT02765243 | 2016 |
| GD2 | Relapsed or refractory neuroblastoma | Sinobioway Cell Therapy Co., Ltd. | NA | NCT02919046 | 2016 |
| GD2 | Solid tumor | Shenzhen Geno-Immune Medical Institute | Phase 1/Phase 2 | NCT02992210 | 2016 |
| GPC3 | Hepatocellular carcinoma | RenJi Hospital | Phase 1 | NCT02395250 | 2015 |
| GPC3 | Hepatocellular carcinoma and liver metastases | Shanghai GeneChem Co., Ltd. | Phase 1/Phase 2 | NCT02715362 | 2016 |
| GPC3 | GPC3-positive hepatocellular carcinoma | Funda Cancer Hospital, Guangzhou | Phase 1/Phase 2 | NCT02723942 | 2015 |
| GPC3 | Lung squamous cell carcinoma | Carsgen Therapeutics, Ltd. | Phase 1 | NCT02876978 | 2016 |
| GPC3 | Hepatocellular carcinoma | Shanghai GeneChem Co., Ltd. | Phase 1/Phase 2 | NCT03130712 | 2017 |
| GPC3 | Advanced hepatocellular carcinoma | Xinqiao Hospital of Chongqing | Phase 1/Phase 2 | NCT03084380 | 2017 |
| GPC3 | Hepatocellular carcinoma, squamous cell lung cancer | Second Affiliated Hospital of Chengdu Medical University | Phase 1 | NCT03198546 | 2017 |
| GPC3 | Hepatocellular carcinoma | RenJi Hospital | - | NCT03146234 | 2017 |
| GPC3/MSLN/ CEA | Hepatocellular, pancreatic cancer, colorectal cancer | Shanghai GeneChem Co., Ltd. | Phase 1/Phase 2 | NCT02959151 | 2016 |
| HER2 | Tumors refractory to chemotherapy and/or HER2 antibody therapy, advanced HER2-positive solid tumors | Chinese PLA General Hospital | Phase 1/Phase 2 | NCT01935843 | 2013 |
| HER2 | Breast cancer, ovarian cancer, lung cancer, gastric cancer, colorectal cancer, glioma, pancreatic caner | Southwest Hospital | Phase 1/Phase 2 | NCT02713984 | 2016 |
| HER2 | Breast cancer | Fudan Cancer Hospital | Phase 1/Phase 2 | NCT02547961 | 2015 |
| HerinCAR-PD1 | Advanced malignancies | Ningbo Cancer Hospital | Phase 1/Phase 2 | NCT02873390 | 2016 |
| HerinCAR-PD1 | Advanced solid tumors (lung, liver, and stomach) | Shanghai International Medical Center | Phase 1/Phase 2 | NCT02862028 | 2016 |
| MSLN | Malignant mesothelioma, pancreatic cancer; ovarian tumor; triple negative breast cancer; endometrial cancer; other mesothelin-positive tumors | Chinese PLA General Hospital | Phase 1 | NCT02580747 | 2015 |
| MSLN | Pancreatic cancer | Shanghai GeneChem Co., Ltd. | Phase 1 | NCT02706782 | 2016 |
| MSLN | Mesothelin-positive tumors | China Meitan General Hospital | Phase 1 | NCT02930993 | 2016 |
| MSLN | Solid tumors, adult advanced cancer | Ningbo Cancer Hospital | Phase 1/Phase 2 | NCT03030001 | 2017 |
| MSLN | Advanced solid tumors | Shanghai Cell Therapy Research Institute | Phase 1/Phase 2 | NCT03182803 | 2017 |
| MG7 | Liver metastases | Xijing Hospital | Phase 1/Phase 2 | NCT02862704 | 2016 |
| MUC1 | Malignant glioma of the brain; colorectal carcinoma; gastric carcinoma | PersonGen Biomedicine (Suzhou) Co., Ltd. | Phase 1/Phase 2 | NCT02617134 | 2015 |
| MUC1 | Advanced refractory solid tumors (hepatocellular carcinoma, NSCLC, pancreatic carcinoma, triple-negative invasive breast carcinoma) | PersonGen Biomedicine (Suzhou) Co., Ltd. | Phase 1/Phase 2 | NCT02587689 | 2015 |
| MUC1 | Advanced solid tumors | Shanghai Cell Therapy Research Institute | Phase 1/Phase 2 | NCT03179007 | 2017 |
| NY-ESO-1 | Advanced NSCLC | Guangzhou Institute of Respiratory Disease | Phase 1 | NCT03029273 | 2017 |
| LMP1 | Nasopharyngeal neoplasms | The Second Hospital of Nanjing Medical University | Phase 1/Phase 2 | NCT02980315 | 2016 |
| PD-L1 CSR | Glioblastoma multiforme | Beijing Sanbo Brain Hospital | Phase 1 | NCT02937844 | 2016 |
| PSCA/MUC1/ PD-L1/CD80/86 | Advanced lung or other cancers | Second Affiliated Hospital of Guangzhou Medical University | Phase 1 | NCT03198052 | 2017 |
| PSMA/FRα | Bladder cancer, urothelial carcinoma bladder | Shenzhen Geno-Immune Medical Institute | Phase 1/Phase 2 | NCT03185468 | 2017 |
| Zeushield | NSCLC | Second Xiangya Hospital of Central South University | Phase 1 | NCT03060343 | 2016 |
*The clinical trials are collected from clinicaltrials.gov
Overview of CAR-T techniques
Chimeric antigen receptor (CAR) is the core component of CAR-T, which endows T cells with the ability to recognize tumor antigens in a HLA-independent manner and enables them to recognize more extensive target antigens than natural T cell surface receptor (TCR) [7]. A basic CAR includes a tumor-associated antigen (TAA) binding domain (usually from the scFv fragment of antigen-binding region of the monoclonal antibody), an extracellular hinge domain, a transmembrane domain and an intracellular signal domain [7, 8].
The activation of T cells mediated by the first generation of CAR is accomplished by the tyrosine activation motif on CD3ζ chain or FcεRIγ [9]. CD3ζ chain can provide “signal I” for T cell activation, cytolysis, regulation of IL-2 secretion and anti-tumor activity in vivo [10, 11]. However, the anti-tumor activity of the first generation of CAR modified T cells is limited in vivo, and the decreased proliferation of T cells ultimately leads to apoptosis [12, 13]. The second generation of CAR adds a new costimulatory signal in the intracellular region, which enlarges the original “signal I” derived from TCR/CD3 complex [14, 15]. Many studies have shown that compared with the first generation of CAR, the second generation of CAR carrying “signal II” has the same antigen specificity, increased T cell proliferation and cytokine secretion, enhanced secretion of anti-apoptotic proteins, and delayed cell death. The ubiquitously used costimulatory molecule is CD28 [16, 17], which have been gradually been replaced with CD137 (4-1BB) [18]. In addition, an idea of using NK cell receptor CD244 has also been proposed to promote sustained activation and proliferation of CAR-T cells [19].
In order to further improve the design of CAR, many studies began to focus on the development of the third generation of CAR, including not only “signal I”, “signal II”, but also additional costimulatory signals. Studies using different targets and costimulatory signals were conducted to compare the results of the second and third generation of CARs and obtained quite encouraging experimental results [20, 21]. Zhong et al. combined CD28 and 4-1BB costimulatory signaling domains to construct a CAR specific for prostate-specific membrane antigen (PSMA). This group demonstrated that the CAR they constructed was able to induce strongest PI3K/Akt activation and Bcl-XL expression in vitro, and the least apoptosis in transduced peripheral blood CD8+ T cells [20]. Targeting a different tumor marker, MUC1, Wilkie et al. designed a CAR containing a fused CD28/OX40/CD3ζ endodomain. Using the engineered CAT-T cells and upon MUC1 stimulation, they showed that these CAR-T cells were able to secrete proinflammatory cytokines indicative of both type-1 (IFN-γ) and Th17 (IL-17) differentiation in vitro. It was noteworthy that IL-17 has been known as tissue destructive cytokine in autoimmune disease animal models, although its anti-tumor effect is still to be elucidated [20, 21].
Up to date, it is still uncertain which design is better between the second generation and the third generation of CAR. Additionally, the second and third generations of CARs have their own on-going clinical trials in the US, China and Europe, and the development and outcome of these clinical trials are being closely watched.
Obstacles in treating solid tumors with CAR-T cells
1. CAR-T cells traffick to the tumor sites
To bind to their target proteins on the surface of tumors, CAR-T cells first need to traffick to tumor sites. This is the fundamental prerequisite for T cell immunotherapy to work properly. Unlike the hematological malignancies, in the case of solid tumors, T cells trafficking to and infiltrating into tumor sites are oftentimes greatly limited by the immunosuppresive microenvironment [22]. Also unlike hematological malignancies that are easy to be targeted and reached by CAR-T cells, some of chemokines such as CXCL1 [23], CXCL12, and CXCL5 [24, 25] secreted by solid tumors prevent T cells from trafficking toward and infiltrating into the tumor lesions. Due to lack of corresponding chemokine receptors expressed on T cells, it is difficult for them to traffic and infiltrate into tumor sites, drastically impeding the ability of CAR-T cells for their designed immuno-cytotoxicity for killing tumor cells. Therefore, in order to overcome this obstacle, T cell will need to be modified to express chemokine receptor that matches the corresponding tumor-derived chemokine. An early study conducted by Kershaw et al. demonstrated that chemokine receptor (CXCL1 receptor)-engineered T cells can greatly drive themselves to migrate toward melanoma cells [23]. This study demonstrated the feasibility that T cell traffic can be re-directed to tumor sites by chemokines secreted by tumor cells.
2. CAR-T cells infiltrate into tumors
Once CAR-T cells successfully traffic to tumor sites, infiltrating into tumor microenvironment is a required critical step to exert anti-tumor effects. This is a very highly dynamic and regulated complex step: it involves rolling, adhesion, extravasation and chemotaxis [26]. Solid tumors have unique histopathological features, such as concentrated blood vessels [27], as well as tumor-associated fibroblasts and myeloid cells forming extracellular matrix (ECM). While these features favor the growth of solid tumors, they impose the difficulty for T cell infiltration in tumor sites [27], thus preventing the continuous contact between T cells and tumor cells that is necessary for T cells to exert cytotoxic antitumor effects.
3. The immunosuppressive state of microenvironment in solid tumors
The immunosuppressive microenvironment within solid tumor has special histopathological features as manifested by high density blood vessels, extensive vascular leakage, poor integrity of tissue structure, and others [2]. These changes result in hypoxia, low pH, immune suppressor cells, augment of inhibitory checkpoint, and more tumor-derived cytokines [28-30]. Following are three major factors that affect the antitumor effects of T cells:
3.1 Immune suppressor cells
Solid tumors generally consist of a large number of immune suppressor cells, such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages. These cells all play roles to protect solid tumor cells from being killed by the host's immune system. Theoretically, costimulatory molecules, such as CD28 and CD137, may benefit CAR-T cell activation and survival in tumor sites [31, 32]. In addition, blocking granulocyte-macrophage colony stimulating factor (GM-CSF) dependent MDSC expansion and PD-L1 expression on MDSC may be a potential approach to enhance the antitumor effects of CAR-T cells [33].
3.2 Tumor-derived cytokines
Tumor-derived cytokines are soluble factors that deter the efficacy of cancer immunotherapy for solid tumors. Transforming growth factor-β (TGF-β) is an inhibitory tumor cytokine that plays a major role in alleviating the antitumor response. TGF-β downregulates CD8+ effector T cell function and upregulates Treg maturation [27]. In contrast, neutralizing TGF-β by antibody or small molecular drugs improves CD8+ T cell functions and enhances the antitumor response by increasing the number, infiltration and persistence of adoptively transferred T cells. Moreover, the activation of cytokines, such as interleukin (IL)-2 and IL-15, improves the antitumor effects of CAR-T cells [34]. IL-12, in particular, alters the tumor microenvironment [35], eliminates antigen-negative tumor cells [36], and prolongs the survival of T cells to increase efficacy of immunotherapy through recruiting and activating macrophages and other innate immune cells [27]. In addition, to alter the tumor microenvironment, armored CAR-T cells have been generated to secrete proinflammatory cytokines to improve CAR-T cell efficacy within the tumor microenvironment [37].
3.3 Checkpoint inhibitory ligands
Inhibitory immune-checkpoint ligands induce the suppressive function of the immune response and are usually overexpressed in solid tumors. For example, PD-L1 is ligand for PD-1 receptor and inhibits CAR-T cell activation by binding to PD-1. PD-L1 has the two function motifs, immune-receptor tyrosine-based inhibitor motif (ITIM) and immune-receptor tyrosine-based switch motif (ITSM), which are involved in dephosphorylation of TCR-associated CD3 and zeta chain-associated protein kinase 70 (ZAP70) [27, 38]. Blocking PD-1 through specific antibody, shRNA, or dominant negative receptor restored the function of Mesothelin-targeting CD28 CAR T cells, and CAR-T cells with constitutive anti-PD-1 secretion were more functional, expandable, and efficient in tumor eradication than parental CAR-T cells in a human lung carcinoma xenograft mouse model [39, 40]. Checkpoint blockades are being applied to many clinical trials in combination treatment with CAR-T cells [41, 42].
4. The immune-related adverse events(irAEs)
One of the major challenges in CAR-T cell therapy for solid tumors is the irAEs [43-46]. In 2010, death of a patient with colon cancer metastasis to the lung and liver following ERBB2-targeting CAR-T cell therapy was reported. The cause may be due to the recognition of CAR-T cells to low levels of ERBB2 on lung epithelial cells and resulted in triggering the release of remarkable amount of cytokines [43]. There are two major mechanisms leading to the increased toxicity observed with CAR-T cell therapy:
4.1 On-target/on-tumor toxicity
The most life-threatening toxicity following CAR-T cell immunotherapy is on-target/on-tumor toxicity, which relates to adverse effects, such as cytokine release syndrome (CRS). Upon binding to antigens on target tumor cells, infused CAR-T cells are extensively activated, leading to the release of a large number of inflammatory cytokines. The CRS symptoms include fever, fatigue, nausea, vomiting, diarrhea, rashes, delirium, hallucinations, hypotension, and even severe multiple organ failure [44, 47]. The CRS has been classified into five grades according to the Common Terminology Criteria for Adverse Events (CTCAE) ranging from a mild reaction/grade 1 to death/grade 5. The level of the CRS severity is believed to be strongly correlated with tumor burden. IL-6 appears to be a major mediator of the CRS [48], and thus the key strategy to ameliorate this side effect is to block IL-6 directly with the IL-6R inhibitor tocilizumab [49]. In addition to tocilizumab, new cytokine-directed approaches might be considered to overcome the CRS.
4.2 On-target/off-tumor toxicity
Another type of toxicity originates from binding of CAR-T cells to target antigen that is also expressed on normal cells. This toxicity may lead to destruction of healthy cells and organs [43]. In the treatment of neuroblastoma, the fatal neurotoxicity was observed in high-affinity GD2-specific CAR-T cell therapy, suggesting that GD2 may be an inappropriate targeting antigen for CAR-T cell therapy [50]. Most of the CAR-T cell target antigens seem not to be tumor specific and shared by normal cells. Therefore, antigen specificity becomes a crucial factor for CAR-T therapy. To reduce the risk of this toxicity, selectivity of a safer antigen for CAR-T cells is critical and may be improved by utilizing dual CAR targeting, and modulating the sensitivity of single-chain variable fragment (scFv) [51]. Toxicity can also be minimized by controlling CAR-T cell activity through manipulating CAR expression and introducing a switch that can be triggered in the severe condition of the irAE [52, 53].
5. Antigen specificity
Tumor cells exhibit different morphologic and phenotypic profiles. One of the predominant biological characteristics of solid tumors is their heterogeneity. Tumor heterogeneity significantly affects immunotherapy efficacy due to the immune target may limit to a certain population of solid tumor cells. Therefore, to improve target specificity and eliminate toxicity are necessary in CAR-T cell therapy. EGFR variant III (EGFRvIII) is believed to be the only tumor-specific antigen (TSA) for CAR-T cells. It is found to be exclusively expressed on human cancer cells rather than normal healthy cells [54-56], EGFRvIII-targeting CAR-T cells precisely target tumor cells and are helpful to increase efficacy and reduce toxicity.
The shortage of TSAs severely limits the use of CAR-T therapy. Therefore, targeting tumor-associated antigens (TAAs) is an alternative method to overcome the shortage of TSAs. Most of ongoing clinical trials of CAR-T cell therapies for solid tumors are using these TAAs [57]. Attention should be paid to use of this CAR-T strategy because it may cause damage of normal tissues as TAAs are not only overexpressed on tumor cells but also on some normal cells.
6. Strategies to refine CAR-T cells for treatment of solid tumors
To overcome the obstacles mentioned above, we need to improve T cell efficacy and adopt new strategies that are discussed as below:
6.1 Improving CAR-T cell trafficking and infiltration
As mentioned earlier, CAR-T cell trafficking requires the establishment of chemotaxis migration between chemokines secreted by tumor cells and chemokine receptors on effector T cells. Different tumor types produce various chemokines so that a corresponding chemokine with its appropriate chemokine receptor is a critical factor for successful trafficking of T cells to tumor sites [22]. T cells engineered with the chemokine receptor CXCR2, which binding to the ligand CXCL1 on tumor cells, have been demonstrated to effectively traffick toward melanoma (Figure 1A) [23]. Di Stasi et al. demonstrated that the anti-lymphoma effects of T cells were improved by co-expression of CCR4. CCR4 can enhance the homing of CAR-CD30-modified T cells to CD30+ Hodgkin lymphoma cells by its secreted CCL17 (ligand for CCR4) [58]. Similarly, the antitumor effects towards malignant pleural mesothelioma and neuroblastoma were improved by CCR2b expression of mesothelin- and GD2-targeting CAR-T cells, respectively [59]. The local delivery of IL-7 and CCL19 by CAR-T cells improved immune cell infiltration and CAR-T cell survival in the tumor [60]. Conversely, tumor cells mediate the secretion of chemokines from suppressive immune cells, such as CXCL5, and the lack of suitable chemokine receptors on T cells, decrease migration of CAR-T cells into tumors (Figure 1A) [24, 25].
Regional delivery of T cell through intraperitoneal and intra-tumoral injection is also a direct way to deliver CAR-T cells. In a Phase I clinical trial study, ErbB-targeting CAR-T cells were delivered via intra-tumoral injection to patients with head and neck squamous cell carcinoma [61]. Local delivery is also suitable for other cancers, such as ovarian cancer and malignant pleural mesothelioma via the peritoneal and pleural cavities [22]. However, innovative deliveries are needed to be developed for those patients with tumors that are not easy to access by regional delivery.
Stroma is mainly composed of ECM whose primary ECM component is heparan sulfate proteoglycan (HSPG) [22]. Degradation of HSPG is the first step for T cells to pass stroma-rich solid tumors. Heparanase (HPSE) is an enzyme that degrades HSPG. Reduction of HPSE mRNA expression has been found in in vitro-expanded CAR-T cells, thus restricting their anti-tumor function in solid tumors due to the abundance of stroma [62]. Caruanu, et al. had engineered CAR-T cells that express HPSE to promote T cell infiltration and antitumor activity. Their study showed improved capability of CAR-T cells to degrade the EMC [62]. Wang et al. demonstrated that fibroblast activation protein (FAP)-targeted CAR-T cells enhanced cytotoxic function by decreasing tumor fibroblasts in animal models [27]. VEGF receptor-2, highly expressed on tumor-associated endothelial cells, has been selected as another candidate for facilitating T cell infiltration [26]. CAR-T cells engineered with VEGF receptor-2 generated a durable and increased tumor infiltration and enhanced antitumor effect (Figure 1A) [63]. With anti-angiogenic pharmacologic intervention, tumor infiltration of CD8+ T cell is increased by blocking VEGF receptor-2 to achieve long-term therapeutic efficacy [64]. It is clear that the complexity of genetic modification and long-term period of in vitro culture may also limit the clinical application of CAR-T cell therapy.
6.2 Reversal of the immunosuppressive microenvironment
Preclinical data have shown that incorporation of costimulatory molecules into CARs helps CAR-T cells to reverse the immunosuppressive tumor microenvironment, for example, CD28 co-stimulation overcomes TGF-β-mediated repression of proliferation and enhances T-cell resistance to Treg cells [31, 32, 65]. Burga et al. showed that MDSCs are responsible for liver metastases and inhibition of CEA-targeted CAR-T cells. Following MDSC depletion in a mouse model, the antitumor activity of CAR-T cells was rescued [33]. During MDSC recruitment, tumor cells secrete high levels of granulocyte-macrophage colony-stimulating factor (GM-CSF) in vivo. Thus, GM-CSF neutralization might be an alternative method to inhibit MDSC expansion (Figure 1) [66, 67].
A. Improving CAR-T cell trafficking and infiltration. 1) Targeting the tumor stroma or vasculature: tumor fibroblasts are depleted by FAP-targeting CAR-T cells to inhibit tumor growth. 2) ECM consumption by a secreted enzyme: the ECM (heparan sulfate proteoglycans) can be disintegrated by HPSE. 3) Homing to tumors expressing chemokine receptors (CCR4, CCR2b): genetically modified CAR-T cells express chemokine receptor(s) matching the tumor chemokine to facilitate migration to the tumor cells. B. Overcoming the immunosuppressive tumor microenvironment. 1) Anti-PD-L1-secreting CAR-T cells: human NK cells are recruited to the tumor site through secretion of anti-PD-L1 antibodies from CAR-T cells. 2) CAR-T cell therapy is potently enhanced by PD-1 blockade. 3) Blockade with the IL-10/TGFβ receptor. 4) Proinflammatory cytokines secreted by armored-CARs and TRUCKS (IL-12 showed an increased antitumor efficacy). C. Multiplex CAR-T cells to target the tumor profile. 1) Pooled CAR-T cells: multiple single-targeting CAR-T cells are mixed together. 2) Multi-specific CAR-T cells: one bispecific CAR-T cell consists of two specific CARs. 3) Tandem CAR-T cells: two different CARs connected in tandem possessing a common intracellular domain. 4) Conditional CAR-T cells: one CAR has a CD3ζ signaling domain, and the other has a costimulatory domain. D. Minimizing CAR-T cell toxicity. 1) EGFRvIII CAR: EGFRvIII is the only truly tumor-specific antigen that is completely restricted to human cancer, such as glioblastoma. 2) Dual CAR: the first CAR activates T cell function through the CD3 signaling domain, and the second CAR contains CD28/CD137 to co-stimulate the signaling function. 3) Affinity-tuned CAR. 4) Inhibitory CAR (iCAR): normal cells are maintained safely because of negative signaling by iCARs that only have antigens that are expressed on normal cells. 5) 'ON' switch CAR-T cells: a CAR molecule is attached with a costimulatory CD3ζ signaling domain that can only be activated in the presence of a small molecule acting as an 'ON' switch. FAP: fibroblast activation protein; ECM: extracellular matrix; HPSE: heparanase; TRUCKs: T cells redirected for universal cytokine killing; CAR: chimeric antigen receptor. Note: The lines represent what A, B, C and D include respectively.
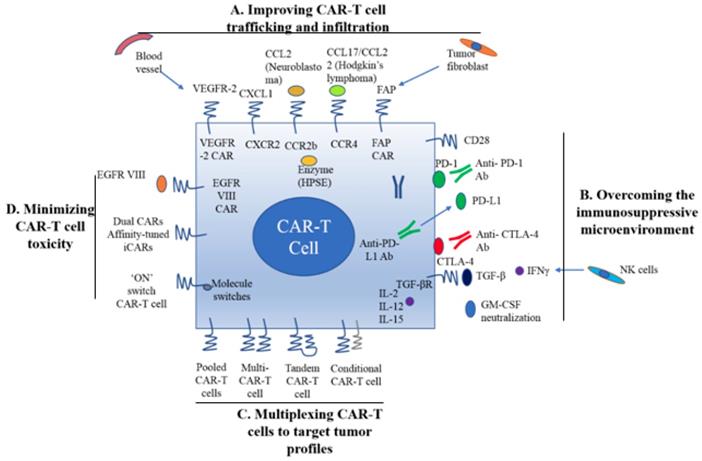
Inhibition of immunosuppressive cytokines by introducing a dominant-negative TGF receptor on CAR-T cells also improves the efficacy of CAR-T cells [68]. In the tumor microenvironment, cytokine (e.g., IL-2, IL-12, and IL-15) activation could antagonize the effects of immunosuppressive factors and improve CAR-T cell efficacy. Studies have shown that the antitumor function is enhanced by CAR-T cells that co-express IL-12 (Figure 1B) [35, 69]. Equally, IL-12 secretion by CAR-T cells has been shown to destroy antigen-negative cancer cells that may escape from the therapy [36]. Other studies have confirmed that the antitumor effects of CAR-T cells are enhanced by IL-2 and IL-15 production [70-74]. To rebalance the tumor microenvironment, armored CAR-T cells or redirected T cells for universal cytokine killing (TRUCKs) have been studied in preclinical trials. Koneru M et al. demonstrated that these armored CARs and TRUCKs secreted proinflammatory cytokines that induced transformation of the tumor microenvironment in mice with human ovarian cancer xenografts [75].
For treatment of cancers such as melanoma and renal cancer, the application of checkpoint inhibitors, such as anti-PD1, anti-CTLA-4 and anti-PD-L1, improves T cell responses in patients [41, 76]. Preclinical data showed that blocking PD1-mediated immunosuppression also boosts the therapeutic effects of CAR-T cells (Figure 1B) [41]. In a study of CAR-T cells with PD-1 blockade in a mouse model, Moon EK et al. found that PD-1 blockade improved the antitumor activity of human mesothelin-targeting CAR-T cells (Figure 1B) [77]. HER2-targeted CAR-T cells in combination with anti-PD-1 significantly eliminated tumor cells in a mouse model [41]. Suarez ER et al. engineered CAR-T cells to secrete anti-PD-L1 antibodies instead of administering anti-PD-L1 antibody [78]. This approach not only reduced tumor progression but also enabled human NK cells to migrate to the tumor sites in a mouse model of renal carcinoma. NK cells exert the anti-tumor efficiency through antibody-dependent cell-mediated cytotoxicity (ADCC) and IFNγ stimulation of CD8+ T cells [22]. Therefore, CAR-T cell therapy for solid tumors can be improved by infiltration of other immune cell subsets into the tumor microenvironment through local anti-PD-L1 antibody secretion. Interestingly, the number of MDSCs was also significantly diminished in the mouse tumor microenvironment. In addition, certain molecules, such as IL-6, may play double-sided roles in tumor microenvironment [79].
6.3 Multiplexing CAR-T cells to target tumor profiles
Given by tumor heterogeneity and antigen escape variants, the next development in CAR-T cell therapy is to target more than one antigens, similar to the combinatorial strategy of traditional chemotherapy [80]. This approach increases the chances of eliminating multiple sub-clonal populations simultaneously by targeting multiple TAAs or other factors in the tumor microenvironment.
There are various ways to create multi-specific CAR-T cells. The basic approach is to construct a pool with two unispecific CAR-T cell products, namely, a 'CAR pool', for simultaneous co-administration (Figure 1C) [81]. A strategy of using combination targeting of HER2 and IL13Rα2 by bispecific T cells in glioblastoma was shown to be more efficient in eliminating tumor cells and showed less antigen escape variants compared with the CAR-T cells targeting HER2 alone in both in vitro and in vivo mouse xenograft models [82]. When treating lung cancer, a similar approach was applied to pool EphA2-targeted CAR and FAPα-targeted CAR to target the tumor microenvironment. This combination showed significant tumor killing in vitro and prolonged the survival of mouse xenografts compared with application of either CAR alone [83].
A single T cell platform can also possess dual antigen targeting when two (bispecific [bi]CARs)[83] or more (triCARs) [84] unispecific CARs are expressed in T cells (Figure 1C) [81]. In breast cancer, the proliferation of biCAR-T cells targeting HER2 and MUC1 in vitro was dependent on contact with both antigens simultaneously. biCAR-T cells coexpressing HER2 and IL13Rα2 CAR molecules showed significant potential of eliminating tumor cells compared with unispecific CARs alone or pooled products in a glioblastoma model and the antigen escape variants also decreased substantially [83]. In a non-small cell lung cancer model, the combination of PSCA- and MUC1-targeting CAR-T cells synergistically eliminated PSCA and MUC1 positive tumor cells [84]. However, another study reported the biCAR-T cells did not enhance the level of IL-2, a marker of T cell activation compared with single CAR-T cells. This is possibly caused by the steric hindrance during simultaneously binding both antigens [85].
CAR-T function can be improved by alteration of the CAR configuration. In a proof-of-concept study, a tandem (tan) CAR was designed by Grada et al. that had two different scFv domains in tandem linked by a spacer (Figure 1C) [81]. The tanCAR individually recognized each target antigen, and its function was strengthened when both antigens interacted with their scFv domain simultaneously in vitro and in vivo [85]. This phenomenon suggests co-docking occurring to both target molecules. This concept was also established in an inducible in vitro targeting model and in vitro and in vivo antigen escape models [86]. In addition to biCARs, triCARs, and tanCARs, neoantigen-like tissue factors (TFs) become promising novel antigens. In certain types of lung cancer, melanoma and other cancers, TFs are overexpressed on the surface of tumor cells, and TF-CAR-T cells were significantly activated to show specific cytotoxicity to TF-positive tumor cells in vitro [87].
Multispecific CAR-T cells have a potential advantage over unispecific CAR-T cells in avoiding on-target/off-tumor toxicities [49]. In lymphoreticular malignancies, instead of CRS, multispecific CAR-T cells led to specific tissue cholangitis [88]. A conditional biCAR was developed to minimize tissue damage (Figure 1C) [81]. Different endomembrane signaling domains are included in the two CARs of T cells, and T cell activation depends on simultaneous interaction of both CARs with their specific TAAs. For example, in prostate cancer, a biCAR was constructed with two prostate cancer antigens (prostate-specific membrane antigen and prostate stem cell antigen), the expression of which are also found on normal tissues. There is a CD3ζ intracellular domain and a CD28-4-1BB domain that constitute a costimulatory intracellular domain. This special biCAR effectively targeted tissues expressing both antigens but not tissues positive with single antigen [89]. Another example is that a conditional biCAR of mesothelin and α-folate receptor (FRα) with intracellular signaling domains of separated CD3ζ and CD28 are fully activated only when both antigens are present simultaneously in ovarian cancer. When multi-specific CAR-T cells were activated, cytokines were released and the tumor burden was reduced in both in vitro and in vivo experiments, similar to the single second generation of mesothelin-targeting CAR-T cells [90]. An approach of adding a further layer of titratable control to activate the CAR-T cells was recently described by Wu et al. The structure contained a small molecule that forms a heterodimerized biCAR, consisting of a binding domain for the antigen and a domain for intracellular signaling (Figure 1C) [81, 91].
Multitargeting CARs have several potential advantages over conventional unispecific CAR molecules. Through cytokine release and cytolysis in vitro and decreasing the tumor burden in vivo, the antitumor effects of CAR-T cells may be further enhanced. Other structures, such as the tanCAR, may strengthen functionality by changing steric interactions with tumor antigens [81]. However, the percentage of targeted tumor cells within tumor tissues should be concerned with multitargeting CARs [89, 92]. There are still growing demands to find novel antigens as potent targets.
6.4 Minimizing CAR-T cell toxicity
As mentioned above, to date, EGFRvIII is exclusively the tumor-specific CAR antigen, and it is fully restricted to human cancers and mostly found in glioblastoma [54, 55]. EGFRvIII-positive tumor cells can be precisely targeted by specific CAR-T cells to enhance therapeutic efficacy and decrease the corresponding toxicity. In an animal model study of glioblastoma, EGFRvIII CAR treatment produced very promising results, and clinical trials are underway to test it on glioblastoma patients [93, 94].
Various targeting strategies have been developed to increase the specificity and safety of CAR-T cell therapy (Figure 1D) [27]. One strategy is to modify T cells with two different CARs, and thus, tumor cells and normal cells can be induced to differentiate effectively. The first CAR molecule consists of the CD3ζ signaling domain, and the second CAR molecule contains the CD28 or CD137 costimulation signaling domain [27, 89, 90]. Only when two CARs expressed in the same cells, the CAR-T cells can be fully activated. The on-target/off-tumor toxicities caused by TAA expression on normal tissues are limited by the CAR costimulatory domain on the same T cells. Preclinical data demonstrated that targeting multiple TAAs helps to minimize the possibility of antigen escape variant and efficiently target tumor subclones [81].
Furthermore, tumor specificity can be enhanced by an affinity-tuned CAR. In recent years, studies have demonstrated that adjusting the CAR affinity of CAR-T cells and maintaining potent antitumor efficacy in vivo, could distinguish tumor cells from normal cells with the expression of the same antigens at lower levels [51, 95]. Thus, tuning CAR sensitivity through high scFv affinity in CAR-T cell therapy for overexpressed antigens in solid tumors provides a promising approach. In addition, an alternative strategy called inhibitory CAR (iCAR) can reduce unwanted off-target effects. iCARs recognize specific antigens that are expressed only on normal cells, thus protects CAR-T cell from attacking normal cells by induction of the negative signaling. Fedorov et al. demonstrated that an iCAR possessing PD-1 and CTLA-4 inhibition ability prevented normal tissues from off-target effects in mouse models, applying the principle of checkpoint inhibition to an antigen in the normal tissue but not in the tumor [96]. However, due to the risk of iCAR to completely abolish T cell function, updated modifications, such as suicide genes, may be useful to control the unwanted toxicity [97].
The introduction of suicide switches, such as herpes simplex thymidine kinase (HSV-TK), inducible caspase 9 (iCasp9) and CD20, has already been clinically tested to control CAR-T cells [53, 98, 99]. This smart “tumor sensing” strategy, with the balance of two-antigen recognition, potentially diminishes on-target/off-tumor toxicity [100]. However, one disadvantage is the unintended elimination of the modified T cells. Transient RNA expression of CAR offers temporary redirected T cell activity and limits toxic side effects, even though, in solid tumor models, RNA CAR-T cell activity is limited by insufficient tumor infiltration [22].
Companion diagnostics for CAR-T cells
Many reviews have exclusively discussed strategies to improve the efficacy of CAR-T cell therapy against solid tumors, while less attention has been paid to companion diagnostics and related detections. These diagnostics and detections are now becoming indispensable for CAR-T cell therapies as they are playing important roles in patient selection, disease outcome prediction, treatment regimen decisions, efficacy evaluation, and relapse prevention. In these regards, we would like to focus on the topics of detection of target antigen expression and the properties of CARs.
Since the death report of a metastatic colon cancer patient caused by on-target/off-tumor effects of ERBB2-targeted CAR-T cell therapy in 2010, the side effects of CAR-T cell therapy for solid tumors have received extensive attention [22, 43]. The on-target/off-tumor effects of CAR-T cells are mainly caused by T cells attacking normal cells due to their expression of target antigens [50, 100]. Thus, there is the compelling need for the identification of TSAs for treating solid tumors with CAR-T cells. Unless the depletion of those normal cells is tolerable, similar to CD19+ B cells, CARs with moderate affinity are considered to improve the effect of CAR-T cell therapy [2, 27, 46, 101, 102]. Despite the lack of an ideal TSA, scientists have attempted to reduce the side effects of CAR-T cell therapies by investigating the extracellular domain of CARs, especially the affinity of the scFv domains. Studies have reported that reducing the affinity of CARs could spare normal cells that express the target antigen at a low level, thus avoiding the on-target/off-tumor effect [51, 95, 103]. With detail comparisons between the T cell receptors and CARs, more precise adjustments have been made to CARs [104, 105]. A CD38-targeted CAR with ~1,000-fold reduced affinity was confirmed to be more suitable for treating multiple myeloma, and an ICAM-1-targeting CAR with micromolar affinity was more appropriate than nanomolar counterparts for treating solid tumors [102, 106]. In addition, the density of CARs on T cells is another factor that should be considered along with the affinity. Altogether, CARs with moderate affinity are suitable for targeting TAAs in solid tumors. However, there is still an important factor that impacts the affinity choice of CARs and should be considered: the density or expression level of the target antigens on tumor cells [107, 108].
To test the expression level of the target antigens on tumor cells, immunohistochemical (IHC) staining is ubiquitously utilized for regimen decisions. Another point that should be highlighted is the differences between the antibodies used for IHC and those for CARs, within which the scFv domains are responsible for binding the target antigens. Thus, using an antibody with the same clone origin for IHC and scFv for CARs has been emphasized [108, 109]. Nevertheless, peptides may form different spatial structures under different circumstances, and this may hide some of the essential binding sites of the scFv domains compared with the related antibodies for IHC [110-113]. Thus, the optimal antibody for IHC tests should bind the same epitopes as the relative CAR, or be in form of custom-made scFv peptide by coupling with a tag for secondary antibody detection.
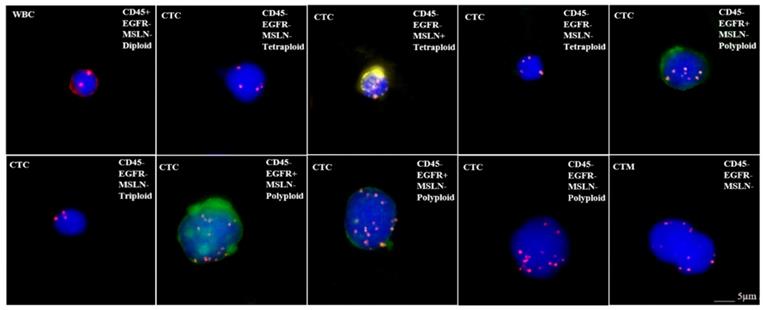
When discussing the detection of target antigen expression on tumor tissues using IHC assays, a question should be raised about the patients who do not have tumor tissue samples available such as in the situation of early stage of disease or metastasis, and minimal residual disease (MRD). In order to test whether those patients are suitable for respective CAR-T treatement, an alternative approach may be found by detecting the expression of target antigens on circulating tumor cells (CTC). We have tested the applicability of this protocol by examining the expression of EGFR and mesothelin on CTCs for CAR-T cell therapy, because these antigens are frequently targeted in clinical trials (Figure 2) [114]. Our preliminary results as shown in Figure 2 demonstrated the feasibility for adapting CTC as one of the companion diagnostic and monitoring tool for CAR-T therapy. However, there are key factors that should be considered with this approach before it can become a reliable clinical testing utility. First, the number of CTCs varies with cancer types and stages. In order to assure sufficient CTCs are obtained and analyzed, the optimal sample blood volume still remain to be determined because too few CTCs may lead to a false negative result. Second, the epithelial-mesenchymal phenotype of CTCs must be considered because the epithelial-mesenchymal transition (EMT) process may hide the expression of target antigens [115, 116]. Many currently available methods for CTCs enrichment do not take this into account, thus lead to another possibility for negative results. Finally, the correlation between the numbers of target antigen-positive CTCs and the therapeutic efficacy needs further investigation after treatment (Table 2). Beside IHC and CTC detection, many other methods and technologies, such as circulating tumor DNAs, circulating miRNAs, T-cell receptor sequencing/profiling and tumor mutation burdens, are currently being developed for companion diagnostics and efficacy monitoring for CAR-T cell therapy.
Microscope images showing CTC analysis results in a bladder cancer patient. CTCsare characterized as two mesothelin (MSLN)+ CTCs (expressing yellow), three EGFR+ CTCs (expressing green), MSLN-/EGFR- CTCs and circulating tumor microemboli (CTM). In addition, each CTC is identified as triploid, tetraploid or polyploid by a FISH probe for CEP8. WBC: white blood cell; CTC: circulating tumor cell; MSLN: mesothelin; CTM: circulating tumor microemboli; EGFR: epidermal growth factor receptor
Summary of application of companion diagnostics
| Diagnostic Assays | Intentions | Related Issues | Suggestions |
|---|---|---|---|
| Immunohistochemistry (IHC) | Detecting the expression of targeting antigens for CAR-T cells | Different binding epitopes with scFv in CAR | Apply the same clone origin of the antibody for IHC and scFv for CAR; Custom synthesis of the scFv peptide by coupling with the substrate for the second antibodies. |
| Affinity tests | Reduce the side effects of CAR-T cells for solid tumors | Too high will cause on-target/off-tumor effects; Too low will cause inefficacy in killing cancer cells. | Moderate affinity is proven to be a better choice; Balance the affinity and density of CAR on the T cells; Balance the affinity of CAR and density of the antigens on the tumor cells. |
| Circulating tumor cells (CTCs) | Provide alternative for patients without tumor tissue; Evaluate therapeutic efficacy of CAR-T cells; Real-time monitoring of antigen-positive tumor cell relapse. | CTCs are rare in blood; CTCs with a mesenchymal phenotype may hide the expression of antigens. | Confirm the correlation between the positivity of the antigen-repressing CTCs and tumor tissues; Extract more blood than the traditional volume; Investigate the correlation between the number of targeting antigen-positive CTCs and the therapeutic efficacy needs. |
Conclusions
In recent years, strategies have been applied to improve the efficacy and safety of CAR-T cell treatments for solid tumors, mainly through overcoming obstacles caused by the characteristics of T cells and tumor environment. Companion diagnostics, including IHC and CTC detection assays, can be applied to ameliorate the treatment of solid tumors with CAR-T cells.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fournier C, Martin F, Zitvogel L, Kroemer G, Galluzzi L, Apetoh L. Trial watch: adoptively transferred cells for anticancer immunotherapy. Oncoimmunology. 2017;6:e1363139
2. Zhang H, Ye ZL, Yuan ZG, Luo ZQ, Jin HJ, Qian QJ. New strategies for the treatment of solid tumors with CAR-T cells. Int J Biol Sci. 2016;12:718-29
3. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B. et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol Ther. 2017;25:2214-24
4. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA. et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7:737
5. Hegde UP, Mukherji B. Current status of chimeric antigen receptor engineered T cell-based and immune checkpoint blockade-based cancer immunotherapies. Cancer Immunol Immunother. 2017;66:1113-21
6. Luo C, Wei J, Han W. Spotlight on chimeric antigen receptor engineered T cell research and clinical trials in China. Sci China Life Sci. 2016;59:349-59
7. Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35-45
8. Bridgeman JS, Hawkins RE, Hombach AA, Abken H, Gilham DE. Building better chimeric antigen receptors for adoptive T cell therapy. Curr Gene Ther. 2010;10:77-90
9. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720-4
10. Heuser C, Hombach A, Losch C, Manista K, Abken H. T-cell activation by recombinant immunoreceptors: impact of the intracellular signalling domain on the stability of receptor expression and antigen-specific activation of grafted T cells. Gene Ther. 2003;10:1408-19
11. Haynes NM, Snook MB, Trapani JA, Cerruti L, Jane SM, Smyth MJ. et al. Redirecting mouse CTL against colon carcinoma: superior signaling efficacy of single-chain variable domain chimeras containing TCR-zeta vs Fc epsilon RI-gamma. J Immunol. 2001;166:182-7
12. Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature. 1992;356:607-9
13. Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233-58
14. Bretscher P, Cohn M. A theory of self-nonself discrimination. Science. 1970;169:1042-9
15. Bretscher PA. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci U S A. 1999;96:185-90
16. Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N. et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995-1004
17. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G. et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822-6
18. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D. et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453-64
19. Altvater B, Landmeier S, Pscherer S, Temme J, Juergens H, Pule M. et al. 2B4 (CD244) signaling via chimeric receptors costimulates tumor-antigen specific proliferation and in vitro expansion of human T cells. Cancer Immunol Immunother. 2009;58:1991-2001
20. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18:413-20
21. Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L. et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180:4901-9
22. Zhang BL, Qin DY, Mo ZM, Li Y, Wei W, Wang YS. et al. Hurdles of CAR-T cell-based cancer immunotherapy directed against solid tumors. Sci China Life Sci. 2016;59:340-8
23. Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM. et al. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther. 2002;13:1971-80
24. Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S. et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016;6:80-95
25. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS. et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110:20212-7
26. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T cells into tumors. Cancer Res. 2014;74:7168-74
27. Xia AL, Wang XC, Lu YJ, Lu XJ, Sun B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: challenges and opportunities. Oncotarget. 2017;8:90521-31
28. Beatty GL, Moon EK. Chimeric antigen receptor T cells are vulnerable to immunosuppressive mechanisms present within the tumor microenvironment. Oncoimmunology. 2014;3:e970027
29. Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, Rooney CM. et al. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol Ther. 2017;25:249-58
30. Anderson KG, Stromnes IM, Greenberg PD. Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer cell. 2017;31:311-25
31. Koehler H, Kofler D, Hombach A, Abken H. CD28 costimulation overcomes transforming growth factor-beta-mediated repression of proliferation of redirected human CD4+ and CD8+ T cells in an antitumor cell attack. Cancer Res. 2007;67:2265-73
32. Loskog A, Giandomenico V, Rossig C, Pule M, Dotti G, Brenner MK. Addition of the CD28 signaling domain to chimeric T-cell receptors enhances chimeric T-cell resistance to T regulatory cells. Leukemia. 2006;20:1819-28
33. Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA. et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother. 2015;64:817-29
34. Wallace A, Kapoor V, Sun J, Mrass P, Weninger W, Heitjan DF. et al. Transforming growth factor-beta receptor blockade augments the effectiveness of adoptive T-cell therapy of established solid cancers. Clin Cancer Res. 2008;14:3966-74
35. Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP. et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther. 2011;19:751-9
36. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71:5697-706
37. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. 2015;4:e994446
38. Abate-Daga D, Hanada K, Davis JL, Yang JC, Rosenberg SA, Morgan RA. Expression profiling of TCR-engineered T cells demonstrates overexpression of multiple inhibitory receptors in persisting lymphocytes. Blood. 2013;122:1399-410
39. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR. et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130-44
40. Li S, Siriwon N, Zhang X, Yang S, Jin T, He F. et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin Cancer Res. 2017;23:6982-92
41. John LB, Kershaw MH, Darcy PK. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology. 2013;2:e26286
42. Chen N, Morello A, Tano Z, Adusumilli PS. CAR T-cell intrinsic PD-1 checkpoint blockade: A two-in-one approach for solid tumor immunotherapy. Oncoimmunology. 2017;6:e1273302
43. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843-51
44. Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther. 2010;18:666-8
45. DeFrancesco L. Juno's wild ride. Nat Biotechnol. 2016;34:793
46. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL. et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15:47-62
47. Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20:119-22
48. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M. et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188-95
49. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR. et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509-18
50. Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z. et al. High-Affinity GD2-Specific CAR T Cells Induce Fatal Encephalitis in a Preclinical Neuroblastoma Model. Cancer Immunol Res. 2018;6:36-46
51. Liu Y, Cao X. Immunosuppressive cells in tumor immune escape and metastasis. J Mol Med (Berl). 2015
52. Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E. et al. Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8:355ra116
53. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C. et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673-83
54. Li G, Wong AJ. EGF receptor variant III as a target antigen for tumor immunotherapy. Expert Rev Vaccines. 2008;7:977-85
55. Choi BD, Archer GE, Mitchell DA, Heimberger AB, McLendon RE, Bigner DD. et al. EGFRvIII-targeted vaccination therapy of malignant glioma. Brain Pathol. 2009;19:713-23
56. Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK. et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2(+) Breast Cancer Metastasis to the Brain. Clin Cancer Res. 2018;24:95-105
57. Castellarin M, Watanabe K, June CH, Kloss CC, Posey AD Jr. Driving cars to the clinic for solid tumors. Gene ther. 2018;25:165-75
58. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE. et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113:6392-402
59. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM. et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33:780-8
60. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018;36:346-51
61. van Schalkwyk MC, Papa SE, Jeannon JP, Guerrero Urbano T, Spicer JF, Maher J. Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurrent head and neck cancer. Hum Gene Ther Clin Dev. 2013;24:134-42
62. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES. et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21:524-9
63. Chinnasamy D, Yu Z, Theoret MR, Zhao Y, Shrimali RK, Morgan RA. et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest. 2010;120:3953-68
64. Manning EA, Ullman JG, Leatherman JM, Asquith JM, Hansen TR, Armstrong TD. et al. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res. 2007;13:3951-9
65. Lee JC, Hayman E, Pegram HJ, Santos E, Heller G, Sadelain M. et al. In vivo inhibition of human CD19-targeted effector T cells by natural T regulatory cells in a xenotransplant murine model of B cell malignancy. Cancer Res. 2011;71:2871-81
66. Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F. et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012;72:876-86
67. Schmidt K, Zilio S, Schmollinger JC, Bronte V, Blankenstein T, Willimsky G. Differently immunogenic cancers in mice induce immature myeloid cells that suppress CTL in vitro but not in vivo following transfer. Blood. 2013;121:1740-8
68. Bollard CM, Rossig C, Calonge MJ, Huls MH, Wagner HJ, Massague J. et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99:3179-87
69. Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z. et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010;70:6725-34
70. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J. et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24:1160-70
71. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA. et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106-15
72. Nishio N, Dotti G. Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. Oncoimmunology. 2015;4:e988098
73. Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA. et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261-71
74. Wang W, Ma Y, Li J, Shi HS, Wang LQ, Guo FC. et al. Specificity redirection by CAR with human VEGFR-1 affinity endows T lymphocytes with tumor-killing ability and anti-angiogenic potency. Gene Ther. 2013;20:970-8
75. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-23
76. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443-54
77. Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J. et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014;20:4262-73
78. Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti S. et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7:34341-55
79. Fisher DT, Appenheimer MM, Evans SS. The two faces of IL-6 in the tumor microenvironment. Semin Immunol. 2014;26:38-47
80. Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA. et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109-13
81. Navai SA, Ahmed N. Targeting the tumour profile using broad spectrum chimaeric antigen receptor T-cells. Biochem Soc Trans. 2016;44:391-6
82. Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y. et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. 2013;21:2087-101
83. Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF. et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. 2013;21:1611-20
84. Wei X, Lai Y, Li J, Qin L, Xu Y, Zhao R. et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology. 2017;6:e1284722
85. Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS. et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol Ther Nucleic Acids. 2013;2:e105
86. Hegde M, Wakefield A, Brawley VS, Grada Z, Byrd TT, Chow KK. et al. Genetic modification of T cells with a novel bispecific chimeric antigen receptor to enhance the control of high-grade glioma (HGG). J Clin Oncol. 2014;32:10027 -
87. Zhang Q, Wang H, Li H, Xu J, Tian K, Yang J. et al. Chimeric antigen receptor-modified T Cells inhibit the growth and metastases of established tissue factor-positive tumors in NOG mice. Oncotarget. 2017;8:9488-99
88. Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R. et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20-2
89. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71-5
90. Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH. et al. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. 2013;1:43-53
91. Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350:aab4077
92. Gross G, Eshhar Z. Therapeutic Potential of T Cell Chimeric Antigen Receptors (CARs) in Cancer Treatment: Counteracting Off-Tumor Toxicities for Safe CAR T Cell Therapy. Annu Rev Pharmacol Toxicol. 2016;56:59-83
93. Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA. et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23:1043-53
94. Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE. et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7:275ra22
95. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S. et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 2015;75:3505-18
96. Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5:215ra172
97. Minagawa K, Zhou X, Mineishi S, Di Stasi A. Seatbelts in CAR therapy: How Safe Are CARS?. Pharmaceuticals (Basel). 2015;8:230-49
98. Ciceri F, Bonini C, Stanghellini MTL, Bondanza A, Traversari C, Salomoni M. et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase III study. Lancet Oncol. 2009;10:489-500
99. Marin V, Cribioli E, Philip B, Tettamanti S, Pizzitola I, Biondi A. et al. Comparison of different suicide-gene strategies for the safety improvement of genetically manipulated T cells. Hum Gene Ther Methods. 2012;23:376-86
100. Lynn RC, Feng Y, Schutsky K, Poussin M, Kalota A, Dimitrov DS. et al. High-affinity FRbeta-specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia. 2016;30:1355-64
101. Han S, Latchoumanin O, Wu G, Zhou G, Hebbard L, George J. et al. Recent clinical trials utilizing chimeric antigen receptor T cells therapies against solid tumors. Cancer Lett. 2017;390:188-200
102. Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J. et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol Ther. 2017;25:1946-58
103. Song DG, Ye Q, Poussin M, Liu L, Figini M, Powell DJ Jr. A fully human chimeric antigen receptor with potent activity against cancer cells but reduced risk for off-tumor toxicity. Oncotarget. 2015;6:21533-46
104. Harris DT, Kranz DM. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol Sci. 2016;37:220-30
105. Oren R, Hod-Marco M, Haus-Cohen M, Thomas S, Blat D, Duvshani N. et al. Functional comparison of engineered T cells carrying a native TCR versus TCR-like antibody-based chimeric antigen receptors indicates affinity/avidity thresholds. J Immunol. 2014;193:5733-43
106. Park S, Shevlin E, Vedvyas Y, Zaman M, Park S, Hsu YS. et al. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep. 2017;7:14366
107. Arcangeli S, Rotiroti MC, Bardelli M, Simonelli L, Magnani CF, Biondi A. et al. Balance of Anti-CD123 Chimeric Antigen Receptor Binding Affinity and Density for the Targeting of Acute Myeloid Leukemia. Mol Ther. 2017;25:1933-45
108. Kunkele A, Taraseviciute A, Finn LS, Johnson AJ, Berger C, Finney O. et al. Preclinical Assessment of CD171-Directed CAR T-cell Adoptive Therapy for Childhood Neuroblastoma: CE7 Epitope Target Safety and Product Manufacturing Feasibility. Clin Cancer Res. 2017;23:466-77
109. Han X, Cinay GE, Zhao Y, Guo Y, Zhang X, Wang P. Adnectin-Based Design of Chimeric Antigen Receptor for T Cell Engineering. Mol Ther. 2017;25:2466-76
110. Wörn A, Plückthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol. 2001;305:989-1010
111. Belicky S, Damborsky P, Zapatero-Rodriguez J, O'Kennedy R, Tkac J. Full-length antibodies versus single-chain antibody fragments for a selective impedimetric lectin-based glycoprofiling of prostate specific antigen. Electrochim Acta. 2017;246:399-405
112. Perchiacca JM, Tessier PM. Engineering aggregation-resistant antibodies. Annu Rev Chem Biomol Eng. 2012;3:263-86
113. Bezverbnaya K, Mathews A, Sidhu J, Helsen CW, Bramson JL. Tumor-targeting domains for chimeric antigen receptor T cells. Immunotherapy. 2017;9:33-46
114. Jiang H, Song B, Wang P, Shi B, Li Q, Fan M. et al. Efficient growth suppression in pancreatic cancer PDX model by fully human anti-mesothelin CAR-T cells. Protein Cell. 2017;8:926-31
115. Chen L, Bode AM, Dong Z. Circulating Tumor Cells: Moving Biological Insights into Detection. Theranostics. 2017;7:2606-19
116. Mohme M, Riethdorf S, Pantel K. Circulating and disseminated tumour cells - mechanisms of immune surveillance and escape. Nat Rev Clin Oncol. 2017;14:155-67
Author contact
![]() Corresponding authors: Qijun Qian, Shanghai Baize Medical Laboratory, 75 Qianyang Road, Shanghai 201805, China. Email: qianorg; Zhenlong Ye, Shanghai Baize Medical Laboratory, 75 Qianyang Road, Shanghai 201805, China. Email: yezlorg
Corresponding authors: Qijun Qian, Shanghai Baize Medical Laboratory, 75 Qianyang Road, Shanghai 201805, China. Email: qianorg; Zhenlong Ye, Shanghai Baize Medical Laboratory, 75 Qianyang Road, Shanghai 201805, China. Email: yezlorg
Received 2019-2-18
Accepted 2019-7-16
Published 2019-9-7