ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2020; 16(3):447-459. doi:10.7150/ijbs.38672 This issue Cite
Research Paper
Cdh1 functions as an oncogene by inducing self-renewal of lung cancer stem-like cells via oncogenic pathways
Ting Ye1,2,4*, Jingyuan Li1,2*, Zhiwei Sun1,2, Doudou Liu1,2, Bin Zeng1,2, Qiting Zhao1,2, Jianyu Wang1,2 ![]() , H. Rosie Xing2,3
, H. Rosie Xing2,3 ![]()
1. Institute of Life Sciences, Chongqing Medical University, Chongqing, China
2. Laboratory of Translational Cancer Stem Cell Research, Chongqing Medical University, Chongqing, China
3. College of Biomedical Engineering, State Key Laboratory of Ultrasound Engineering in Medicine, Chongqing Medical University, Chongqing, China
4. Department of Laboratory Medicine, the Affiliated Hospital of Southwest Medical University, Luzhou, China
*these authors were contributed to the study equally
Abstract

The mortality rate of lung cancer remains the highest amongst all cancers despite of new therapeutic developments. While cancer stem cells (CSCs) may play a pivotal role in cancer, mechanisms underlying CSCs self-renewal and their relevance to cancer progression have not been clearly elucidated due to the lack of reliable and stable CSC cellular models. In the present study, we unveiled the novel oncogene function of cadherin 1 (Cdh1) via bioinformatic analysis in a broad spectrum of human cancers including lung adenocarcinoma (LUAD), adding a new dimension to the widely reported tumor suppressor function of Cdh1. Experimentally, we show for the first time that Cdh1 promotes the self-renewal of lung CSCs, consistent with its function in embryonic and normal stem cells. Using the LLC-Symmetric Division (LLC-SD) model, we have revealed an intricate cross-talk between the oncogenic pathway and stem cell pathway in which Cdh1 functions as an oncogene by promoting lung CSC renewal via the activation of the Phosphoinositide 3-kinase (PI3K) and inhibition of Mitogen-activated protein kinase (MAPK) pathways, respectively. In summary, this study has provided evidence demonstrating effective utilization of the normal stem cell renewal mechanisms by CSCs to promote oncogenesis and progression.
Keywords: cancer stem cells, Cdh1, oncogene, self-renewal, oncogenic pathways
Introduction
Lung cancer is currently the most common cause of cancer related death worldwide [1]. Among all primary lung cancers, non-small cell lung cancer (NSCLC) accounts for 85% of all lung carcinomas and lung adenocarcinoma (LUAD) is the main pathological subtype [2, 3]. Despite of therapeutic developments, the prognosis and treatment efficacy of LUAD have not been substantially improved, generally attributed to late diagnosis and tumor metastasis. As such, the 5-year survival rate remains less than 15% [4, 5]. Thus, search for new molecular mechanisms associated with LUAD oncogenesis and progression is ongoing. Accumulating evidence suggest lung cancer stem-like cells (CSCs), a group of cells with self-renewal capability, may play a role in lung cancer initiation and progression [6, 7]. However, the cancer biology characteristics of CSCs have not been well elucidated due to the lack of stable cellular models of CSCs. Consequently, mechanisms that regulate CSC self-renewal are poorly defined [8, 9].
Cdh1 gene coding E-cadherin is a transmembrane calcium-dependent adhesion molecule expressed in almost all epithelial cells [10]. In addition, E-cadherin is highly conserved evolutionarily, and is crucial for embryonic stem cell pluripotency, self-renewal and differentiation [11-16]. In early cancer literature, Cdh1 is widely regarded as a tumor suppressor gene [17]. It's down-regulation or silencing by DNA methylation is associated with loss of epithelial morphology and increased invasiveness through epithelial-mesenchymal transition (EMT) [18-20] and is correlated with high grade, advanced stage, and poor prognosis [21, 22]. Noteworthy, in recent years, few studies showed a positive correlation between Cdh1 expression and metastasis [23-25], though the mechanisms explored appeared to involve the reserve process of EMT - MET (mesenchymal to epithelial transition) [26-28]. The discrepancies between these findings and those on Cdh1 as a tumor suppressor have not been resolved. In addition, whether Cdh1 may regulate the self-renewal of CSCs as it does in normal stem cells has not been examined at the mechanistic level.
Bioinformatics has been widely applied in cancer research. In the present study, through bioinformatics analyses of Oncomine, Gene Expression Omnibus (GEO), The Cancer Genome Atlas (TCGA) and Gene Expression Profiling Interactive Analysis (GEPIA) databases, we uncovered that Cdh1 gene expression was elevated in human cancer tissues compared with normal counterparts in 17 types of cancers analyzed, including LUAD. Moreover, in LUAD, Cdh1 expression correlated with clinicopathological features and prognosis. This clinical finding has added a new dimension to our knowledge about Cdh1 in addition to its role as a tumor suppressor. Moreover, Cdh1 expression was increased in the mouse LLC-SD lung adenocarcinoma CSC cellular model we generated. Using the LLC-SD model, we have revealed an intricate cross-talk between the oncogenic pathway and stem cell pathway in which Cdh1 functions as an oncogene by promoting lung CSC renewal via the activation of the PI3K and inhibition of MAPK pathways, respectively. Further, we show for the first time that Cdh1 promotes the self-renewal of lung CSCs, consistent with its function in embryonic and normal stem cells. In summary, this study has provided new evidence demonstrating the effective utilization of the normal stem cell renewal mechanisms by CSCs to promote oncogenesis and progression.
Materials and Methods
Bioinformatics analysis of Cdh1
The mRNA expression of Cdh1 in a variety of tumor types was analyzed by GEPIA database (http://gepia.cancer-pku.cn/index.html). The Oncomine datasets (https://www.oncomine.org/) were used to analyze the expression of Cdh1 in LUAD tumors. Students' t-test was used, and two-times of fold change with the P-value of <0.0001 was defined as clinically significant. All of the data from the TCGA-LUAD datasets (https://cancergenome.nih. gov/) were downloaded including the Cdh1 mRNA expression levels and clinicopathological features of tumor staging. Human lung adenocarcinoma data was extracted from the GEO database, accession numbers GSE32867 [29] (n = 57 patients) dataset. Meanwhile, the following datasets were included as Bild dataset [30] and Selamat dataset [29] from the Oncomine database. The association between the expression of Cdh1 and survival, including overall survival (OS), progression-free survival (PFS) and post-progression survival (PPS) was assessed through analysis in the Kaplan-Meier plotter (http://kmplot. com/analysis/).
Cell culture and cell lines
LLC-Parental cell line was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in Dulbecco modified Eagle medium (DMEM) high glucose medium (Hyclone, USA) containing 10 % FBS (Gibco, USA). LLC-SD cells, the stem-cell component of the LLC-Parental [31], were maintained in serum-free DMEM-F12 medium (Hyclone, USA) containing B27 Supplement (Gibco, USA).
Reverse transcription and quantitative real-time polymerase chain reaction (RT-qPCR)
For RT-qPCR experiments, total RNA was isolated using TRIZOL (Takara, Japan) and reverse-transcribed into cDNA following to the manufacturer's instructions. Relative expression was normalized to that of TBP internal control. The following PCR condition was used on the Light Cycler: 39 cycles of 95°C for 30s, 95°C for 5s, followed by 60°C for 30s in a 10µl reaction volume. The primer sequences for RT-qPCR are listed in Table 3.
Western blotting (WB)
Cells were lysed in RIPA (Radio Immunoprecipitation Assay) buffer (Beyotime, China) containing 1% PMSF. Extracted protein concentration was measured by BCA (bicinchoninic acid) method and stored at -80°C. 25 µg of protein sample was resolved by 8-10% SDS-PAGE and transferred to a PVDF membrane (Bio-Rad, USA). Afterwards, the membrane was blocked in 5% nonfat milk (BD, USA) and incubated with primary antibodies at 4°C overnight. Finally, the membrane was incubated with secondary antibodies and developed under the gel electrophoresis imager (Bio-Rad, USA). GAPDH protein was used as a reference control. The following primary antibodies were used: anti-E-cadherin (Proteintech, USA), anti-PI3K, anti-Akt, anti-mTOR, anti-S6K, anti-Eif4e, anti-Erk1/2, anti-p-Mek1/2, anti-p-Erk1/2, anti-p-Akt, anti-p-mTOR, anti-p-S6k (Cell Signaling Technology, USA), anti-Mek1 (Upstate, USA) and anti-GAPDH (Proteintech, USA).
Primers for RT-qPCR
| Gene name | Forward primers | Reverse primers |
|---|---|---|
| mouse Cdh1 | AGCCATTGCCAAGTACATCC | TCTGGCCTGTTGTCATTCTG |
| mouse PI3K | GCAGAGGGCTACCAGTACAGA | CTGAATCCAAGTGCCACTAAGG |
| mouse Mtor | AGACCTTGAGTTGGCTGTGC | CCTCTGCTTGGATGTGATGA |
| mouse Akt | ACTCATTCCAGACCCACGAC | CACAATCTCCGCACCATAGA |
| mouse Mek1 | GTGCAGTCGGACATCTGGAG | CCACATGGCATCCAAACAGT |
| mouse Mek2 | ACATGGATGGTGGCTCACTG | CTGGTGCTTCTCTCGGAGGT |
| mouse Eif4e | TGTGGGTAGCAGAGTGGAAA | CAACAAGAGCAGGCGGTTAT |
| mouse Mapk1 | CTTCCAACCTCCTGCTGAAC | TCTGTCAAGAACCCTGTGTGA |
| mouse Sox2 | AGGGCTGGGAGAAAGAAGAG | ATCTGGCGGAGAATAGTTGG |
| mouse Nanog | TTAGAAGCGTGGGTCTTGGT | CCCTCA AACTCCTGGTCCTT |
| mouse CD133 | CTCCCATCAGTGGATAGAGAACT | ATACCCCCTTTTGACGAGGCT |
| mouse Aldh1a1 | ATACTTGTCGGATTTAGGAGG CT | GGGCCTATCTTCCAAATGAAC A |
| mouse Nr5a2 | AAACGGGCAGTAACCCTCTT | CCACATTTCAGCAACAGCAG |
| mouse Tbx3 | CGGAAGTCCCATTATCCTCA | CCCTCTACAAGCGCTCAGAT |
| mouse p110 | TAAAGGCCGAAAGGGTGCTA | GCGGTACAGGCCAGAGATTC |
| mouse Tbp | AGGGATTCAGGAAGACCACA | ATGCTGCCACCTGTAACTGA |
siRNA transient interference assay
The siRNAs were synthesized and purchased from GenePharma Company (Shanghai, China). 1×106 LLC-SD cells were seeded in each well of a six-well culture plate (Thermo, USA). 5µl of siRNA combined with 5µl of Lipofectamine 2000 (Thermo, USA) were diluted in 200µl MEM medium (Gibco, USA), respectively, and incubated for 5 min. The mixture was then used for transient interference assay following the manufacturer's protocol. The sequences of siRNAs are as follows: 5'-UUCUCCGAACGUGUCACGUTT-3' (siN.C.), 5'-AGUCGGACAUCUGGAGCAUTT-3'(siMek1/2), 5'-GGAGAGACAUCUACGAAAUTT-3'(sip110), 5'-GGGUCAGGAAAUCACAUCUTT-3'(siCdh1).
Soft agar colony formation assay
Cells were seeded into the medium containing 0.2% agar (Sigma, USA) in 6-well cell culture plates at a density of 200 cells/well and cultured for 1 weeks. The colonies that consisted of more than 50 cells were then stained with 0.05% crystal violet, counted and recorded.
Single cell colony formation assay
Single cell suspension was prepared and the concentration was adjusted to 10 cells/ml. One hundred microliters of cell suspensions were seeded into each well of 96-well plates (Thermo, USA). Single-cell seeding was confirmed using microscope and wells containing one single cell were marked. After cultured at 37°C and 5% CO2 for 10 days, colonies exceeding 50 cells were counted.
Protein-protein interaction (PPI) network analysis
The 50 most frequently altered neighbor genes analyzed from cBioportal of Cdh1. STRING database version 11.0 (http://www.string-db.org) was used to analyze PPI network of the neighbor genes of Cdh1. PPI refers to the protein complex formed by two or more proteins through covalent bond. The most important protein and connected nodes were analyzed by R-3.51 software.
Gene Ontology (GO) enrichment analysis
To investigate the intrinsic mechanisms of Cdh1 expression in LUAD tissues, we acquired Cdh1-associated co-expression genes from cBioPortal database, the GEPIA database and the UALCAN database (http://ualcan.path.uab.edu/index.html). This study selected genes with an absolute value of the Pearsons' correlation coefficient >0.4 in LUAD. Finally, the sum aggregate results among cBioPortal, GEPIA and UALCAN were obtained and used for further analyses. GO enrichment analysis and functional annotation for co-expression genes were performed using KOBAS 3.0 (https:// kobas.cbi.pku.edu.cn/), which is used to evaluate the significance of enrichment of pathways in human tumors.
Gene set enrichment analysis (GSEA)
GSEA was performed using GSEA 3.0 software. 594 LUAD samples in TCGA were divided into a low expression group and high expression group using Cdh1 expression median level as a cut-off point. In order to identify the potential functions of Cdh1, GSEA was used to determine whether a series of previously-defined gene sets were enriched in different phenotypes. The expression level of Cdh1 (high versus low) was used as the phenotypic label and the number of permutations was 1000. The gene expression datasets were used collections H (Hallmark gene set) and C2 (curated gene set: KEGG), publicly available at MsigDB (http://www.broad.mit.edu/gsea/msigdb/index.jsp). All other parameters were set to default values. A nominal P-value of P < 0.05 and false discovery rate (FDR) < 0.25 were chosen as significance cut-off criterion.
Serial spheroid formation assay
For spheroid formation, 1000 cells were mixed with 2ml culture medium, and plated into each well of 6-well plate. Cells were photographed and counted after 5 days of growth and diluted to a density of 500 cells/ml for reseeding. The assay was repeated for three rounds.
Construction and transfection of lentiviral vectors expressing shRNA
The shRNA that targets the sequence of the optimal siRNA was designed and synthesized. The annealed oligonucleotide fragment was cloned into the lentivirus plasmid PLL3.7(Addgene, USA) to establish the shRNA lentiviral vector. The recombinant plasmid DNA was identified by sequencing. When the cultured 293T cells reached a 70-80% density, Lipofectamine 2000 was used to mediate transfection of shRNA lentiviral plasmid DNA with the right sequence, medium was renewed at 12h. The incubation was continued for 48h, when the supernatant of each culture was collected and filtered by 0.22 µm filter (Millipore, USA). Following concentration, the supernatant was stored in aliquots at -80°C. Lentivirus infections of LLC-SD cell lines were carried out with the presence of 8 µg/ml of polybrene.
Animals
BALB/c nude mice (6-8 weeks old) were obtained from Huafukang bioscience company (Beijing, China). C57BL/6 mice (6-8 weeks old) were obtained from Chongqing national biological industry base experimental animal center of Chongqing Medical University. All animal studies were conducted in accordance with the approved protocol by the Institutional Review of Chongqing Medical University and carried out in accordance with the animal welfare and institutional ethical guidelines of Chongqing Medical University.
Subcutaneous tumor transplantation assay in BALB/c nude mice
For transplantation analysis uaing BALB/c nude mice, approximately 1 × 103 single cell suspensions were mixed with 50μL Matrigel Matrix (Corning) at a 1:1 ratio and 100μL mixture was subcutaneously injected into the both insides of the hind legs of mice. Tumor size was assessed every two days and tumor volume was calculated using V= (length x width2)/2. The mice were euthanized and killed when the tumor volume reached about 1000mm3.
Orthotopic tumor transplantation in C57BL/6 mice
Single cell suspensions (1×105) mixed with 12.5μL Matrigel Matrix (Corning) at a 1:1 ratio was injected subcutaneously into left lobe of the lungs of C57BL/6 mice as we previously described [31]. For tumorigenesis analysis, the mice were sacrificed, orthotopic and metastatic lung nodules were counted and immunohistochemistry was evaluated after 14 days. For the survival assay, the death time of each mouse was monitored and recorded after tumor transplantation.
Statistical analysis
Data was analyzed by Student's independent t-test of variance using GraphPad Prism and R-3.51 software and described as mean ± SEM indicated in figure legends. Differences were considered statistically significant when P < 0.05. *P < 0.05, **P < 0.01, *** P < 0.001.
Results
The expression of Cdh1 is elevated in human cancer tissues including lung adenocarcinoma
We first compared the transcriptional levels of Cdh1 in 33 types of cancers with their normal counterparts by using GEPIA database (http://gepia.cancer- pku.cn/). Cdh1 expression was increased in 17 types of cancers, decreased in 3 types of cancers, and there was no significant difference in 13 types of cancers (Figure 1A-i). These findings, for the first time revealed that elevated Cdh1 expression is a rather common feature of human cancer that is currently under appreciated. We conduced in-depth analysis in LUAD. For LUAD, Cdh1 transcriptional level in tumor tissues (483 cases) was significantly elevated compared with the normal lung tissues (347 cases) (Figure 1A-ii). To validate the results of the GEPIA database, we first conducted Cdh1 expression analyses using the Oncomine (www.oncomine.org) database for the expression of Cdh1 in lung adenocarcinoma. Four datasets were included in the analysis: the 'Landi Lung' [32], 'Selamat Lung' [29], 'Stearman Lung' [33] and 'Su Lung' [34]. Cdh1 was significantly overexpressed in LUAD compared to normal individuals (Figure 1B). Similar findings in LUAD were obtained from additional analyses performed using the TCGA database (LUAD group: n = 535 cases; normal group: n = 59 cases) (Figure 1C-i), and the TCGA and GEO database (LUAD tissues: n = 57 cases; adjacent normal tissues: n = 57 cases) (Figure 1C-ii and Figure 1D). These results indicated that Cdh1 expression is elevated in human cancer, including lung adenocarcinoma.
The bioinformatic data mining analysis of prognostic significance of Cdh1 expression. (A)(i) Cdh1 gene expression profile across all types of tumor samples and normal tissues from GEPIA analysis. (ii) The differential expression of Cdh1 mRNA between LUAD tissues (n=483) and corresponding control samples (n=347) from GEPIA analysis. Red box is LUAD tissues (T), the gray box is the normal tissues (N). (B) The combined results of Cdh1 expression across four studies in ONCOMINE datasets, the redder the square, the more is the correlation with cancer state. (C)(i-ii) Cdh1 expression level in LUAD cancer tissues is higher than that in the normal or pericarcinomatous tissues. The mRNA levels of Cdh1 in unmatched LUAD and matched LUAD were download from TCGA datasets. (D) The mRNA levels of Cdh1 in LUAD and matched non-cancerous lung tissue based on GEO LUAD dataset GSE32867. E(i-ii) Correlation between Cdh1 expression and tumor stage in LUAD patients by analysis of the Oncomine datasets (Bild and Selamat dataset). (F) Differential expression of Cdh1 in early and late tumor stage of LUAD patients based on GSE32867. (G) (i-iii) Elevated expression of Cdh1 indicated poor clinical outcome for LUAD patients. The overall survival (OS), progression-free survival (PFS) and post-progression survival curves of LUAD patients with high (red) or low (black) Cdh1 expression were plotted from Kaplan-Meier plotter database. (* p < 0.05, ** p < 0.01, *** p < 0.001).
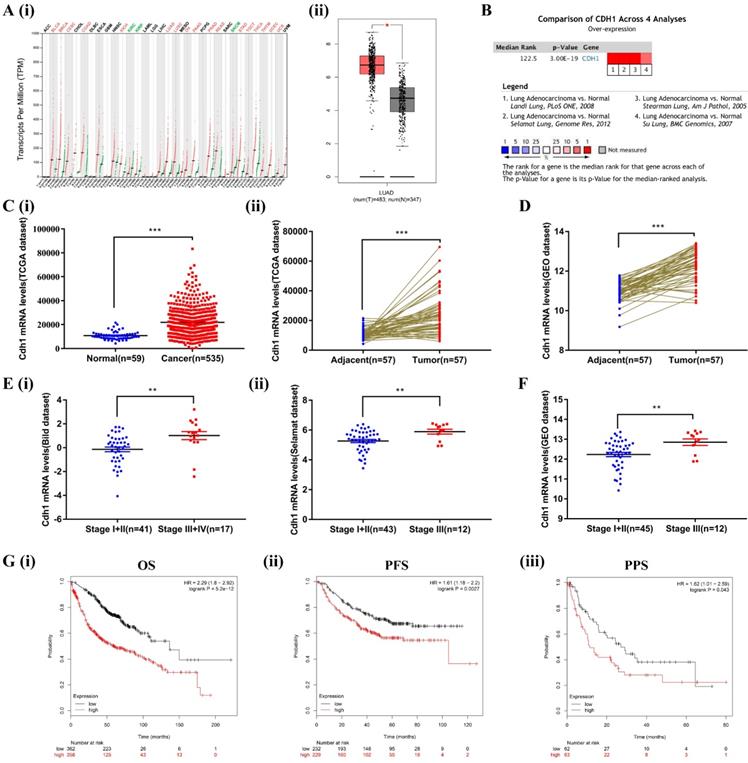
Increased expression of Cdh1 is associated with TNM stage and poor prognosis in human lung adenocarcinoma
To verify the relationship between the level of expression of Cdh1 and clinicopathological features of patients with LUAD, we further analyzed the expression of Cdh1 with TNM using Oncomine (the Bild dataset[30] and the Selamat dataset[29]) and the GEO database. Interestingly, Cdh1 expression level was much higher in patients with advanced stage (stageⅢ~Ⅳ) disease compared with those with early stage (stageⅠ~Ⅱ) disease (Figure 1E and Figure 1F). Kaplan-Meier Plotter tool (http://kmplot.com/ analysis/) was used to analyze the correlation between the Cdh1 expression and LUAD survival. LUAD patients with a low Cdh1 expression exhibited a better overall survival (OS) [HR = 2.29, P < 0.001] (Figure 1G-i), progression-free survival (PFS) [HR = 1.61, P = 0.0027] (Figure 1G-ii) and post-progression survival (PPS) [HR = 1.62, P = 0.043] (Figure 1G-iii) compared with patients with a high Cdh1 expression. Thus, a low Cdh1 expression appears to be a protective factor and a favorable prognostic marker for patients with LUAD.
Cdh1 regulates self-renewal of LLC-SD cells in vitro
Cdh1 encodes E-cadherin, which was shown to enhance the maintenance of embryonic stem (ES) cells pluripotency and self-renewal. However, it was unclear whether Cdh1 may regulate the self-renewal of CSCs as it does in normal stem cells. Here, the expression of Cdh1 in LLC-Parental and its CSC derivative LLC-SD cell line was detected by RT-qPCR and WB. And the results showed that LLC-SD CSC cells had elevated Cdh1 mRNA (Figure 2A-i) and protein expression (Figure 2A-ii) in comparison with the LLC-Parental cells.
To investigate the relationship between elevated Cdh1 and changes in the stem cell properties of LLC-SD cells, we inhibited Cdh1 expression by transient siRNA interference and confirmed its effective silencing at the transcriptional level (Figure 2B). The effect of Cdh1 silencing on stem cell self-renewal was measured by the soft agar colony formation assay and the single-cell cloning formation assay in vitro. Transient silencing of Cdh1 expression in LLC-SD-siCdh1 cells significantly hindered the self-renewal capability (Figure 2C) and clonogenic efficiency (Figure 2D) of LLC-SD cells. These observations indicated that Cdh1 promotes the self-renewal of LLC-SD lung CSC cells in vitro. Based on the bioinformatic analyses on the clinical datasets and these observations, we hypothesized that Cdh1 may function as an oncogene by promoting the CSC stemness.
Cdh1 regulates LLC-SD self-renewal in vitro. (A)(i) mRNA expression of Cdh1 in LLC-Parental and LLC-SD cells. (ii) Protein expression of Cdh1 in LLC-Parental and LLC-SD cells. (B) mRNA expression of Cdh1 in LLC-SD when the Cdh1 was silenced, N.C. was the negative control si-RNA. C(i) The morphology of soft agar spheroid formation assay with crystal violet-stained using LLC-SD-siN.C. and LLC-SD-siCdh1 cells. Where the red arrow is pointing. (ii) Quantification of spheroid formation ratio. Data are presented as the mean ± S.E.M. of three independent experiments. D(i) The morphology of single-cell cloning formation from LLC-SD-siN.C. and LLC-SD-siCdh1 cells. Scale bars, 120μm. (ii) Quantification of single-cell cloning ratio. Data are presented as the mean ± S.E.M. of three independent experiments. (* p < 0.05, ** p < 0.01).
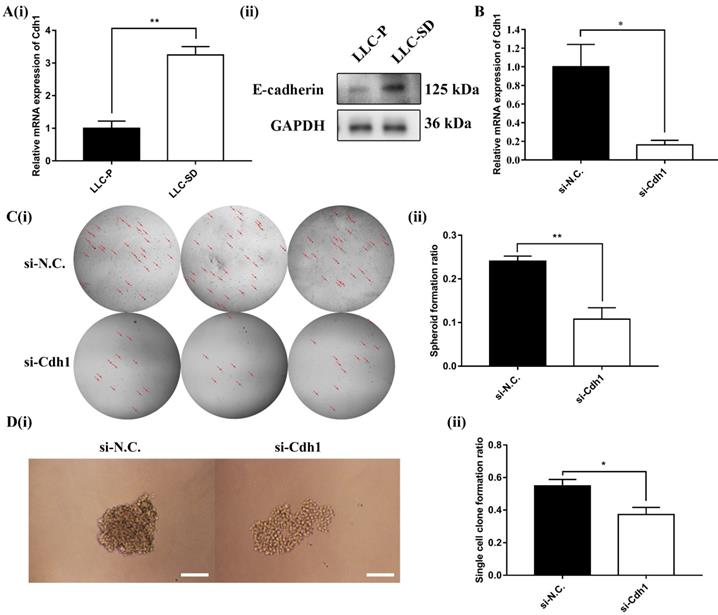
Assessment of the function of Cdh1 as an oncogene in vivo
In order to explore the role of oncogene of Cdh1 in vivo, we generated LLC-SD cell line in which Cdh1 was stably and effectively inhibited using the lentivirus expression system (Methods, Figure 5A and Figure 5B). 103 LLC-SD-shCdh1 and control cells were injected subcutaneously into each side of the hinder leg of nude mice, respectively. Measurable tumors were formed after 16 days of tumor cell inoculation (8/8 sites). Stable inhibition of Cdh1 expression in vivo resulted in lower tumor incidence and smaller tumor burden compared with the control group (Figure 5C and Figure 5D).
Prior to this study, we have developed a clinically relevant syngeneic orthotopic mouse model of lung cancer which allowed the characterization of tumorigenicity and metastasis of LLC-SD CSC cells in vivo in C57BL/6 mice (Methods)[31]. Upon Cdh1 stable depletion, 105 LLC-SD-shN.C. cells and LLC-SD-shCdh1 cells were injected in the left lung of C57BL/6 mice, respectively. All mice were sacrificed on day 14 post tumor cell injection. Orthotopic tumor growth and metastatic progression in the thoracic cavity were examined and recorded. At the site of tumor cell injection, tumor nodules were developed in 8/8 mice injected with LLC-SD-shN.C. cells and only in 3/8 mice injected with LLC-SD-shCdh1 cells. In mice injected with LLC-SD-shN.C. cells, visible metastatic foci were found at right lung (1/8 mice), left thoracic cavity (5/8 mice), right thoracic cavity (1/8 mice) and mediastinal lymph nodes (3/8 mice). In contrast, no thoracic metastases were observed mice injected with LLC-SD-shCdh1 cells (Table 2 and Figure S1). LLC-SD-shN.C. and LLC-SD-shCdh1 tumors on the lung were removed and analyzed by hematoxylin and eosin (HE) staining. Histology of the left lung (the orthotopic tumor) and the right lung (metastatic foci) from LLC-SD-shN.C. tumor showed destroyed alveoli and poorly differentiated hyperchromatic tumor cells with prominent nucleoli, as well as the presence of more severe right lung metastases. However, no typical lung carcinoma-like morphology was observed in the left lung tissue and no tumor cells were found in the right lung from LLC-SD-shCdh1 group (Figure 5E-i and Figure 5E-ii). Survival assay was also carried out and the time of death was recorded when it occurred until termination of the assay on Day 60 post tumor cell inoculation. Mice injected with LLC-SD-shN.C. cells died from 11th day until 27th day. Inhibition of Cdh1 delayed the onset of death and prolonged the course of the survival assay which is evident from the right-shift of the survival curve (Figure 5F). Based on the tumorigenesis xenograft assay in nude mice, the orthotopic tumorigenesis and progression assay as well as the survival assay in the syngeneic mice, we conclude that Cdh1 enhanced oncogenic properties of LLC-SD cells in vivo.
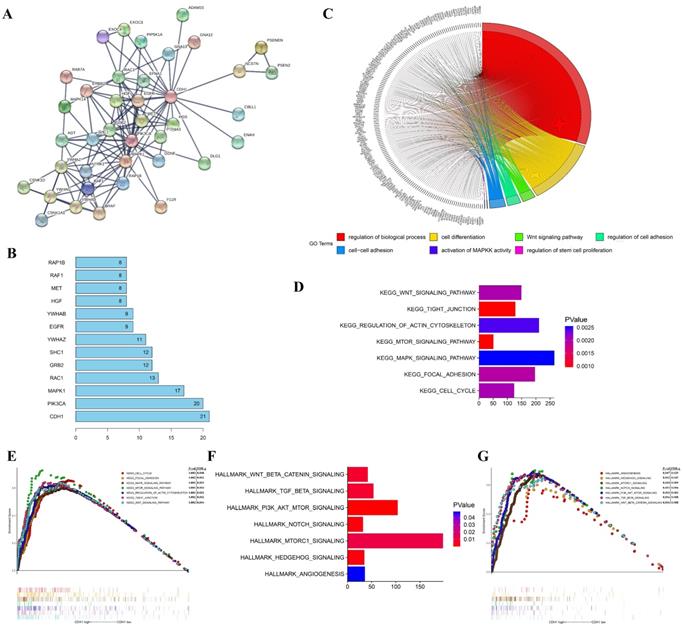
Integrative bioinformatics analysis of Cdh1-associated signaling pathways in human lung adenocarcinoma
To investigate the molecular mechanisms of the oncogene activity of Cdh1 in human lung adenocarcinoma, we conducted additional bioinformatics analyses. We firstly acquired Cdh1-associated 50 most frequently altered neighbor genes from cBioportal databases. Then PPI analyses of these neighbor genes were performed in STRING (http://www.string-db.org/) to validate the protein interaction association to predict pathways likely mediating the oncogenic function of Cdh1 carries in lung adenocarcinoma (Figure 3A). The results of the top 10 overall score of key proteins from PPI network demonstrated that PIK3CA, MAPK1, EGFR and RAF1 represented key genes in these pathway (Figure 3B). Notably, these genes are key components of the oncogenic receptor tyrosine kinase-MAPK and PI-3K signaling pathways.
To explore the gene-enrichment and functional annotation analyses of Cdh1-related genes, we performed analysis of Gene Ontology (GO) and Gene Set Enrichment Analysis (GSEA) (Methods). The results from GO analysis revealed that target genes in the LUAD group mainly participated in regulation of biological process (GO:0050789), cell differentiation (GO:0030154), Wnt signaling pathway (GO:0016055), regulation of cell adhesion (GO:0030155), cell-cell adhesion (GO:0098609), activation of MAPKK activity (GO:0000186) and regulation of stem cell proliferation (GO:0072091) (Figure 3C and Table 1). The results of Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis indicated that Cdh1-related genes were enriched in the following pathway in LUAD group, including Wnt signaling pathway, Tight junction, regulation of actin cytoskeleton, mTOR signaling pathway, MAPK signaling pathway, focal adhesion and cell cycle pathway (Figure 3D and Figure 3E) which are well characterized oncogenic pathways for human cancer, including lung cancer. We then evaluated the oncogenic pathway more likely to be Cdh1-related by GSEA, listing those genes in the categories described as hallmarks of gene expression and found that they are related to Wnt signaling pathway, TGF-β signaling pathway, PI3K signaling pathway, Notch signaling pathway and Hedgehog signaling pathway (Figure 3F and Figure 3G).
Taken together, bioinformatics analysis results collectively predicted the close association of the oncogenic pathways, such as the PI3K and MAPK pathways and the oncogene phenotypes of Cdh1 in human LUAD. Based on the findings of these analyses and the promotion of self-renewal in LLC-SD CSCs, we next explored the possibility of a cross-talk between the oncogenic pathways and the stem cell pathway as the mechanism that underlies the oncogenic function of Cdh1.
Enriched GO terms of the genes co-expressed with Cdh1
| GO ID | Term | Count | P-value |
|---|---|---|---|
| GO:0050789 | regulation of biological process | 157 | 4.84E-40 |
| GO:0030154 | cell differentiation | 60 | 1.73E-14 |
| GO:0016055 | Wnt signaling pathway | 12 | 2.42E-05 |
| GO:0030155 | regulation of cell adhesion | 12 | 3.61E-04 |
| GO:0098609 | cell-cell adhesion | 14 | 6.48E-04 |
| GO:0000186 | activation of MAPKK activity | 2 | 0.038452263 |
| GO:0072091 | regulation of stem cell proliferation | 2 | 0.047918442 |
The PI3K and MAPK signaling pathways had an opposing effect on Cdh1 expression and its regulation of CSC self-renewal in vitro
We first analyzed mRNA and protein expression of key signaling components of the PI3K and MAPK pathway by RT-qPCR and WB in LLC-Parental and LLC-SD cell lines which differs in Cdh1 expression. The results showed that the expression of the signaling molecules on the MAPK signaling pathway including Mapk1, Mek1 and Eif4e was robustly upregulated in LLC-SD cells, while the expression of signaling molecules of the PI3K pathway including PI3K, Akt and Mtor was higher in LLC-Parental cells (Figure 4A). Altered protein expression by confirmed by WB was in agreement with the RT-qPCR results (Figure 4B). This set of observations indicate that in LLC-SD CSC cells, while the MAPK pathway is upregulated in the LLC-SD cells, the PI3K pathway is inhibited.
The functions of Cdh1 and genes significant associated with Cdh1 were predicted by analysis of integrative bioinformatics in human lung adenocarcinoma. (A) PPI network of the 50 most frequently altered neighbor genes associated with Cdh1 from cBioportal. (B) Top 10 of key proteins from the PPI network. The vertical coordinate is the name of genes, and the horizontal coordinate represents the number of gene connections. (C) Chord plot displayed gene ontology (GO) terms of Cdh1 co-expression genes in LUAD by KOBAS 3.0. (D-E) The GSEA results showed the KEGG pathway enrichment results using TCGA-LUAD datasets. (F-G) The GSEA results demonstrated the enriched hallmark gene sets results with correlation of Cdh1 mRNA levels using TCGA-LUAD datasets.
The PI3K and MAPK signaling pathway mediate Cdh1 regulation of CSC self-renewal in vitro. (A) mRNA expression and (B) Protein expression of related genes of PI3K and MAPK signaling pathways in LLC-Parental and LLC-SD cells. (C) mRNA expression of Cdh1 in LLC-SD cells with treatment of LY294002 and PD98059 by RT-qPCR. DMSO was used as control. D(i) The morphology of spheroid formation in LLC-SD cells treated with LY294002 and PD98059. DMSO was used as control. Scale bars, 120μm. (ii) Expression of stem genes in LLC-SD cells by RT-qPCR, the DMSO was used as control. E(i) The morphology of single-cell cloning formation in LLC-SD cells treated with LY294002 and PD98059. DMSO was used as control. Scale bars, 120μm. (ii) Quantitative analysis of single-cell cloning ratio. Data are presented as mean ± S.E.M. of three independent experiments. (F)(i-ii) Efficiency of p110 and Mek1/2 siRNAs (si-p110 and si-Mek1/2) interference determined by RT-qPCR. G(i) The morphology of soft agar spheroid formation assay with crystal violet-stained using LLC-SD-siN.C., LLC-SD-sip110 and LLC-SD-siMek1/2 cells. Where the red arrow is pointing. (ii) Quantification of spheroid formation ratio. Data are presented as mean ± S.E.M. of three independent experiments. H(i) The morphology of single-cell cloning formation in LLC-SD upon p110 and Mek1/2 knock down, LLC-SD-siN.C. was the negative control. Scale bars, 120μm. (ii) Quantification of single-cell cloning efficiency in which 96 wells was analyzed. Data are presented as mean ± S.E.M. of three independent experiments. (* p < 0.05, ** p < 0.01, *** p < 0.001).
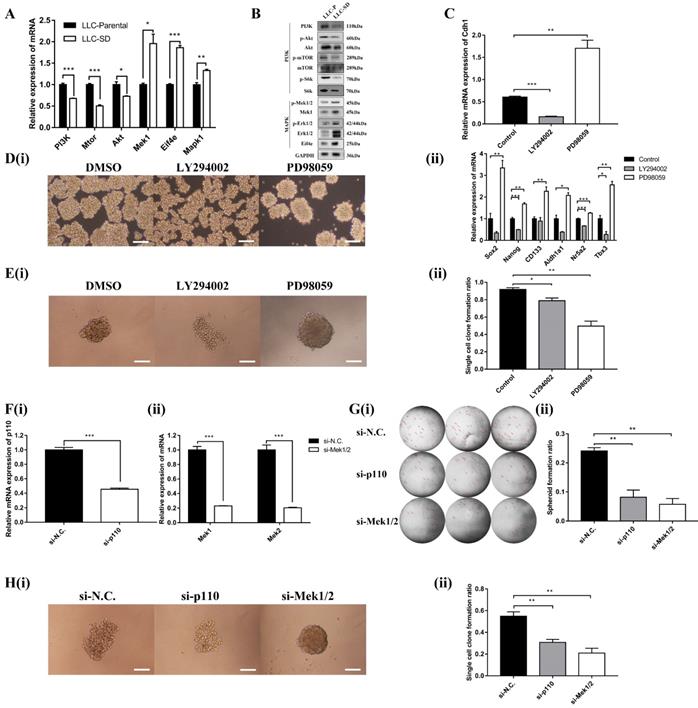
We next examined whether MAPK and/or PI3K activation are required for the Cdh1 expression, LLC-SD cells were treated with PI3K inhibitor LY294002 or MEK inhibitor PD98059, respectively. Shown in Figure 4C, inactivation of PI3Kand MAPK pathway by their respective synthetic inhibitor had an opposite effect on Cdh1 expression. LY294002 markedly reduced Cdh1 expression, whereas PD98059 significantly increased Cdh1 levels. Meanwhile, LLC-SD cells exhibited the irregular morphology with a loose structure after the inhibition of PI3K signaling pathway by LY294002 (Figure 4D-i). Conversely, LLC-SD cells exhibited the stem-like spheroid morphology with a compact structure after the inhibition of MAPK signaling pathway by PD98059 (Figure 4D-i). We further examined the expression of the candidate stemness genes, Sox2, Nanog, CD133, Aldh1a1, Nr5a2 and Tbx3 by RT-qPCR. And the results demonstrated that the expression of Nanog, Nr5a2 and Tbx3 were significantly lower in the LLC-SD treated with LY294002, whereas the expression of Sox2, Nanog, CD133, Aldh1a1, Nr5a2 and Tbx3 were highly upregulated in the LLC-SD treated with PD98059 than that in the negative control (Figure 4D-ii). The single-cell cloning assay in 96-well plate showed that colonies treated with LY294002 exhibited loosely irregular morphology and the colonies treated with PD98059 exhibited regular compact spheroid (Figure 4E-i). However, the single-cell cloning rate of SD cells with inhibitors treatment were both lower than that of control cells. This may have to do with inhibition of both self-renewal and proliferation activities upon inhibition of the PI3K and MAPK signaling (Figure 4E-ii).
Subsequently, successful PI3K silencing by targeting the p110 catalytic unit and Mek1/2 silencing by siRNA was confirmed by RT-qPCR analysis (Figure 4F-i and Figure 4F-ii). The effect of p110 and Mek1/2 silencing on the stemness was measured by the soft agar colony formation assay and the single cell cloning assay and yielded very comparable findings (Figures 4G and H) as using the inhibitors (Figure 4E). Taken together, our data supports that inactivation of the MAPK pathway and activation of the PI3K pathway correlates with Cdh1 up-regulation in LLC-SD cells and Cdh1 promotes self-renewal in LLC-SD cells.
The PI3K and MAPK signaling pathways mediate LLC-SD oncogenesis in vivo. (A) The analysis of mRNA expression of shRNA in the three experimental groups of LLC-SD cells by RT-qPCR, sh-N.C. was the negative control, sh-Cdh1 was the stable silencing of Cdh1, sh-Mek1/2 was the silencing of Mek1 and Mek2, and sh-p110 was the silencing of p110. (B) Efficiency of shRNAs (sh-Cdh1, sh-Mek1/2 and sh-p110) interference determined by Western blot analysis. (C) Tumor formation in nude mice following injection of control cells and genetically modified LLC-SD (sh-Cdh1, sh-Mek1/2 and sh-p110) cells respectively. (D) Tumor growth curves of LLC-SD-shRNAs (sh-Cdh1, sh-Mek1/2 and sh-p110) and control cells (sh-N.C.) in nude mice. (i) Tumor volume. (ii) Tumor weight. E(i) Images of left lung orthotopic nodules in C57BL/6 mice injected with LLC-SD-shRNAs (sh-Cdh1, sh-Mek1/2 and sh-p110) or control cells. (ii) Immunohistochemistry analysis of the left and right lung harvested from C57BL/6 mice injected with control or experimentally modified LLC-SD cells (sh-Cdh1, sh-Mek1/2 and sh-p110) respectively. Scale bars = 120μm, the black box indicates the enlarged area, bar = 60μm. (F) The survival curve of C57BL/6 mice injected with 1×105 control and LLC-SD-shRNAs (sh-Cdh1, sh-Mek1/2 and sh-p110) cells respectively. (** p < 0.01, *** p < 0.001).
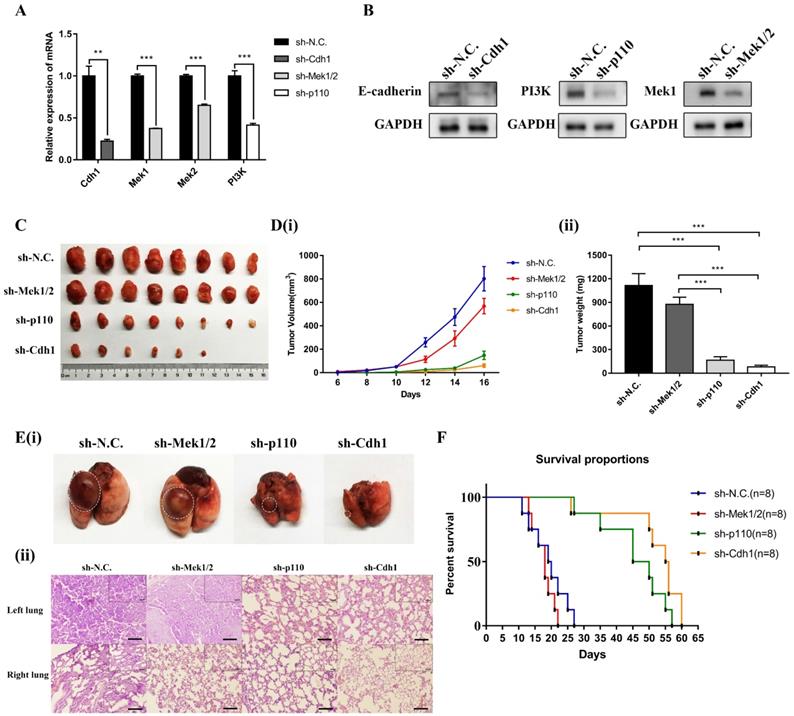
The PI3K and MAPK signaling pathway mediates oncogenesis of LLC-SD cells in vivo
Given the essential role of PI3K and MAPK signaling pathway mediated Cdh1 in regulating LLC-SD self-renewal in vitro, we first generated LLC-SD derivative cell lines in which p110 and Mek1/2 expression was stably inhibited by RNA interference (sh-p110 and sh-Mek1/2). Effective inhibition of p110 and Mek1/2 expression was confirmed by RT-qPCR (Figure 5A) and western blot (Figure 5B). Then we conducted xenograft transplantation assay in nude mice to investigate the importance of MAPK and PI3K pathway in LLC-SD lung cancer tumorigenesis. 103 LLC-SD-shMek1/2, LLC-SD-shp110 and LLC-SD-shN.C. cells were injected subcutaneously into each side of the hinder leg of nude mice respectively. The lower tumor incidence and smaller tumor burden were measured by tumor growth and tumor weight in vivo upon inactivation of the PI3K pathway by p110 knockdown. However, inhibition of Mek1/2 expression and the resultant inactivation of the MAPK pathway produced no significant difference in terms of tumor volume and weight compare with LLC-SD-shN.C. cells (Figure 5C and Figure 5D).
As for orthotopic tumor transplantation of C57BL/6 mice assay, 105 LLC-SD-shN.C. cells, LLC- SD-shMek1/2 and LLC-SD-shp110 cells were injected in the left lung of C57BL/6 mice, respectively. All mice were sacrificed on day 14 post tumor cell injection. The orthotopic tumor growth and metastatic progression in the mediastinal lymph nodes and thoracic cavity showed no significant difference in LLC-SD-shMek1/2 cells than that of LLC-SD-shN.C. cells. However, it was reduced upon inactivation of the PI3K pathway by p110 knockdown, which was consistent with the LLC-SD-shCdh1 group cells (Table 2 and Figure S1). Histology of the left lung (the orthotopic tumor) and the right lung (metastatic foci) confirmed the presence of more severe right lung metastases in mouse received LLC-SD-shN.C. and LLC-SD-shMek1/2 cells (Figure 5E). Survival assay was carried out and the time of death was recorded when it occurred. While the survival of mice receiving Mek1/2 knockdown LLC-SD cells and LLC-SD-shN.C. cells were comparable and mice incurred rapid death. In contrast, inactivation of PI3K by p110 silencing delayed the onset of death and prolonged the course of the survival as evident by the right-shifted survival curve. Inactivation of PI3K produced similar effect on in vivo survival as seeing in Cdh1 silenced LLC-SD- shCdh1 group (Figure 5F). These sets of observations are consistent with the in vitro findings, confirming the importance of PI3K and MAPK pathway of CSC self-renewal regulation in oncogenesis and tumor progression in vivo.
Characterization of orthotopic LLC-SD tumorigenesis and metastatic progression
| 105 cells(n=8) | In situ Metastasis | ||||
|---|---|---|---|---|---|
| Lung(left) | Mediastinal lymph | Lung(right) | Thoracic cavity(left) | Thoracic cavity(right) | |
| sh-N.C. | 8/8 | 3/8 | 1/8 | 5/8 | 1/8 |
| sh-Mek1/2 | 8/8 | 4/8 | 0/8 | 3/8 | 1/8 |
| sh-p110 | 3/8 | 0/8 | 0/8 | 0/8 | 0/8 |
| sh-Cdh1 | 3/8 | 0/8 | 0/8 | 0/8 | 0/8 |
Discussion
In cancer literature, the role of E-cadherin, encoded by Cdh1 gene, that functions as a tumor suppressor is widely reported. Downregulation or loss of the E-cadherin is reported to be involved in the invasion and metastatic progression of many malignancies [20, 35-37]. The main mechanism underlying the tumor suppression function of Cdh1 is the inhibition of epithelial-mesenchymal transition (EMT), a key process for tumor progression [17]. However, small bodies of reported studies have demonstrated the pro-oncogenic activity of Cdh1. Cdh1 upregulation and the subsequent promotion of the mesenchyme-to-epithelial transition (MET) for colonization phase of metastasis have been implicated [23-25]. In addition, our knowledge of the promoting effect of Cdh1 on stem cell self-renewal comes mostly from studies involving normal stem cells [14-16]. A recent study reported that Cdh1 can substitute for Oct4 during somatic cell reprogramming and is required to maintain the undifferentiated state of mouse embryonic stem cells (mESCs) [12]. However, the role of Cdh1 in regulating CSCs function has not been established prior to the present study [26, 38]. This study has made the following novel findings that an alternative mechanism underlying the oncogenic activity of Cdh1 may be realized via its promotion of CSC self-renewal, similar to its function in normal stem cells:
First, the oncogene function of Cdh1 overexpression in most cancers including lung adenocarcinoma has been uncovered by bioinformatic analysis and data mining of publically available cancer clinical databases, including GEPIA, Oncomine, TCGA, GEO and Kaplan-Meier plotter. Prior to this study, the clinical relevance of Cdh1 has not been well explored and established. Cdh1 expression is elevated in 17/33 human cancer types anaylazed, including lung adenocarcinoma (Figure 1A-F). Thus, high Cdh1 expression is a rather general feature of human cancer and is not limited to lung cancer. In depth analysis of lung adenocarcinoma data sets show the diagnostic and prognostic significance of Cdh1 high expression which merits further investigation to establish whether it is a new prognostic marker for lung cancer. We also found that a low Cdh1 expression may be a protective factor in patients with LUAD (Figure 1G). Further in-depth mechanistic characterization will answer whether Cdh1 is a novel target for therapeutic development.
Second, utilizing the stable lung CSCs cellular models and syngeneic orthotopic lung cancer model we established and characterized[31, 39, 40], we provide convincing experimental evidence supporting a rather definitive role of Cdh1 in regulating self-renewal of LLC-SD in vitro (Figure 2) as well as promoting lung cancer oncogenesis and progression in vivo (Figure 5, Figure S1 and Table 2). These observations provide new mechanistic understanding other than EMT/MET to confer the oncogenic activity of Cdh1 by regulating the self-renewal of CSCs.
Third, this study was the first attempt to predict the potential molecular mechanisms underlying the oncogenic activity of Cdh1 in LUAD using bioinformatics analyses. We performed PPI analyses on Cdh1-associated 50 most frequently altered neighbor genes from cBioportal databases and demonstrated that PIK3CA, MAPK1, EGFR and RAF1 served as core genes in the Cdh1 PPI network (Figure 3A-B). Furthermore, GO, KEGG and Hallmarks gene sets analysis showed that the Cdh1-associated 50 genes were mostly associated with PI3K and MAPK signaling pathways in caner (Figure 3C-G and Table 1). PI3K/AKT and MAPK signaling pathways are the two most important intracellular signal transduction pathways that are activated to mediate the activity of receptor tyrosine kinases [41-43]. These two signaling pathways also regulate normal stem cell self-renewal and differentiation [44, 45]. We confirmed these predictions with experimentation.
Fourth, we have identified and verified an intricate cross-talk between the oncogenic pathways and stem cell pathway using our stable lung CSCs model both in vitro and in vivo. We provided convincing evident for the positive regulation of PI3K and negatively regulation of MAPK on Cdh1 in the maintenance of lung CSC self-renewal and stem cell properties (Figure 4). Further, using the nude mouse xenograft and orthotopic lung transplantation model, characterized the effect of this cross-talk on promoting lung cancer oncogenesis and progression in vivo (Figure 5 and Table 2). Noteworthy, the inconsistency of function experiments upon MAPK inhibition in vitro and in vivo might be ascribed to in the growing LLC-SD tumors, inactivation of MAPK pathway lead to the concomitant inhibition of both stem cell self-renewal and the proliferative activity of the LLC-SD cells for tumor expansion. Moreover, there might be a mechanism for feedback regulation between Cdh1 and PI3K and MAPK signaling [46, 47]. This merits further investigations.
In conclusion, to the best of our knowledge, our study is the first to show that Cdh1 functions as an oncogene by inducing self-renewal of lung cancer stem-like cells via the opposing effect of PI3K and MAPK signaling pathways. Therefore, these results show the potential of Cdh1 as a new clinical target for diagnosis, prognosis and treatment of LUAD which will be explored in our future studies.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank all the subjects who volunteered to participate in the study. This work was supported by National Natural Science Foundation of China (81672959); National Natural Science Youth Fund of China (81602596); the Science and Technology Project Affiliated to the Education Department of Chongqing (Grant No. KJQN201800426).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA: a cancer journal for clinicians. 2017;67:7-30
2. Ridge CA, McErlean AM, Ginsberg MS. Epidemiology of lung cancer. Semin Intervent Radiol. 2013;30:93-8
3. Gridelli C, Rossi A, Carbone DP, Guarize J, Karachaliou N, Mok T. et al. Non-small-cell lung cancer. Nat Rev Dis Primers. 2015;1:15009
4. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA: a cancer journal for clinicians. 2010;60:277-300
5. Lennon FE, Cianci GC, Cipriani NA, Hensing TA, Zhang HJ, Chen CT. et al. Lung cancer-a fractal viewpoint. Nat Rev Clin Oncol. 2015;12:664-75
6. Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A. et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell death and differentiation. 2008;15:504-14
7. MacDonagh L, Gray SG, Breen E, Cuffe S, Finn SP, O'Byrne KJ. et al. Lung cancer stem cells: The root of resistance. Cancer Lett. 2016;372:147-56
8. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell stem cell. 2014;14:275-91
9. Doherty MR, Smigiel JM, Junk DJ, Jackson MW. Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers (Basel). 2016:8
10. Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science (New York, NY). 1991;251:1451-5
11. Chen T, Yuan D, Wei B, Jiang J, Kang J, Ling K. et al. E-cadherin-mediated cell-cell contact is critical for induced pluripotent stem cell generation. Stem cells (Dayton, Ohio). 2010;28:1315-25
12. Redmer T, Diecke S, Grigoryan T, Quiroga-Negreira A, Birchmeier W, Besser D. E-cadherin is crucial for embryonic stem cell pluripotency and can replace OCT4 during somatic cell reprogramming. EMBO reports. 2011;12:720-6
13. Larue L, Antos C, Butz S, Huber O, Delmas V, Dominis M. et al. A role for cadherins in tissue formation. Development (Cambridge, England). 1996;122:3185-94
14. Karpowicz P, Willaime-Morawek S, Balenci L, DeVeale B, Inoue T, van der Kooy D. E-Cadherin regulates neural stem cell self-renewal. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:3885-96
15. Li L, Wang S, Jezierski A, Moalim-Nour L, Mohib K, Parks RJ. et al. A unique interplay between Rap1 and E-cadherin in the endocytic pathway regulates self-renewal of human embryonic stem cells. Stem cells (Dayton, Ohio). 2010;28:247-57
16. Li Z, Qiu D, Sridharan I, Qian X, Zhang H, Zhang C. et al. Spatially resolved quantification of E-cadherin on target hES cells. The journal of physical chemistry B. 2010;114:2894-900
17. Rodriguez FJ, Lewis-Tuffin LJ, Anastasiadis PZ. E-cadherin's dark side: possible role in tumor progression. Biochimica et biophysica acta. 2012;1826:23-31
18. Berx G, Becker KF, Hofler H, van Roy F. Mutations of the human E-cadherin (CDH1) gene. Human mutation. 1998;12:226-37
19. Machado JC, Soares P, Carneiro F, Rocha A, Beck S, Blin N. et al. E-cadherin gene mutations provide a genetic basis for the phenotypic divergence of mixed gastric carcinomas. Laboratory investigation; a journal of technical methods and pathology. 1999;79:459-65
20. Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer metastasis reviews. 2009;28:151-66
21. Guilford P. E-cadherin downregulation in cancer: fuel on the fire?. Molecular medicine today. 1999;5:172-7
22. Stemmler MP. Cadherins in development and cancer. Molecular bioSystems. 2008;4:835-50
23. Spaderna S, Schmalhofer O, Hlubek F, Jung A, Kirchner T, Brabletz T. Epithelial-mesenchymal and mesenchymal-epithelial transitions during cancer progression. Verhandlungen der Deutschen Gesellschaft fur Pathologie. 2007;91:21-8
24. De Marzo AM, Knudsen B, Chan-Tack K, Epstein JI. E-cadherin expression as a marker of tumor aggressiveness in routinely processed radical prostatectomy specimens. Urology. 1999;53:707-13
25. Chao YL, Shepard CR, Wells A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol Cancer. 2010;9:179
26. Bae KM, Parker NN, Dai Y, Vieweg J, Siemann DW. E-cadherin plasticity in prostate cancer stem cell invasion. Am J Cancer Res. 2011;1:71-84
27. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-15
28. Abell AN, Johnson GL. Implications of Mesenchymal Cells in Cancer Stem Cell Populations: Relevance to EMT. Current pathobiology reports. 2014;2:21-6
29. Selamat SA, Chung BS, Girard L, Zhang W, Zhang Y, Campan M. et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome research. 2012;22:1197-211
30. Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D. et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353-7
31. Wang J, Sun Z, Liu Y, Kong L, Zhou S, Tang J. et al. Comparison of tumor biology of two distinct cell sub-populations in lung cancer stem cells. Oncotarget. 2017;8:96852-64
32. Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A. et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One. 2008;3:e1651
33. Stearman RS, Dwyer-Nield L, Zerbe L, Blaine SA, Chan Z, Bunn PA Jr. et al. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. The American journal of pathology. 2005;167:1763-75
34. Su LJ, Chang CW, Wu YC, Chen KC, Lin CJ, Liang SC. et al. Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC genomics. 2007;8:140
35. Yun JA, Kim SH, Hong HK, Yun SH, Kim HC, Chun HK. et al. Loss of E-Cadherin expression is associated with a poor prognosis in stage III colorectal cancer. Oncology. 2014;86:318-28
36. Qureshi R, Arora H, Rizvi MA. EMT in cervical cancer: its role in tumour progression and response to therapy. Cancer Lett. 2015;356:321-31
37. Kim SA, Inamura K, Yamauchi M, Nishihara R, Mima K, Sukawa Y. et al. Loss of CDH1 (E-cadherin) expression is associated with infiltrative tumour growth and lymph node metastasis. Br J Cancer. 2016;114:199-206
38. Farmakovskaya M, Khromova N, Rybko V, Dugina V, Kopnin B, Kopnin P. E-Cadherin repression increases amount of cancer stem cells in human A549 lung adenocarcinoma and stimulates tumor growth. Cell cycle (Georgetown, Tex). 2016;15:1084-92
39. Wang J, Zhou T, Sun Z, Ye T, Zhou S, Li J. et al. Zeb1 Regulates the Symmetric Division of Mouse Lewis Lung Carcinoma Stem Cells through Numb mediated by miR-31. Int J Biol Sci. 2018;14:1399-410
40. Ye T, Li J, Sun Z, Liu Y, Kong L, Zhou S. et al. Nr5a2 promotes cancer stem cell properties and tumorigenesis in nonsmall cell lung cancer by regulating Nanog. Cancer Med. 2019
41. Xiang RF, Wang Y, Zhang N, Xu WB, Cao Y, Tong J. et al. MK2206 enhances the cytocidal effects of bufalin in multiple myeloma by inhibiting the AKT/mTOR pathway. Cell Death Dis. 2017;8:e2776
42. Eisenmann KM, VanBrocklin MW, Staffend NA, Kitchen SM, Koo HM. Mitogen-activated protein kinase pathway-dependent tumor-specific survival signaling in melanoma cells through inactivation of the proapoptotic protein bad. Cancer Res. 2003;63:8330-7
43. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nature reviews Cancer. 2013;13:184-99
44. Huang TS, Li L, Moalim-Nour L, Jia D, Bai J, Yao Z. et al. A Regulatory Network Involving beta-Catenin, e-Cadherin, PI3k/Akt, and Slug Balances Self-Renewal and Differentiation of Human Pluripotent Stem Cells In Response to Wnt Signaling. Stem cells (Dayton, Ohio). 2015;33:1419-33
45. Hamilton WB, Brickman JM. Erk signaling suppresses embryonic stem cell self-renewal to specify endoderm. Cell reports. 2014;9:2056-70
46. Brouxhon SM, Kyrkanides S, Teng X, Athar M, Ghazizadeh S, Simon M. et al. Soluble E-cadherin: a critical oncogene modulating receptor tyrosine kinases, MAPK and PI3K/Akt/mTOR signaling. Oncogene. 2014;33:225-35
47. Tang MKS, Yue PYK, Ip PP, Huang RL, Lai HC, Cheung ANY. et al. Soluble E-cadherin promotes tumor angiogenesis and localizes to exosome surface. Nat Commun. 2018;9:2270
Author contact
![]() Corresponding authors: Jianyu Wang, Institute of Life Sciences, Chongqing Medical University, Chongqing, China. Email: wjy2003123com OR H. Rosie Xing, College of Biomedical Engineering, State Key Laboratory of Ultrasound Engineering in Medicine, Chongqing Medical University, Chongqing, China. Email: xinglab310com
Corresponding authors: Jianyu Wang, Institute of Life Sciences, Chongqing Medical University, Chongqing, China. Email: wjy2003123com OR H. Rosie Xing, College of Biomedical Engineering, State Key Laboratory of Ultrasound Engineering in Medicine, Chongqing Medical University, Chongqing, China. Email: xinglab310com
Received 2019-7-23
Accepted 2019-11-4
Published 2020-1-1