ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2012; 8(3):310-327. doi:10.7150/ijbs.3524 This issue Cite
Research Paper
An Anti-PCSK9 Antibody Reduces LDL-Cholesterol On Top Of A Statin And Suppresses Hepatocyte SREBP-Regulated Genes
Liwen Zhang1 ![]() , Timothy McCabe2, Jon H. Condra2, Yan G. Ni1, Laurence B. Peterson1, Weirong Wang3, Alison M. Strack1, Fubao Wang2, Shilpa Pandit1, Holly Hammond2, Dana Wood2, Dale Lewis1, Ray Rosa1, Vivienne Mendoza1, Anne Marie Cumiskey1, Douglas G. Johns1, Barbara C. Hansen10, Xun Shen1, Neil Geoghagen1, Kristian Jensen1, Lei Zhu5, Karol Wietecha5, Douglas Wisniewski4, Lingyi Huang, Jing Zhang Zhao, Robin Ernst, Richard Hampton, Peter Haytko, Frances Ansbro7, Shannon Chilewski7, Jayne Chin1, Lyndon J. Mitnaul1, Andrea Pellacani6, Carl P. Sparrow1, Zhiqiang An2,8, William Strohl2,9, Brian Hubbard1, Andrew S. Plump1, Daniel Blom1, Ayesha Sitlani1
, Timothy McCabe2, Jon H. Condra2, Yan G. Ni1, Laurence B. Peterson1, Weirong Wang3, Alison M. Strack1, Fubao Wang2, Shilpa Pandit1, Holly Hammond2, Dana Wood2, Dale Lewis1, Ray Rosa1, Vivienne Mendoza1, Anne Marie Cumiskey1, Douglas G. Johns1, Barbara C. Hansen10, Xun Shen1, Neil Geoghagen1, Kristian Jensen1, Lei Zhu5, Karol Wietecha5, Douglas Wisniewski4, Lingyi Huang, Jing Zhang Zhao, Robin Ernst, Richard Hampton, Peter Haytko, Frances Ansbro7, Shannon Chilewski7, Jayne Chin1, Lyndon J. Mitnaul1, Andrea Pellacani6, Carl P. Sparrow1, Zhiqiang An2,8, William Strohl2,9, Brian Hubbard1, Andrew S. Plump1, Daniel Blom1, Ayesha Sitlani1 ![]()
1. Department of Atherosclerosis, Merck Research Laboratories, Rahway, NJ 07065;
2. Department of Biologics Research, Merck Research Laboratories, West Point, PA 19486;
3. Department of Preclinical DMPK, Merck Research Laboratories, West Point, PA 19486;
4. Department of In Vitro Sciences, Merck Research Laboratories, Rahway, NJ 07065;
5. Department of GEM Target Validation, Merck Research Laboratories, Rahway, NJ 07065;
6. Department of MBV and Early Biologics, Upper Gwynedd, PA 19454;
7. Department of Bioprocess and Bioanalytical, Merck Research Laboratories, West Point, PA 19486;
8. Present address: University of Texas Health Science Center, Houston, TX;
9. Present address: Centocor R&D, Inc., Radnor, PA 19087;
10. Departments of Internal Medicine and Pediatric, University of South Florida, Tampa, FL 33612.
Abstract
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a promising therapeutic target for treating coronary heart disease. We report a novel antibody 1B20 that binds to PCSK9 with sub-nanomolar affinity and antagonizes PCSK9 function in-vitro. In CETP/LDLR-hemi mice two successive doses of 1B20, administered 14 days apart at 3 or 10 mpk, induced dose dependent reductions in LDL-cholesterol (≥ 25% for 7-14 days) that correlated well with the extent of PCSK9 occupancy by the antibody. In addition, 1B20 induces increases in total plasma antibody-bound PCSK9 levels and decreases in liver mRNA levels of SREBP-regulated genes PCSK9 and LDLR, with a time course that parallels decreases in plasma LDL-cholesterol (LDL-C). Consistent with this observation in mice, in statin-responsive human primary hepatocytes, 1B20 lowers PCSK9 and LDLR mRNA levels and raises serum steady-state levels of antibody-bound PCSK9. In addition, mRNA levels of several SREBP regulated genes involved in cholesterol and fatty-acid synthesis including ACSS2, FDPS, IDI1, MVD, HMGCR, and CYP51A1 were decreased significantly with antibody treatment of primary human hepatocytes. In rhesus monkeys, subcutaneous (SC) dosing of 1B20 dose-dependently induces robust LDL-C lowering (maximal ~70%), which is correlated with increases in target engagement and total antibody-bound PCSK9 levels. Importantly, a combination of 1B20 and Simvastatin in dyslipidemic rhesus monkeys reduced LDL-C more than either agent alone, consistent with a mechanism of action that predicts additive effects of anti-PCSK9 agents with statins. Our results suggest that antibodies targeting PCSK9 could provide patients powerful LDL lowering efficacy on top of statins, and lower cardiovascular risk.
Keywords: PCSK9, low density lipoprotein cholesterol (LDL-C), hypercholesterolemia, LDL receptor, sterol regulatory element binding protein (SREBP), primary hepatocytes
Introduction
Cardiovascular disease is a significant public health burden in the US and other developed countries. Although statin therapy has been used successfully in treating dyslipidemia and reducing cardiovascular events in humans, a significant number of patients still do not reach their target LDL-C levels, as set out by the Coordinating Committee of the National Cholesterol Education Program(1, 2). Additionally, of those treated that do reach their LDL-C goal, many continue to have cardiovascular events(3). There is also a segment of the patient population that are statin intolerant(4). Therefore, there is a significant unmet medical need to lower LDL-C and cardiovascular risk in these high risk patients.
Proprotein convertase subtilisin/kexin type 9 (PCSK9), which interacts with and promotes cellular degradation of the low density lipoprotein receptor (LDLR), is a promising therapeutic target for treating hypercholesterolemia and coronary heart disease (5-13). A strong link between PCSK9, LDL cholesterol and coronary heart disease has been established in humans. Several large human genetic studies have shown that putative gain- or loss-of function mutants (missense or truncation mutations) correlate with increased or reduced plasma LDL levels and CHD, respectively (14-18). A recent genome-wide association study further bolstered the importance of PCSK9 by establishing a link between a single nucleotide polymorphism at a locus near PCSK9 with early onset myocardial infarction (19). A clear link between PCSK9 and LDL-C is also observed in animal studies. PCSK9 knockout mice have decreased plasma LDL-C (20). In non-human primates, PCSK9 knockdown by siRNA or inhibition by a monoclonal antibody also leads to decreased plasma LDL (21-23).
There is extensive evidence that plasma PCSK9 raises LDL cholesterol levels by binding to cell surface LDLR and targeting the receptor to lysosome for degradation(9-13, 24-35). Statins exert their plasma LDL lowering effect by inhibiting hepatic cholesterol biosynthesis, raising SREBP (Sterol Regulatory Element binding Protein) tone in hepatocytes, and therefore increasing LDLR transcription and protein level (36-40). However, PCSK9, another SREBP responsive gene, is also induced and could attenuate statin's LDLR raising effect (20, 41-44). Therefore, introducing a PCSK9 antagonist on top of a statin is predicted to be additive to statins and further lower LDL-C. Here we describe the characterization of a monoclonal antibody 1B20, which binds to PCSK9 with high affinity, disrupts the PCSK9-LDLR interaction, and inhibits the effect of PCSK9 on cellular LDL uptake. Treatment with the 1B20 anti-PCSK9 monoclonal antibody via either IV or SC dosing in mice and rhesus monkeys led to robust LDL-C lowering in plasma, decreased liver PCSK9 and LDLR mRNAs, and transient increases in total plasma levels of PCSK9. 1B20 and simvastatin showed additive effects in LDL-C lowering in dyslipidemic monkeys with a genetic predisposition to metabolic syndrome. Consistent with in-vivo data in mice and monkeys, in human primary hepatocytes 1B20 treatment reduces PCSK9 and LDLR mRNAs on top of simvastatin, inhibits cellular PCSK9 uptake, and leads to increases in secreted PCSK9 protein.
Results
PCSK9 affinity characterization
1B20 binding kinetics was measured using surface plasmon resonance in a Biacore 2000. Polyclonal anti-human IgG antibody was covalently coupled to the surface and 1B20 was captured. Association and dissociation rates of soluble PCSK9 protein were then determined and used to calculate the affinity constant KD = kd/ka. Repeated determinations were made against recombinant soluble human, mouse, rhesus and rat PCSK9-V5-His proteins. The determined kinetic and equilibrium constants are given in Table 1. 1B20 binds to human, rat, rhesus, and mouse PCSK9 with high affinity at KD of 0.3, 4.5, 0.65, and 1 nM respectively.
Affinity characterization of 1B20 by surface plasmon resonance (Biacore 2000). Values are expressed as arithmetic means ± standard errors.
| Human PCSK9 | Rat PCSK9 | Rhesus PCSK9 | Mouse PCSK9 | |
|---|---|---|---|---|
| ka (1/Ms) | 1.4E05 ± 1.3E04 | 1.5E05 ± 3.6E03 | 1.6E05 ± 4.7E03 | 1.5E05 ± 1.6E04 |
| kd (1/s) | 4.0E-05 ± 5.8E-07 | 7.5E-04 ± 1.4E-05 | 1.0E-04 ± 5.9E-06 | 1.5E-04 ± 7.1E-06 |
| KD (M) | 3.0E-10 ± 3.0E-11 | 4.5E-09 ± 1.3E-10 | 6.5E-10 ± 2.4E-11 | 1.0E-9 ± 6.4E-11 |
Kinetic constants for association rate (ka) and for dissociation rate (kd) were determined and used to calculate the apparent equilibrium affinity constant (KD), using the equation KD = kd/ka. The values represent the averages of three experiments.
In vitro functional efficacy of 1B20 for PCSK9
The biological activity of 1B20 was examined using in vitro assays that measure PCSK9 function. First, 1B20 was tested in a cell-based assay that measures PCSK9-dependent effects on cellular LDL-uptake, where LDL is fluorescently labeled (AF-LDL). Recombinant PCSK9 protein (1 μg/ml, 13 nM) was added exogenously to HepG2 cells in the presence or absence of increasing amounts of the anti-PCSK9 mAb 1B20. This antibody dose-dependently and completely inhibited the effects of PCSK9 on cellular AF-LDL-uptake. The addition of 1B20 to cells in the absence of exogenously added PCSK9 had no effect on cellular AF-LDL uptake (data not shown), indicating that the effects of 1B20 are specific to inhibition of exogenously added PCSK9 protein. In HepG2 cells the regulation of cell surface LDLR presumably is not by secreted PCSK9. One possible explanation is that the level of secreted PCSK9 in HepG2 cells is extremely low (~ 0.03 nM) compared to human plasma PCSK9 level (~5 nM), therefore the minimal level of secreted PCSK9 in this cell line does not have much impact on cell surface LDLR protein. When exogenous PCSK9 was added to these cells, LDL uptake was significantly decreased, and the addition of 1B20 antibody reversed this decrease. We have not fully explored factors that regulate LDLR, but published information from Dong et al (reference#46) has shown that Idol is involved in the regulation of LDLR in HepG2 cells. Because HepG2 is a cancer cell line which does not resemble liver cells very well at cellular or molecular levels, we switched to primary human hepatocytes in later studies.
While the results indicate that the potencies of 1B20 for all species of PCSK9 are equivalent (Table 2), the lowest theoretical IC50 that is measurable in this assay is ~ 4 nM. This assay will not differentiate modest potency differences lower than 4 nM as would be predicted by the binding affinity data (Table 1).
The ability of 1B20 to directly inhibit the PCSK9-LDLR interaction, a critical protein-protein interaction that is required for PCSK9's effects on cellular LDL-uptake, was analyzed by the method of Surface plasmon resonance (SPR) on a Biacore instrument. In this assay, recombinant human LDLR protein was immobilized on a CM5 sensor chip using standard coupling chemistry. Either human, mouse, rhesus or rat PCSK9 were diluted in running buffer to a concentration of around 25 nM and varying amounts of 1B20 were added. As shown in Table 2, 1B20 is a potent antagonist of the interaction of human, mouse, rhesus, rat PCSK9 toward LDLR. The IC50 range is from 6 to 11 nM. The IC50 is less than the PCSK9 concentration and close to the floor of the assay sensitivity (~ 6 nM), consistent with the notion that the true inhibition constant is most likely much lower than the IC50 value.
1B20 is a full antagonist of PCSK9 - LDLR interaction in vitro, and blocks PCSK9 inhibitory effect on cellular LDL uptake in HepG2 cells.
| Protein | IC50 (nM) on PCSK9 - LDLR interactionA | IC50 (nM) on PCSK9 inhibitory effect on cellular LDL uptakeB |
|---|---|---|
| Human PCSK9 | 11.4 ± 1.5 | 4 ± 1 |
| Mouse PCSK9 | 5.8 ± 1.4 | 3 ± 0 |
| Rhesus PCSK9 | 11.4 ± 1.4 | 4 ± 2 |
| Rat PCSK9 | 7.8 ± 1.3 | 3 ± 1 |
APCSK9 - LDLR interaction was quantified on Biacore instrument. Human recombinant LDLR was immobilized on a CM5 sensor chip. PCSK9 alone, or PCSK9 with various concentrations of 1B20 was added, and resonance units were measured. Data are expressed as means ± standard deviation of at least three experiments. BPCSK9 alone or PCSK9 pre-incubated with 1B20, together with AF-546-LDL, was added to cultured HepG2 cells. Cellular uptake of AF546-LDL was measured. Values are expressed as arithmetic means ± standard deviations of at least three experiments.
LDL-lowering efficacy of 1B20 in mice (multiple-dosing)
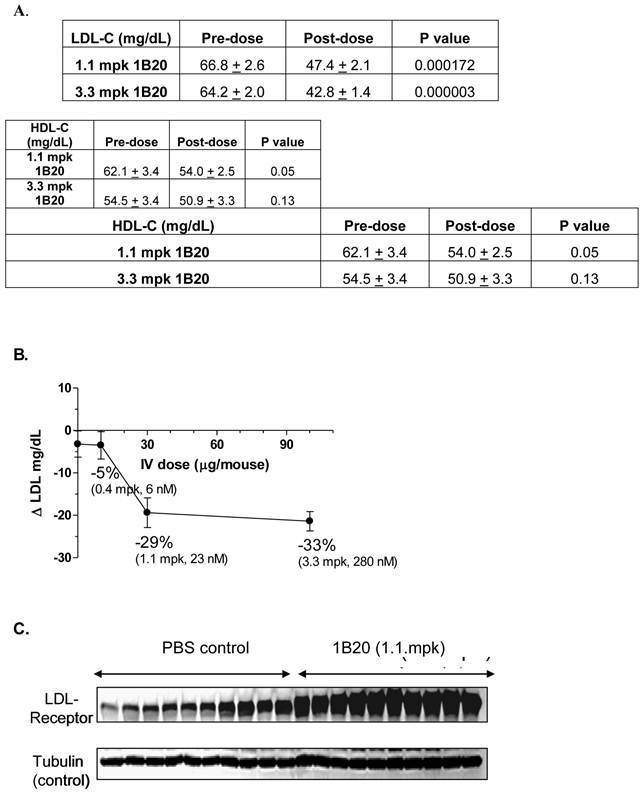
Typically, wild-type mice are not ideal models because their baseline circulating levels of LDL-C are low (~ 10 mg/dl) and the circuitry of their lipid metabolism is such that they are generally unresponsive to HMG-CoA reductase inhibitors (Statins). Therefore, a transgenic mouse model expressing the human CETP transgene and a single allele copy of the LDL-receptor (CETPtg[LDLR+/-]) was utilized (23). By introducing the CETP transgene, mice are able to, like humans, undergo reverse cholesterol metabolism thereby decreasing their steady-state HDL-C levels. The single copy of the LDLR further promotes an increase in circulating LDL-C in these mice and the overall lipid distribution closely mimics that of young healthy humans. Additionally, the CETPtg[LDLR+/-] mice have circulating levels of PCSK9 that are similar to humans (~ 5 nM). A single 1.1 mpk IV dose of 1B20 (in PBS buffer) induced ~29% LDL-C lowering 48 hr post dose. At this time point, the level of serum 1B20 was measured as 3.5 ug/ml (23 nM). Similar LDL lowering effects were obtained with IP dosing of this antibody (data not shown). The PBS-only control and an irrelevant antibody targeting a separate and distinct antigen had no LDL lowering effects (data not shown). Consistent with the proposed mechanism of LDL-lowering, 1B20 markedly increased the steady state levels of hepatic LDL-receptor protein compared to the PBS control, as shown in the Western blot in Figure 1. Other circulating lipid levels including high-density lipoproteins (HDL) and triglycerides (TGs) were not affected by 1B20 treatment (data not shown).
1B20 lowered LDL-C (A, B) and increased hepatic LDL-receptor protein (C) in a transgenic mouse model [CETPtg(LDLR+/-)]. Mice were sacrificed 48 hours after single IV injection of 1B20. Plasma samples were collected for LDL-C and HDL-C analysis (A) and liver samples were collected for western blot (C). Note: Comparing to wild type C57BL/6 mice, the LDL-C level in these CETPtg(LDLR+/-) mice is relatively high (~66 mg/dL) and the HDL-C level is relatively low (~58 mg/dL) (23).
Effects of multiple doses given 14 days apart, of either 3 mpk or 10 mpk IV of 1B20 in CETPtg(LDLR+/-) mice on 1B20 levels (A), %LDL-C changes (B), % TE (C) and total PCSK9 levels (D). The PK profiles were comparable following the first and second doses, suggesting 1B20 does not elicit a strong immune response under this dosing regimen (A).1B20 induced a robust 50-70% LDL-C lowering, and the extent of LDL-C lowering was not significantly different with the second dose (B). The changes in TE were consistent with the time course of changes in plasma LDL-C (B,C). Both doses transiently increased total PCSK9, but no accumulation of this increase was observed with multiple dosing (D).
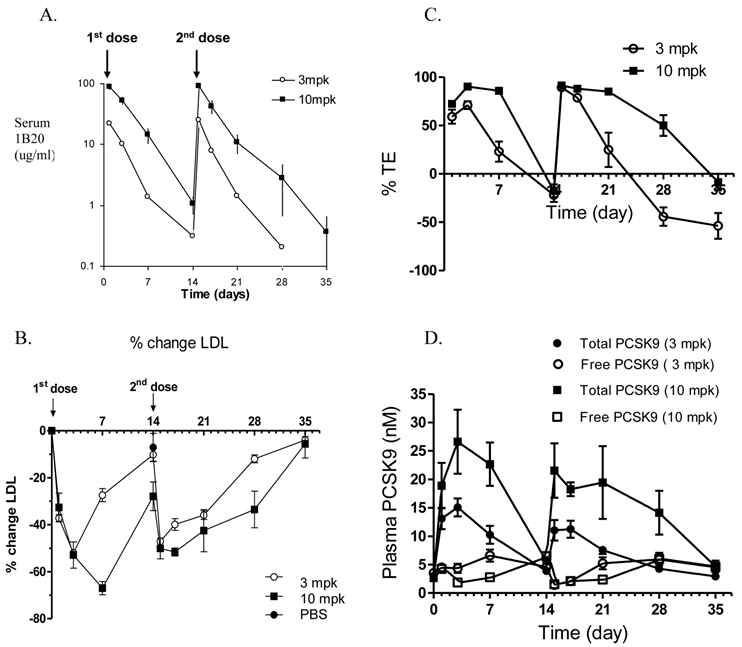
To examine the multiple dose effect of 1B20, a multidose study in CETPtg(LDLR+/-) mice was performed using either 3 or 10 mg/kg (mpk) doses of 1B20, administered 14 days apart (Figure 2). The pharmacokinetics (PK) profiles of 1B20 are comparable following the first and the second doses, suggesting that 1B20 does not elicit a strong immune response under this dosing regime (Figure 2A). Additionally, a robust 50-70% LDL-C lowering was induced by 1B20 and the extent of LDL-C lowering was not significantly different with the second dose (Figure 2B). The 1B20 dose response between 3 mpk and 10 mpk on LDL lowering was seen only at longer time points (day 7 and later, not before day 3). This is likely due to the circulating 1B20 levels dropping to limiting concentrations at longer time points (~1ug/ml or 7 nM at day 7 for the 3 mpk dose) compared to circulating PCSK9 levels (~5 nM) in these mice. To access the occupancy of PCSK9 by 1B20 antibody, two Delfia ELISA assays were performed. One Delfia assay measures plasma total PCSK9 and the other assay measures free (unbound) PCSK9. The 1B20-bound PCSK9 is calculated as the difference between total and free PCSK9. The percent target engagement is defined as the percentage of 1B20-bound PCSK9 in total PCSK9. Importantly, the changes in % target engagement (TE) were consistent with the time-course of changes in plasma levels of LDL-C. The duration of ≥ 25% LDL lowering was 7 days post a 3 mpk dose and ~ 14 days post a 10 mpk dose. Both doses of 1B20 transiently increased total PCSK9 levels significantly; 4.5 fold for the 3 mpk dose and 7.5 fold for the 10 mpk dose with levels returning to baseline at days 14 and day 35.
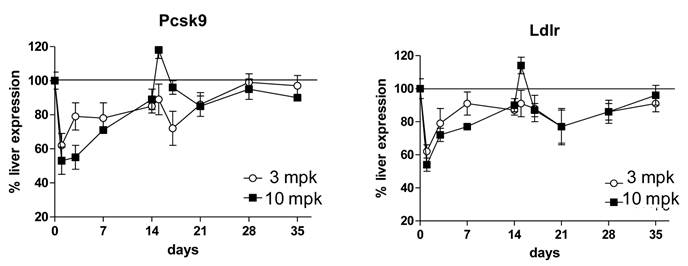
As part of the multidose study, liver tissues were harvested for analysis of gene expression changes. It was anticipated that inhibition of PCSK9 would result in an increase in intracellular liver cholesterol concentrations; this in turn would inactivate the transcriptional protein SREBP-2 and result in a suppression of gene expression of PCSK9 and LDLR. Consistent with expectations, 1B20 promoted a reduction of PCSK9 and LDLR mRNAs that paralleled the time course of plasma LDL-reductions (Figure 3). Despite this reduction in PCSK9 mRNA levels, the total circulating levels of PCSK9 increased with 1B20 treatment. This increase in PCSK9 protein most likely is due to the sequestering of PCSK9 by 1B20 in the circulation, which may lead to slower degradation of PCSK9 and higher steady-state levels, rather than increased synthesis.
In-vivo efficacy in healthy rhesus monkeys (IV & SC dose)
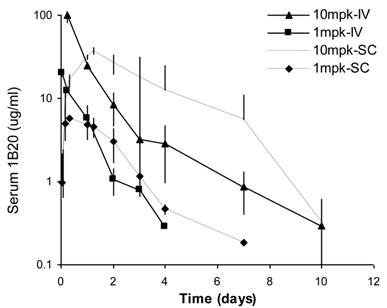
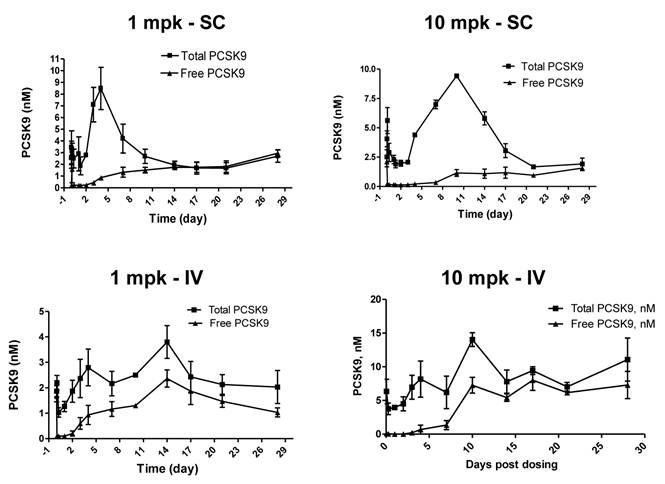
Sub-cutaneous (SC) administration is the preferred route of delivery in humans. To test the feasibility and aid SC dose selection in humans, a study was conducted in healthy rhesus monkeys to evaluate PK, PD (pharmacodynamics) and TE (target engagement) of 1B20 following a single SC or IV dose of 1 and 10 mpk of 1B20. As shown in Figure 4, at 1 mpk, SC and IV dosing led to similar serum 1B20 levels. At 10 mpk, SC dosing of 1B20 led to higher day 2-7 circulating levels of 1B20, possibly due to the extended release of 1B20 into circulation after SC dosing. Consistent with the PK profile, at 1 mpk, SC and IV dosing led to similar time courses of % LDL changes, while at 10 mpk, SC dosing led to longer duration of LDL reduction with LDL returning to baseline around day 28, demonstrating a strong PK-PD relationship (Figure 5). At 1 mpk SC and IV dosing, % TE returned to baseline around day 15, which is consistent with the time course of LDL changes; at 10 mpk SC and IV dosing, %TE returned to baseline around day 28, and is also consistent with the LDL changes, suggesting a good TE-PD correlation. Overall, these data demonstrate a PK-PD-TE correlation. Additionally, similar to what was seen in the mouse study, 1B20 treatment promoted a transient increase in total PCSK9 levels (2-3 fold), which returned to baseline levels during the study (Figure 6). In summary, both IV and SC dosing of 1B20 induced robust LDL-C lowering in healthy, normocholesterolemic monkeys, and the preferred route of administration is SC, which makes it amenable for self-injections in humans.
1B20 treatment led to reductions of liver PCSK9 and LDLR mRNAs in mice that paralleled the time course of LDL-C reductions in the multiple dosing study. First dose was given at day 0, second dose was given at day 14. 1B20 dose response of mRNA decreases correlated with dose response of LDL lowering.
Comparison of serum 1B20 levels as a function of IV or SC administration of 1 and 10 mpk doses of 1B20 in healthy rhesus monkeys. Serum 1B20 levels were higher after 10 mpk dosing than 1 mpk dosing. At 1 mpk, SC and IV dosing led to similar serum 1B20 levels. Comparing to IV dosing, SC dosing of 1B20 (10 mpk) led to higher day 2-7 circulating levels of 1B20, possibly due to the extended release of 1B20 into circulation after SC dosing. The PK profiles are consistent with the time courses of LDL lowering (see below).
Comparison of %TE and LDL-C changes as a function of IV or SC administration of 1 and 10 mpk doses of 1B20 in healthy rhesus monkeys. TE levels after 10 mpk dosing (IV or SC) were higher than TE after 1 mpk. SC or IV dosing of 1 mpk 1B20 had similar effects on TE and LDL. SC dosing of 10 mpk 1B20 led to higher %TE and longer duration of LDL-C lowering than 10 mpk IV dosing.
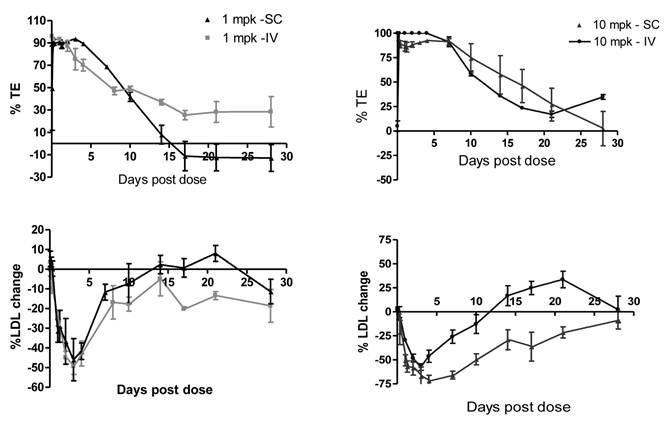
Effects of a single SC or IV dose of 1 and 10 mpk 1B20 in healthy rhesus monkeys on total and free (unbound) levels of plasma PCSK9. In all cases, 1B20 promoted a transient decrease in free PCSK9 and increase in total PCSK9 levels (2-3 fold), which returned to baseline at ~2 weeks post treatment.
Statin additivity study in metabolic syndrome monkeys
To assess the statin additivity effects of 1B20, a study was designed in a cohort of metabolic syndrome monkeys. These monkeys naturally develop metabolic syndrome, are overweight and dyslipidemic and have been reported to respond to statins (45). Detailed characterization of the metabolic syndrome monkeys can be found in reference#45. Briefly, the metabolic syndrome (MS) monkeys have significantly higher level of plasma insulin (906 pM MS vs. 241 pM healthy), HbA1c (6.1% MS vs. 4.3% healthy), and lower insulin-stimulated glucose uptake rate (5.3 vs. 12.9 mg/kg fat-free mass/min, MS vs. healthy).
A multiple arm study was designed to compare the effects of simvastatin, 1B20, and simvastatin plus 1B20 in the same cohort of 10 monkeys. Simvastatin was given at a 30 mg/kg/day dose in food treats and 1B20 was administered IV as a 3 mpk dose. The study design is as follows: Monkeys were treated with vehicle in weeks 1 and 2, simvastatin in weeks 3 and 4 (30mg/kg/day in food), followed by vehicle in weeks 5 and 6. At the start of week 7, a single 3 mpk IV dose of 1B20 was administered to each monkey. Given the result obtained in lean monkeys, it was anticipated that LDL-C would return to baseline levels at 14 days post this first dose. At the start of week 9, a second dose of single 3 mpk IV 1B20 was administered together with daily dosings of simvastatin in food treats. During the study, weekly plasma samples were collected for measurements of LDL-C., total and free PCSK9.
As shown in Figure 7, after 7 days of treatment with simvastatin alone, LDL-C was lowered by ~ 22% (-29 mg/dl). In comparison, 7 days post a single IV 3 mpk dose of 1B20, LDL-C was lowered by ~20% (-23 mg/dl). Note that results obtained with lean monkeys would predict that at 7 days post dosing with 1B20, the LDL-C lowering is past its maximal effect, which would explain the modest 20% lowering observed at this time point in this study. Nevertheless, in the combination 1B20 + simvastatin arm of the study, LDL-C was lowered by ~ 40% (48 mg/dl), suggestive of an additive effect of these agents.
To assess target engagement of PCSK9 by 1B20, levels of total PCSK9 and free (unbound) PCSK9 were measured in this study (Figure 8). At 7 days post 1B20 dosing alone or in combination with simvastatin, ~ 10-11 fold increases in total PCSK9 levels were observed for both conditions; however, the levels of free PCSK9 remained low which corresponded to calculated levels of ~ 90% target engagement. In comparison, in lean healthy monkeys, the increase in total PCSK9 levels with 1B20 treatment was relatively modest (2-3 fold). Additionally, even at 14 days post treatment, despite calculated levels of TE of ~ 50-60%, the levels of total PCSK9 in this study were 3.5 fold higher than untreated levels, suggesting that accumulation of PCSK9 may contribute to the LDL rebound in 1B20-treated monkeys. In comparison, transient increases in the levels of total PCSK9 observed in both the lean monkey and mouse studies returned to untreated levels by 14 days post dosing with 1B20.
Effects of Simvastatin, 1B20 alone and 1B20 + Simvastatin combination on plasma LDL-C in metabolic syndrome monkeys (n = 10). A. Study design. Monkeys were treated with vehicle in weeks 1 and 2, simvastatin in weeks 3 and 4 (30mg/kg/day in food), followed by vehicle in weeks 5 and 6. At the start of week 7, a single 3 mpk IV dose of 1B20 was administered to each monkey. Given the result obtained in lean monkeys, it was anticipated that LDL-C would return to baseline levels at 14 days post this first dose. At the start of week 9, a second dose of single 3 mpk IV 1B20 was administered together with daily dosings of simvastatin in food treats. During the study, weekly plasma samples were collected for measurements of LDL-C. B. At day 7 post dosing with simvastatin alone, LDL-C was lowered by ~22%; at day 7 post dosing with 1B20 alone, LDL-C was lowered by ~20%; at day 7 post dosing with simvastatin/1B20 combination, LDL-C was lowered by ~40%, indicating statin additivity.
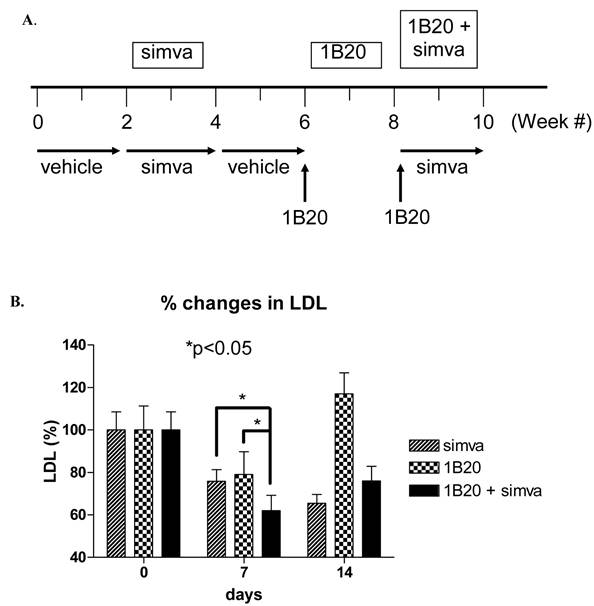
Effects of Simvastatin, 1B20 alone and the combination on levels of plasma total PCSK9, free PCSK9 and calculated target engagement in metabolic syndrome monkeys. At 7 days post dosing with 1B20 or 1B20/simvastatin combination, total PCSK9 levels were increased by ~10 fold, yet free PCSK9 levels remained low, corresponding to ~90% target engagement.
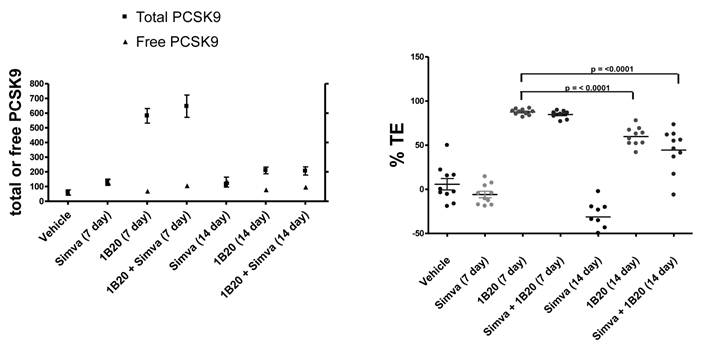
Free and total PCSK9 (secreted) after 1B20 treatment in human primary hepatocytes. 1B20 treatment on human primary hepatocytes increased secreted total PCSK9 protein and decreased free (unbound) PCSK9, with and without statin treatment.
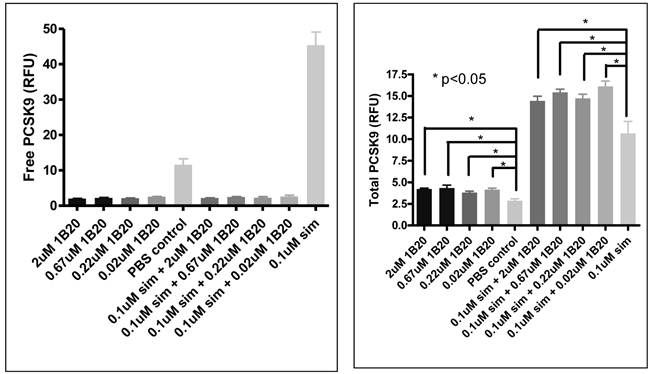
Effects of 1B20 treatment in human primary hepatocytes
In order to assess the mechanism of action of anti-PCSK9 antibodies, a cell-based system was developed using human primary hepatocytes to model the in-vivo observations. 1B20 treatment reduced free PCSK9 levels, and increased total (free + bound) secreted PCSK9 levels in culture medium (Figure 9). Interestingly, these effects of 1B20 were observed with and without simvastatin treatment. These results were consistent with the changes in plasma PCSK9 levels observed in mouse and monkey studies. We hypothesize that the increases in total PCSK9 might be attributed to the tight binding of 1B20 to PCSK9, leading to the inhibition of cellular clearance of PCSK9. Indeed, we observed blockade of PCSK9 uptake by 1B20 in human primary hepatocytes (Figure 10).
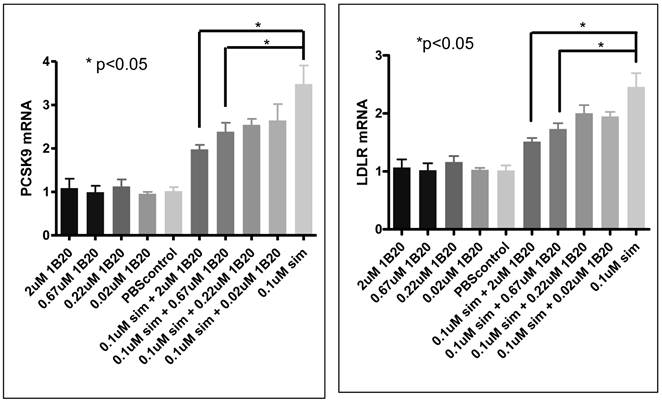
As part of the primary hepatocytes study, we measured PCSK9 and LDLR mRNA to evaluate the possible effect of 1B20 treatment on SREBP tone. As shown in Figure 11, simvastatin treatment induced increases in PCSK9 and LDLR mRNAs, and 1B20 treatment partially reversed this induction in a dose-dependent manner. This is consistent with the notion that PCSK9 inhibition would lead to increased cellular cholesterol uptake and decreased SREBP tone, and also is in agreement with reductions of PCSK9/LDLR mRNA levels in the livers of mice treated with 1B20. To further evaluate the transcriptional effect of 1B20, mRNA levels of genes representing major lipid metabolism pathways were measured. As shown in Table 3, comparing 1B20 + simvastatin combination versus simvastatin alone, the mRNA levels of key genes in both cholesterol and fatty acid synthesis pathways were reduced, consistent with a reduced SREBP activity.
Messenger RNA expression of SREBP-regulated key genes in cholesterol and fatty acid synthesis pathways was reduced in human primary hepatocytes comparing treatment with 1B20 + simvastatin versus simvastatin alone.
| Gene symbol | % Change | P value |
|---|---|---|
| ACSS2 | 34% down | 0.018 |
| FDPS | 37% down | 0.002 |
| IDI1 | 41% down | 0.005 |
| MVD | 40% down | 0.002 |
| HMGCR | 22% down | 0.049 |
| CYP51A1 | 25% down | 0.05 |
| SCD | 36% down | 0.02 |
| FADS1 | 22% down | 0.01 |
| FADS2 | 32% down | 0.007 |
| PCSK9 | 20% down | 0.04 |
| LDLR | 27% down | 0.01 |
Genes involved in cholesterol synthesis: ACSS2, FDPS, IDI1, MVD, HMGCR, and CYP51A1. Genes involved in fatty acid synthesis: ACSS2, SCD, FADS1, and FADS2.
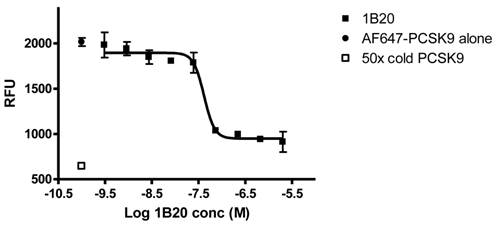
1B20 blocks PCSK9 uptake in human primary hepatocytes. AF647-labeled PCSK9, alone or pre-mixed with antibody, was incubated with human primary hepatocytes for 5 hr. Cells were washed and cellular fluorescent levels were quantified on ArrayScan. 1B20 blocks PCSK9 uptake, suggesting that the increases in plasma total PCSK9 might be due to decreased cellular clearance of PCSK9 by hepatocytes.
PCSK9/LDLR mRNA expression after 1B20 treatment in human primary hepatocytes. 1B20 treatment significantly reduced PCSK9 and LDLR mRNAs in the presence of simvastatin.
Discussion
Although statin therapy has been used successfully in treating hypercholesterolemia, a significant percentage of patients still do not reach the target LDL-C levels and continue to have cardiovascular events. In addition, a segment of the patient population is statin intolerant. Therefore there is a significant unmet medical need for additional therapies to lower LDL-C and cardiovascular risk. PCSK9 has been proven to be a promising new therapeutic target for treating hypercholesterolemia and coronary heart disease by human genetic studies. Here we report an anti-PCSK9 monoclonal antibody 1B20, similar to an internal antibody 1D05 (23) binds to PCSK9 with high affinity, disrupts PCSK9-LDLR protein interaction, and completely blocks PCSK9 inhibition on cellular LDL uptake. More importantly, 1B20 treatment induced robust 50-70% plasma LDL-C lowering in mice in a multiple dosing study, increased hepatic LDLR protein, and reduced hepatic PCSK9 and LDLR mRNAs. In rhesus monkeys, both IV and SC dosing of 1B20 led to robust and long-lasting LDL-C lowering. Furthermore, for the first time, our study in metabolic syndrome monkeys demonstrated that the combination of 1B20 plus simvastatin reduced LDL-C more than simvastatin or 1B20 alone, highlighting the advantage of an anti-PCSK9 antibody/statin combination therapy over either treatment alone. Consistent with in vivo observations, 1B20 treatment in human primary hepatocytes reduced PCSK9 mRNA on top of simvastatin while increasing secreted total PCSK9 protein, indicating that the PCSK9 protein increase is due to decreased PCSK9 clearance rather than increased synthesis.
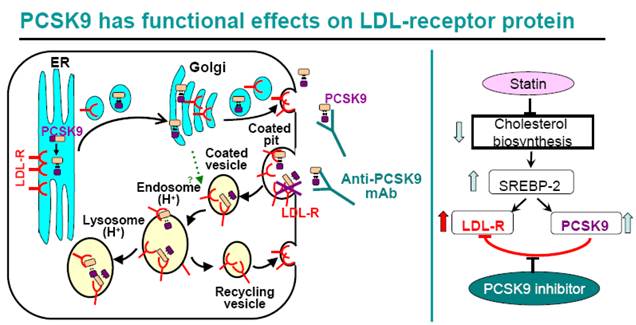
For many high risk CHD patients, statin therapy alone, even at high doses, does not reduce LDL-C to the target goals(1, 2). As shown in Figure 12, statins inhibit cellular cholesterol biosynthesis, which leads to increased SREBP-2 activity, thus turns on the transcription of both LDLR and PCSK9 genes. Therefore statin treatment leads to both a beneficial effect of increased cellular LDLR protein level, increased LDL clearance by liver cells, and a counteractive effect of increased PCSK9 protein level. Indeed, increased circulating PCSK9 protein level after statin treatment has been reported in both humans and animals (20, 41-44), and increased PCSK9 mRNA and protein levels were observed in human primary hepatocytes treated with simvastatin (Figure 9, 11). The statin-induced increase in PCSK9 would attenuate the LDL-lowering efficacy of statins since PCSK9 binds to LDLR and triggers its trafficking to the lysosome for degradation. The introduction of a PCSK9 inhibitor, for example, anti-PCSK9 antibody 1B20, would block PCSK9-mediated LDLR protein degradation, increase hepatic LDLR protein level and LDL clearance, therefore is expected to be additive to statins in LDL lowering. Indeed, in our study in metabolic syndrome monkeys, 1B20 + simvastatin combination treatment lowered LDL cholesterol more than 1B20 or simvastatin alone (Figure 7). Although liver samples could not be obtained from the monkeys, we did analyze mouse livers from 1B20 treatment, and observed an increase in liver LDLR protein (Figure 1). Interestingly, we observed reductions in liver LDLR and PCSK9 mRNAs in 1B20-treated mice (Figure 3), confirming that the increase in liver LDLR protein was not due to increased LDLR mRNA expression, rather due to decreased post-transcriptional LDLR protein degradation. The decreases in liver LDLR and PCSK9 mRNAs were most likely attributed to a feedback response, as cellular SREBP-2 activity in hepatocytes would be reduced due to increased LDLR-mediated LDL uptake leading to increased cellular cholesterol content following 1B20 treatment, and both LDLR and PCSK9 gene transcription are regulated by SREBP-2. Both LDLR and PCSK9 mRNAs eventually returned to baseline. Consistent with decreased LDLR and PCSK9 mRNAs in mouse livers following 1B20 treatment, we observed dose-dependent decreases in LDLR and PCSK9 mRNAs in human primary hepatocytes treated with 1B20 in the presence of simvastatin (Figure 11). In addition, we also observed reductions in other SREBP-inducible genes, including genes involved in cholesterol and fatty acid synthesis (Table 3), suggesting a suppressed SREBP activity.
In mouse and monkey efficacy studies, 1B20 treatment led to decreases in plasma free (unbound) PCSK9 levels, and increases in plasma total PCSK9 levels. We speculate this increase is caused by the sequestering of PCSK9 by 1B20, which blocks PCSK9 uptake and clearance by the liver, the major organ for PCSK9 clearance in the body. Similar to the observations with 1B20, treatment of monkeys with the 1D05 antibody (23) resulted in increases, to a lesser extent, in plasma total PCSK9. Consistent with the in vivo results, we observed decreases in free PCSK9 levels and increases in total PCSK9 (secreted) after 1B20 treatment in human primary hepatocytes, with and without simvastatin. Our finding that 1B20 blocks the internalization of PCSK9 into human primary hepatocytes suggests that this antibody inhibits PCSK9 clearance by hepatocytes, which in turn leads to the accumulation of PCSK9 in the circulation. Increased PCSK9 synthesis is ruled out since PCSK9 mRNA levels were reduced in both mouse study and human primary hepatocytes. It is important to point out the PCSK9 is antibody-bound, therefore functionally inactive in promoting LDLR degradation.
A PCSK9 inhibitor is expected to be additive to statins in LDL lowering. Statins inhibit cholesterol biosynthesis, leading to increased cellular SREBP-2 activity, and increased transcription of SREBP-2-inducible LDLR and PCSK9 mRNAs. Therefore statins increase cellular LDLR protein, leading to increased LDL clearance and lower circulating LDL. At the same time, statins also increase PCSK9 protein which binds to LDLR and triggers LDLR protein trafficking to the lysosome for degradation. Therefore, statin-induced PCSK9 increase limits statin's efficacy of LDL lowering. The introduction of a PCSK9 inhibitor would disrupt the interaction between PCSK9 and LDLR, raise LDLR protein, and be additive to statins in LDL lowering.
We acknowledge that as with all the antibody therapies, the cost is an issue. The proposed strategy is that the mAb therapy would be primarily used in high risk patients who have had cardiovascular event(s), and those who are statin-intolerant. The efficacy of 1B20 in LDL lowering is similar to the other published antibodies (22, 23), considering that they were tested in different animal models. In terms of other therapeutic approaches, RNAi technology targeting PCSK9 was reported in lowering LDL in animals (21), but the feasibility of the long term use of this new technology in humans is currently unknown. EGF-A mimetics are expected to inhibit the interaction between PCSK9 and LDLR, but the issue is that they tend to have very poor pharmacokinetics (short half life) in vivo.
In summary, the data presented here suggest that antibodies targeting PCSK9 could provide patients powerful LDL lowering efficacy on top of statins and would be expected to lower cardiovascular risk. Our findings in human primary hepatocytes demonstrate the critical role of liver cells and provide new insight in PCSK9 biology. The anti-PCSK9 antibody 1B20 can serve as a valuable tool in further elucidating the mechanism of action of PCSK9. Finally, the results from this study will help in the design of next generation PCSK9 inhibitors and clinical trials.
Materials and Methods
Isolation of anti-PCSK9 antibody 1B20
The human combinatorial antibody HuCAL GOLD phage display libraries were panned against recombinant human PCSK9-V5-His protein immobilized on Nunc Maxisorp plates. Initial expression and purification of 1B20 Fab was performed as previously described(28). Full 1B20 IgG2m4 was expressed and purified from CHO cells.
Antibody inhibition of PCSK9 - LDLR interaction
Surface plasmon resonance (SPR) measurements were carried out with a Biacore instrument (Biacore S51, Uppsala, Sweden) at 25oC. Human recombinant LDLR (R&D, Cat # 2148-LD/CF @ 40 ug/ml) was immobilized in 10 mM Na-acetate pH 4.5 on a CM5 sensor chip (GE, BR-1005-31) using standard EDC [1-ethyl-3-(3-dimethyl-aminopropyl)carbodiimide-HCl] and NHS (N-hydroxysuccinimide) coupling chemistry and subsequent blocking with ethanolamine per manufacturer's instructions. The targeted immobilization level was 1000 resonance units (RU). Running buffer was standard HBS-N buffer (GE, BR-1006-70) supplemented with 1 mM calcium chloride (Fluka, # 21114) and 0.005% surfactant P20 (GE, BR-1000-54). Human, rhesus, rat and mouse PCSK9 were diluted in running buffer to a concentration of around 25 nM and varying amounts of 1B20 were added. For analysis typically a sample volume of 120 ul was injected at a flow rate of 30 ul/min. Regeneration of the CM5 chip was accomplished with a short burst of 3 ul of 0.01 N HCL. Data were analyzed using BIAevaluation software.
AF546-labeled LDL Uptake Assay
HepG2 cells were plated in a 96-well poly-D-lysine-coated plate (Corning) at a density of 30,000 cells/well in DMEM containing 10% FBS. After 24 hr, the medium was switched to DMEM lacking serum. After 18 hr, the medium was removed and purified PCSK9 protein was added to the cells in 100 µl of mixture A (DMEM containing 10% lipoprotein-deficient Serum, 10 mg/ml AF-labeled LDL and 2ug/ml PCSK9). 1B20 antibody was titrated in presence of 2ug/ml PCSK9, starting with 100ug/ml. Antibody-PCSK9 mixture was incubated at room temperature for 20 minutes before added to the cells. The plates were incubated at 37 °C for 5 hr, and the cells were washed quickly with Tris-buffered saline (Bio-Rad) containing 2 mg/ml BSA. The wash step was repeated, but this time the wash buffer was incubated for 2 min with the cells. Finally, the cells were quickly washed twice with Tris-buffered saline (without BSA) and lysed in 100µl of RIPA buffer. The lysate was transferred to a 96-well black plate (Thermo LabSystems), and fluorescence was measured using a Spectra-MAX tunable spectrofluorometer (Molecular Devices) at an excitation wavelength of 520nm and an emission wavelength of 580 nm. Total cellular protein was measured in each well using the BCA protein assay, and the fluorescence units were normalized to total protein. The amount of 1B20 required for 50% inhibition of PCSK9 function (IC50) was determined by fitting data to a sigmoidal dose-response curve using nonlinear regression (GraphPad Software Inc.).
1B20 treatment in CETPtg[LDLR+/-] mice
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at Merck Research Laboratories. A transgenic mouse model expressing the human CETP transgene and a single allele copy of the LDL-receptor (CETPtg[LDLR+/-]) was utilized. 12-week old male mice were treated IV with indicated doses of 1B20 and serum samples were collected at indicated time points for measurements of LDL-C, total and free PCSK9, and 1B20 levels. Serum samples were treated with lipase inhibitors and protease inhibitors (Sigma), stored at 4°C and assayed within 7 days of sample collection. LDL-C was measured with LDL Direct Select Cholesterol Reagent (Equal Diagnostics). Liver samples were flash frozen in liquid nitrogen and stored at -80oC until analysis.
Mouse liver LDLR western blot
100mg of mouse liver was homogenized in 500ul RIPA Lysis buffer with 1x PMSF, sodium orthovanadate and protease inhibitors (Santa Cruz, sc-24948) on ice, centrifuged at 10,000g for 30 minutes. 50ug of protein was loaded to Novex Tris-Glycine gel (4-12%, Invitrogen), transferred to nitrocellulose membrane. The membrane was blocked with blocking buffer ( 1xTBS (Fisher ), 1% tween-20 and 5% non fat mik) at RT for an hour, washed 3x with wash buffer (1xTBS,1% tween-20), then incubated with LDLR antibody (R&D Systems, anti-mLDLR goat IgG ,Cat # AF2255, 1:1000 diluted in blocking buffer) and anti-tubulin (Sigma,T5168) at 4 0C overnight. After wash, the membrane was blot with HRP-anti-goat (GE Healthcare) and HRP-anti-mouse antibodies (GE Healthcare) for 45min, then washed 3 times. ELC plus (GE Healthcare, RPN2132) treated membrane was scanned on Typhoon 9400(GE Healthcare).
1B20 treatment in healthy rhesus monkeys
To characterize pharmacokinetics (PK), pharmacodynamics (PD) and target engagement (TE) of 1B20, single dose IV and SC studies were conducted in 15-year old male rhesus monkeys (Macaca mulatta). All rhesus monkeys used in the study were naïve to biologics. All studies were approved by the Merck Research Laboratories Institutional Animal Care and Use Committee (IACUC).
Each animal (n=6/group) received a single intravenous injection of 1B20 at 1 or 10 mg/kg via the cephalic vein. For SC administration, rhesus monkeys (n=3/group) were given a 1 or 10 mg/kg subcutaneous dose of 1B20 between the shoulder blades. In all studies, blood samples were collected from the saphenous/femoral vessel at designated time points post dosing and the resulting plasma/serum was used for PK (1B20 levels), PD (LDL-C level) and target engagement (free and total PCSK9) measurements.
A 1B20 specific, enzyme-linked immunosorbent assay (ELISA) was used to quantify 1B20. Briefly, ELISA plates are coated with an anti-PCSK9 capture antibody, which does not compete with 1B20 for binding to PCSK9, followed by recombinant PCSK9 immobilization. Serum samples containing 1B20 are applied to the plates pre-coated with the anti-PCSK9 antibody and PCSK9. Bound 1B20 is detected with an anti-human IgG2(Fc)-specific detection antibody.
Pharmacokinetic parameters were estimated using a non-compartmental method with WinNonlin 5.01 (Pharsight Inc). Clearance (CL) was calculated as the dose divided by the area under the serum concentration-time curve from zero to infinity (AUC0-∞). The apparent terminal half-life (t1/2) was estimated from the slope of the terminal phase of the log serum concentration-time data. The volume of distribution at steady state (Vdss) was determined using non-compartmental method: Vdss = (Dose iv x AUMC0-∞)/ (AUC0-∞)2, where AUMC0-∞ is the total area under the first moment of the drug concentration-time curve from time zero to infinity.
Rhesus target engagement Delfia assay
96-well Immulon 4HBX plates (Thermo Lab systems part # 3855) were coated with 5ug/ml of anti-PCSK9 antibody E07 at 4C overnight, washed 3 times with 300ul wash buffer (1x TBST buffer, Sigma, T9039) and then blocked 1h at RT with blocking buffer ( 1X TBS (Fisher Scientific,BP2472), 1% BSA, 0.05% Tween-20). Rhesus plasma (1:4 or 1:8) and rhesus PCSK9 standards were diluted in assay buffer (1% BSA in PBS). After 3 x wash with wash buffer, 50ul of diluted plasma samples or PCSK9 standards were added on the plate and incubated at 37 C for 1h, followed by 3x 300ul wash. 50ul of either biotinylated 1H23 (detects total PCSK9) or biotinylated 1B20 (detects free PCSK9) at 1ug/ml in Defila assay buffer (Perkin Elmer, 1244-111) were added to the plate and incubated at RT for 1h. After 3x wash with wash buffer, 50ul of 1:1000 Streptavidin/Europium (Perkin Elmer, 1244-360) in Defila assay buffer was added on the plate, incubated for 20min at RT, followed by 3x wash and 100ul of DELFIA Enhance (Perkin Elmer part # 1244-105). Plate was kept at RT, protected from light for 1h and read on Europium reader (Envision 2103). The bound PCSK9 is calculated as the difference between total and free PCSK9. The percentage of target engagement is defined as the percentage of bound PCSK9 in total PCSK9.
1B20 treatment in metabolic syndrome monkeys with and without a statin
To assess the statin additivity effects of 1B20, a study was designed in collaboration with B. C. Hansen in a cohort of metabolic syndrome monkeys. This multiple arm study was designed to compare the effects of simvastatin, 1B20, and simvastatin plus 1B20 in the same cohort of 10 monkeys. Simvastatin was given at a 30 mg/kg/day dose in food treats and 1B20 was administered IV as a 3 mpk dose. The study design is as follows: 19-year old male monkeys were treated with vehicle in weeks 1 and 2, simvastatin in weeks 3 and 4 (30mg/kg/day in food), followed by vehicle in weeks 5 and 6. At the start of week 7, a single 3 mpk IV dose of 1B20 was administered to each monkey. Given the result obtained in lean monkeys, it was anticipated that LDL-C would return to baseline levels at 14 days post this first dose. At the start of week 9, a second dose of single 3 mpk IV 1B20 was administered together with daily dosings of simvastatin in food treats. During the study, weekly plasma samples were collected for measurements of LDL-C., total and free PCSK9.
Lipoprotein analysis (RoboFplcLipidANalysis)
To generate lipoprotein profiles, plasma was fractionated by chromatography over Superose-6 size exclusion column (GE LifeSciences, Inc.) on an Ultimate 3000 Series HPLC system (Dionex Corporation). Total cholesterol levels in the column effluent were continuously measured via in-line mixture with a commercially available enzymatic colorimetric cholesterol detection reagent (Total Cholesterol E, cat #439-17501; Wako USA) followed by downstream spectrophotometric detection of the reaction products at 600nm absorbance. The first peak of cholesterol eluted from the column was attributed to VLDL, the second peak to LDL and the third to HDL; the area under each peak was calculated using software provided with the HPLC. The cholesterol concentrations for each lipoprotein fraction were calculated by extrapolating the ratio of the corresponding peak areas to total peak areas and multiplying by the total cholesterol concentration measured in the samples. Total cholesterol was measured in a microtiter plate from a 1:1 mix of 100ul Cholesterol E reagent and 2.5ul plasma diluted in PBS to 100ul, incubated at 37oC for 30 minutes and then read at 600nm in a SpectraMAX plate reader (Molecular Devices). Cholesterol standards were provided in the kit as a 200mg/dl Cholesterol stock and serially diluted to provide a standard curve. Non-HDL measures were calculated using the precipitation method detailed in the Wako HDL- E kit (cat# 431-52501).
Human primary hepatocytes model system
Cryopreserved plateable human primary hepatocytes were purchased from CellzDirect (Durham, NC) (cat#HMCPIS), and cultured by following supplier's instructions. On day 1, cells were thawed, recovered in cryopreserved hepatocytes recovery medium (CHRM, cat#CM7000) and plated in Plating medium (Williams E medium plus serum-containing thawing/plating supplement, cat#CM3000) at 50,000 cells/well/200ul on collagen-coated 96-well plates (Becton Dickinson, cat#356649), cultured in 37oC/5%CO2 incubator. On day 2, plating medium was removed and replaced with Maintenance medium (Williams E medium plus maintenance supplement, CellzDirect cat#CM4000). This medium was then replaced with Maintenance medium containing various concentrations of 1B20 antibody (starting at 300ug/ml) with or without simvastatin. On day 6, culture medium was harvested and the secreted free and total PCSK9 protein were measured using the same Delfia ELISA method as that used in rhesus monkey target engagement assay. mRNA was isolated from the cells using mRNA Catcher Plus kit (Invitrogen, cat#K1570-03) or Qiagen Rneasy mini kit, converted to cDNA, and mRNA levels of specific genes were measured using taqman quantitative PCR, with reagents from Applied Biosystems. A custom designed PCR array for genes in key lipid metabolism pathways was developed in collaboration with SABiosciences-Qiagen.
PCSK9 uptake in human primary hepatocytes
On day 1, cryopreserved plateable human primary hepatocytes were thawed and plated in Plating medium (above) at 40,000 cells/well/100ul on collagen-coated 96-well plates (Nunc, cat#152036), and cultured in 37oC/5%CO2 incubator. Six hours after plating, medium was removed, wells washed with PBS, replaced with cold Maintenance medium (above) containing 0.25 mg/ml Geltrex (Invitrogen, cat#12760-021), and returned to the 37oC incubator. On day 2 and 3, medium was removed and replaced with warm Maintenance medium. On day 4, medium was removed and replaced with Maintenance medium containing 15 ug/ml AF647-labeled human PCSK9 alone (protect from light) or with titrations of 1B20 starting at 300 ug/ml. AF647-PCSK9 and 1B20 were pre-mixed for 20 min at room temperature. Cells were incubated at 37oC/5%CO2 for 5 hours. Cells were washed with TBS containing 2 mg/ml BSA, then TBS. To exclude dead cells, cells were stained with LIVE/DEAD fixable Dead Cell green dye (Invitrogen, cat#L23101) for 20 min at room temperature by following manufacturer's instructions, and washed with TBS. Cell nuclei were stained and cells were fixed in TBS containing 1 ug/ml Hoechst's 33342 dye (Invitrogen, cat#H3570) and 4% paraformaldehyde (Polysciences, cat#18814) for 20 min at room temperature. Plates were washed twice in TBS, sealed and stored overnight at 4oC. On day 5, cells were analyzed and cellular content of AF647-PCSK9 were measured on ArrayScan.
Statistics
Student's t-tests (two-tailed and unpaired) were performed and P<0.05 was considered statistically significant. The error bars are SEMs.
Conflict of Interests
Disclosure: Authors are/were employed by Merck and Co.
References
1. Ferrieres J, Berard E, Crisan O, Bongard V. Residual dyslipidaemia after statin treatment in France: Prevalence and risk distribution. Arch Cardiovasc Dis. 2010;103:302-309
2. Franks P, Tancredi D, Winters P, Fiscella K. Cholesterol treatment with statins: who is left out and who makes it to goal?. BMC Health Serv Res. 2010;10:68
3. Toutouzas K, Drakopoulou M, Skoumas I, Stefanadis C. Advancing therapy for hypercholesterolemia. Expert Opin Pharmacother. 2010;11:1659-1672
4. Eckel R.H. Approach to the patient who is intolerant of statin therapy. J Clin Endocrinol Metab. 2010;95:2015-2022
5. Abifadel M, Pakradouni J, Collin M, Samson-Bouma M.E, Varret M, Rabes J.P, Boileau C. Strategies for proprotein convertase subtilisin kexin 9 modulation: a perspective on recent patents. Expert Opin Ther Pat. 2010;20:1547-71
6. Costet P. Molecular pathways and agents for lowering LDL-cholesterol in addition to statins. Pharmacol Ther. 2010;126:263-278
7. Li H, Ziegler N, Cui R, Liu J. Recent patents on PCSK9: a new target for treating hypercholesterolemia. Recent Pat DNA Gene Seq. 2009;3:201-212
8. Cao G, Qian Y.W, Kowala M.C, Konrad R.J. Further LDL cholesterol lowering through targeting PCSK9 for coronary artery disease. Endocr Metab Immune Disord Drug Targets. 2008;8:238-243
9. Seidah N.G. PCSK9 as a therapeutic target of dyslipidemia. Expert Opin Ther Targets. 2009;13:19-28
10. Costet P, Krempf M, Cariou B. PCSK9 and LDL cholesterol: unravelling the target to design the bullet. Trends Biochem Sci. 2008;33:426-434
11. Lambert G, Charlton F, Rye K.A, Piper D.E. Molecular basis of PCSK9 function. Atherosclerosis. 2009;203:1-7
12. Horton J.D, Cohen J.C, Hobbs H.H. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci. 2007;32:71-77
13. Duff C.J, Scott M.J, Kirby I.T, Hutchinson S.E, Martin S.L, Hooper N.M. Antibody-mediated disruption of the interaction between PCSK9 and the low-density lipoprotein receptor. Biochem J. 2009;419:577-584
14. Cohen J.C, Boerwinkle E, Mosley T.H Jr, Hobbs H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264-1272
15. Cohen J, Pertsemlidis A, Kotowski I.K, Graham R, Garcia C.K, Hobbs H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161-165
16. Kotowski I.K, Pertsemlidis A, Luke A, Cooper R.S, Vega G.L, Cohen J.C, Hobbs H.H. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet. 2006;78:410-422
17. Abifadel M, Varret M, Rabes J.P, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D. et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154-156
18. Huang C.C, Fornage M, Lloyd-Jones D.M, Wei G.S, Boerwinkle E, Liu K. Longitudinal association of PCSK9 sequence variations with low-density lipoprotein cholesterol levels: the Coronary Artery Risk Development in Young Adults Study. Circ Cardiovasc Genet. 2009;2:354-361
19. Kathiresan S, Voight B.F, Purcell S, Musunuru K, Ardissino D, Mannucci P.M, Anand S, Engert J.C, Samani N.J, Schunkert H. et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334-341
20. Rashid S, Curtis D.E, Garuti R, Anderson N.N, Bashmakov Y, Ho Y.K, Hammer R.E, Moon Y.A, Horton J.D. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci U S A. 2005;102:5374-5379
21. Frank-Kamenetsky M, Grefhorst A, Anderson N.N, Racie T.S, Bramlage B, Akinc A, Butler D, Charisse K, Dorkin R, Fan Y. et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc Natl Acad Sci U S A. 2008;105:11915-11920
22. Chan J.C, Piper D.E, Cao Q, Liu D, King C, Wang W, Tang J, Liu Q, Higbee J, Xia Z. et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci U S A. 2009;106:9820-9825
23. Ni Y.G, Di Marco S, Condra J.H, Peterson L.B, Wang W, Wang F, Pandit S, Hammond H.A, Rosa R, Cummings R.T. et al. A proprotein convertase subtilisin-like/kexin type 9 (PCSK9)-binding antibody that structurally mimics the EGF(A) domain of LDL-receptor reduces free circulating PCSK9 and LDL-cholesterol. J Lipid Res. 2011;52:78-86
24. Fasano T, Sun X.M, Patel D.D, Soutar A.K. Degradation of LDLR protein mediated by 'gain of function' PCSK9 mutants in normal and ARH cells. Atherosclerosis. 2009;203:166-171
25. Zhang D.W, Garuti R, Tang W.J, Cohen J.C, Hobbs H.H. Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc Natl Acad Sci U S A. 2008;105:13045-13050
26. Zhang D.W, Lagace T.A, Garuti R, Zhao Z, McDonald M, Horton J.D, Cohen J.C, Hobbs H.H. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007;282:18602-18612
27. Fisher T.S, Lo Surdo P, Pandit S, Mattu M, Santoro J.C, Wisniewski D, Cummings R.T, Calzetta A, Cubbon R.M, Fischer P.A. et al. Effects of pH and low density lipoprotein (LDL) on PCSK9-dependent LDL receptor regulation. J Biol Chem. 2007;282:20502-20512
28. Ni Y.G, Condra J.H, Orsatti L, Shen X, Di Marco S, Pandit S, Bottomley M.J, Ruggeri L, Cummings R.T, Cubbon R.M. et al. A proprotein convertase subtilisin-like/kexin type 9 (PCSK9) C-terminal domain antibody antigen-binding fragment inhibits PCSK9 internalization and restores low density lipoprotein uptake. J Biol Chem. 2010;285:12882-12891
29. Fan D, Yancey P.G, Qiu S, Ding L, Weeber E.J, Linton M.F, Fazio S. Self-association of human PCSK9 correlates with its LDLR-degrading activity. Biochemistry. 2008;47:1631-1639
30. Poirier S, Mayer G, Poupon V, McPherson P.S, Desjardins R, Ly K, Asselin M.C, Day R, Duclos F.J, Witmer M. et al. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J Biol Chem. 2009;284:28856-28864
31. Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, Mayer H, Nimpf J, Prat A, Seidah N.G. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem. 2008;283:2363-2372
32. Cameron J, Holla O.L, Ranheim T, Kulseth M.A, Berge K.E, Leren T.P. Effect of mutations in the PCSK9 gene on the cell surface LDL receptors. Hum Mol Genet. 2006;15:1551-1558
33. Lagace T.A, Curtis D.E, Garuti R, McNutt M.C, Park S.W, Prather H.B, Anderson N.N, Ho Y.K, Hammer R.E, Horton J.D. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006;116:2995-3005
34. Maxwell K.N, Fisher E.A, Breslow J.L. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci U S A. 2005;102:2069-2074
35. Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, Asselin M.C, Hamelin J, Varret M, Allard D. et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. 2004;279:48865-48875
36. Salek L, Lutucuta S, Ballantyne C.M, Gotto Jr A.M, Marian A.J. Effects of SREBF-1a and SCAP polymorphisms on plasma levels of lipids, severity, progression and regression of coronary atherosclerosis and response to therapy with fluvastatin. J Mol Med. 2002;80:737-744
37. Fiegenbaum M, Silveira F.R, Van der Sand C.R, Van der Sand L.C, Ferreira M.E, Pires R.C, Hutz M.H. Determinants of variable response to simvastatin treatment: the role of common variants of SCAP, SREBF-1a and SREBF-2 genes. Pharmacogenomics J. 2005;5:359-364
38. Matsuyama H, Sato K, Nakamura Y, Suzuki K, Akiba Y. Modulation of regulatory factors involved in cholesterol metabolism in response to feeding of pravastatin- or cholesterol-supplemented diet in chickens. Biochim Biophys Acta. 2005;1734:136-142
39. Scharnagl H, Schinker R, Gierens H, Nauck M, Wieland H, Marz W. Effect of atorvastatin, simvastatin, and lovastatin on the metabolism of cholesterol and triacylglycerides in HepG2 cells. Biochem Pharmacol. 2001;62:1545-1555
40. Mascaro C, Ortiz J.A, Ramos M.M, Haro D, Hegardt F.G. Sterol regulatory element binding protein-mediated effect of fluvastatin on cytosolic 3-hydroxy-3-methylglutaryl-coenzyme A synthase transcription. Arch Biochem Biophys. 2000;374:286-292
41. Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah N.G, Bernier L, Prat A. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24:1454-1459
42. Dong B, Wu M, Li H, Kraemer F.B, Adeli K, Seidah N.G, Park S.W, Liu J. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J Lipid Res. 2010;51:1486-1495
43. Welder G, Zineh I, Pacanowski M.A, Troutt J.S, Cao G, Konrad R.J. High-dose atorvastatin causes a rapid sustained increase in human serum PCSK9 and disrupts its correlation with LDL cholesterol. J Lipid Res. 2010;51:2714-2721
44. Dubuc G, Tremblay M, Pare G, Jacques H, Hamelin J, Benjannet S, Boulet L, Genest J, Bernier L, Seidah N.G. et al. A new method for measurement of total plasma PCSK9: clinical applications. J Lipid Res. 2010;51:140-149
45. Ding S.Y, Tigno X.T, Hansen B.C. Nuclear magnetic resonance-determined lipoprotein abnormalities in nonhuman primates with the metabolic syndrome and type 2 diabetes mellitus. Metabolism. 2007;56:838-846
46. Dong B, Wu M, Cao A, Li H, Liu J. Suppression of Idol expression is an additional mechanism underlying statin-induced up-regulation of hepatic LDL receptor expression. Int J Mol Med. 2011;27:103-110
Author contact
![]() Corresponding author: Liwen Zhang, PhD, and Ayesha Sitlani, PhD, Merck and Co., 126 E. Lincoln Avenue, Rahway, NJ 07065. E-mail: liwennet; ayesha_sitlanicom.
Corresponding author: Liwen Zhang, PhD, and Ayesha Sitlani, PhD, Merck and Co., 126 E. Lincoln Avenue, Rahway, NJ 07065. E-mail: liwennet; ayesha_sitlanicom.
Received 2011-9-16
Accepted 2011-12-23
Published 2012-2-9