Impact Factor ISSN: 1449-2288
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Pathogenesis
Risk genetic loci
Epigenetics and T1DM
Environmental factors
Other related genes
Treatment and prevention trials
Challenges
Future directions for treatment...
Abbreviations
Acknowledgements
References
Author biography

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(7):666-679. doi:10.7150/ijbs.6610 This issue Cite
Research Paper
Risk Factors and Primary Prevention Trials for Type 1 Diabetes
Yan-Ling Wu1 ![]() , Yan-Ping Ding1, Jian Gao1, Yoshimasa Tanaka2, Wen Zhang3
, Yan-Ping Ding1, Jian Gao1, Yoshimasa Tanaka2, Wen Zhang3 ![]()
1. Virus Inspection Department, Zhejiang Provincial Center for Disease Control and Prevention, 630 Xincheng Road, Hangzhou, 310051, PR China.
2. Center for Innovation in Immunoregulative Technology and Therapeutics, Graduate School of Medicine, Kyoto University, Kyoto, 606-8501, Japan.
3. Department of Biopharmaceutical Sciences, College of Pharmaceutical Sciences, Zhejiang University of Technology, 18 Chaowang Road, Hangzhou, 310014, PR China.
Received 2013-5-4; Accepted 2013-7-9; Published 2013-7-18
Abstract
Type 1 diabetes mellitus (T1DM) is a chronic autoimmune disease resulting in the designated immune destruction of insulin producing β-cells, usually diagnosed in youth, and associated with important psychological, familial, and social disorders. Once diagnosed, patients need lifelong insulin treatment and will experience multiple disease-associated complications. There is no cure for T1DM currently. The last decade has witnessed great progress in elucidating the causes and treatment of the disease based on numerous researches both in rodent models of spontaneous diabetes and in humans. This article summarises our current understanding of the pathogenesis of T1DM, the roles of the immune system, genes, environment and other factors in the continuing and rapid increase in T1DM incidence at younger ages in humans. In addition, we discuss the strategies for primary and secondary prevention trials of T1DM. The purpose of this review is to provide an overview of this disorder's pathogenesis, risk factors that cause the disease, as well as to bring forward an ideal approach to prevent and cure the disorder.
Keywords: Type 1 diabetes, pathogenesis, susceptibility gene, epigenetics, environmental factors, c-kit, PD-1, prevention trials.
Introduction
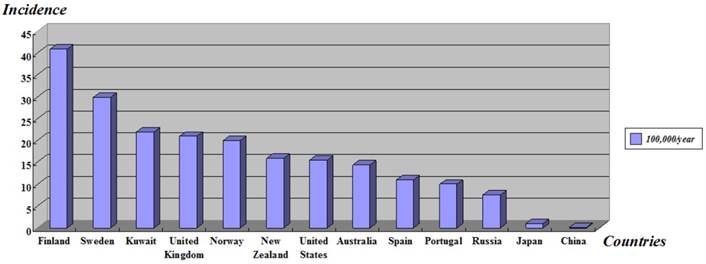
Type 1 diabetes mellitus (T1DM) or insulin-dependent diabetes mellitus (IDDM) is a lifelong metabolic disorder which is caused by insulin deficiency with secondary autoimmune destruction of the insulin producing pancreatic β-cells [1], usually diagnosed among children, adolescents and young adult people, and bounded up with important psychological, familial and social disorders. More than 346 million people worldwide have diabetes [2] and the number of new cases of T1DM is increasing at rate of approximately 3% per year in different countries around the world, particularly among younger children [3, 4]. Epidemiological investigations show the geographical differences of T1DM incidence, the age-adjusted incidence of Type 1 diabetes (T1D) varied from 0.1/100,000 per year in China to 40.9/100,000 per year in Finland [5] (Figure 1). This organ-specific destruction is mediated by T helper-1 (Th1) lymphocyte and develops owing to the interaction between susceptibility genes, environmental factors [6] and some other risk factors.
Pathogenesis
Type 1 diabetes is characterized by a lack of insulin production caused by a cellular-mediated autoimmune destruction of pancreatic islet β-cells, the only cells in the body for a crucial hormone insulin production. The β-cells function as a glucose sensor, playing vital roles in detecting glucose and releasing insulin to maintain physiologic glucose levels within a relatively narrow range. They thus make up much more than just an insulin factory. Once these cells are destroyed, blood-glucose control is lost, resulting in acute conditions such as ketoacidosis [7] and secondary complications (e.g. heart disease, blindness and kidney failure). The autoimmune destruction is irreversible, and the disease is incurable.
Biomarkers of the immune destruction of the β-cells include autoantibodies to islet cells, glutamic acid decarboxylase (GADA), insulin (IAA), the tyrosine phosphatases IA-2 and IA-2β, and zinc transporter 8 (ZnT8A). 85-90% of the newly diagnosed T1D patients are positive for one or more of these autoantibodies, but the proportion depends on patient's age, the number and quality of the assays used, and ethnicity. A small number of T1D patients may be negative for all islet autoantibodies at diagnosis, despite presence before or after diagnosis [8].
In this form of diabetes, the rate of β-cell destruction is rapid in infants and children, and slow in adolescents and adults with lower-risk human leukocyte antigen (HLA) genotypes. Some patients may exhibit ketoacidosis as the first manifestation of the disease. Others show modest hyperglycemia that can rapidly decompensate when faced with infection or other stress. A portion of adult patients may exhibit residual β-cell functions sufficient to prevent ketoacidosis for several years, but eventually need insulin for survival and are at risk for ketoacidosis. No or little insulin is produced at the later stage of the disease, as manifested by low or undetectable levels of plasma C-peptide.
Multiple genes within the major histocompatibility complex (MHC) were recognized over the last two decades as the dominant loci associated with disease in both the no obese diabetic (NOD) model and human disease. MHC is grouped into the class I, II and III regions, and each contains groups of genes with specific functions. The MHC class I and MHC class II genes encode human leukocyte antigens (HLAs), which are proteins that exist on the cell surface and define the tissue type of individuals and play a significant role in the antigen presentation. Proteins inside the cell can function as peptide antigens by MHC proteins when they are broken into short fragments. This will contributes to the immune system to distinguish normal (self) antigens from those that are foreign and potentially dangerous. MHC class III genes encode some components of the complement system, a collection of soluble proteins exist in the blood that target foreign cells and break open their membranes. In both humans and NOD mice, T1DM arises as a complex polygenic trait, and the strongest genetic link with disease susceptibility is certain major MHC class II alleles [9]. NOD mice express only a single unique MHC class II molecule [10] designated I-Ag g7, which is the primary gene conferring diabetes susceptibility.
Age-standardized incidence of type 1 diabetes in children under 14-year-old worldwide. Adapted from The DIAMOND Project Group (2006) [5].
Risk genetic loci
There are at least 20 regions of the genome provisionally assigned as the factors predisposing to T1DM. The genes at the HLA locus on human chromosome 6 play a critical role in helping the immune system to discriminate between 'self' (such as the beta cells of the pancreas) and non-self (such as bacteria and viruses). Inheriting certain HLA alleles increases the probability that immune cells will attack the body's own beta cells, thereby predisposing to type 1 diabetes. Within the HLA region, the genes are divided into three classes: Class I genes (HLA-A, HLA-B and HLA-C), which are located on the surface of all nucleated cells [11], encoding class I HLA antigens; Class II genes (HLA-DR, HLA-DQ and HLA-DP), which can produce class II HLA antigens that are found exclusively on macrophages, B-lymphocytes, activated T-lymphocytes, and epithelial cells of the islets of Langerhans; Class III genes, code for complement components (C2, properdin factor B, C4A and C4B), and products involved in T-cell-mediated inflammation, such as TNF-α, TNF-β, and acute phase protein [12]. The major susceptibility for T1D has been mapped to the HLA class II genes HLA-DQB1, -DQA1 and -DRB1 [13]. Both susceptible and protective DR-DQ haplotypes exist in all populations. In the early 1970s, several groups discovered that there is a relationship between HLA class I and T1D. Later, it was found that lymphocyte-defined HLA-D antigens, HLA class II DR3 (HLA-DRB1*0301, DQB1*0201) and DR4 (HLA-DRB1*04, DQB1*0302) were much more closely associated with T1D [14], accounting for approximately 40 % of the genetic risk for T1DM development, and the DR3/DR4 combination, two susceptible alleles, could produce a higher-risk genetic combination [15, 16]. Children aged under 5-year-old with a family history of T1DM, carrying the highest risk HLA class II genotypes, and persistently positive for two or more autoantibody types, have a considerably high risk of being diagnosed with the disease, for whose lifetime risk is more than 90% [17]. Recently, novel statistical methods have been applied to genetic association data from the HLA region in T1D, and this has made it possible to identify effects of other genes independently of the effects at the classical HLA-DR, -DQ risk loci. These include HLA-B and HLA-A, located in the telomere of the classical loci, and loci within the HLA class III region [18]. Besides HLA, the insulin gene (IDDM2) on chromosome 11 [19], the CTLA4 gene at the IDDM12 susceptibility locus [20], PTPN22 lyp [21] and other susceptibility loci are also strongly associated with the onset of the disease (listed in Table 1).
Susceptibility loci for type 1 diabetes.
| Locus | Chromosome | λs [26] | LOD | Candidate genes | Ref. |
|---|---|---|---|---|---|
| IDDM1 (HLA) | 6p21.3 | 1.7-4.2 | 65.8 | HLADR/DQ | [22] |
| IDDM2 (INS) | 11p15.5 | 1.6 | 4.28 | INSULIN (INS) VNTR | [22] |
| IDDM3 | 15q26 | [23] | |||
| IDDM4 | 11q13 | 1.0-1.5 | 2.7 | LRP5, FADD | [24] |
| IDDM5 | 6q25 | 4.5 | MnSOD, SUMO4 | [25] | |
| IDDM6 | 18q12-q21 | 1.0-1.5 | 1.1 | JK(Kidd), ZNF236, BCL2 | [26] |
| IDDM7 | 2q31-33 | 1.0-1.6 | 1.2 | NEUROD | [27] |
| IDDM8 | 6q25-27 | 1.0-2.1 | 3.6 | [25] | |
| IDDM9 | 3q22-q25 | 1.0-1.7 | 3.4 | [28] | |
| IDDM10 | 10p11-q11 | 1.1-2.2 | 2.8 | GAD2 | [22] |
| IDDM11 | 14q24.3-q31 | 4.0 | ENSA, SEL-1L | [29] | |
| IDDM12 (CTLA-4) | 2q31-q33 | 3.57 | CTLA-4, CD28 | [30], [22] | |
| IDDM13 | 2q34-q35 | [31] | |||
| IDDM15 | 6q21 | 2.36 | [22] | ||
| IDDM16 (IGH) | 14q32 | [32] | |||
| IDDM17 | 10q25 | 2.38 | [33] | ||
| IDDM18 (IL-12p40) | 1q42 | 2.2 | [22] | ||
| 5q31.1-33.1 | IL12B | [34] | |||
| 7q25 | 1.81 | [22] | |||
| 8q22-q24 | 2.4 | [35] | |||
| 16q22-q24 | 1.6 | 3.93 | [22] | ||
| PTPN22 | 1p13 | PTPN22 (LYP) | [36] | ||
| SUMO4 | 6q25 | SUMO4 | [11] |
Notes: IGH-Immunoglobulin heavy chain; p-the long arm of chromosome; q-the short arm of chromosome; LOD-logarithm of the odds: the LOD score has been used as a measure of the statistical evidence for linkage between a marker and a gene; λs values reflect sibling risk of a disease in relation to its population prevalence.
IDDM1. The HLA class II gene, designated as IDDM1, a susceptibility gene in the HLA region of chromosome 6 (6p21.3), accounts for at least 40% of the familial aggregation of this disease [37]. When evaluated as haplotypes, DQA1*0501-DQB1*0201 and DQA1*0301-DQB1*0302 are most tightly associated with T1D in Caucasian populations, in linkage disequilibrium with DRB1*03 and DRB1*04, respectively. Specific DRB1*04 alleles also influence the risk associated with the DQA1*0301-DQB1*0302 haplotype. Other high risk haplotypes for T1DM are also reported, including DRB1*07-DQA1*0301-DQB1*0201 among African Americans, DRB1*09-DQA1*0301-DQB1*0303 among Japanese, and DRB1*04-DQA1*0401-DQB1*0302 among Chinese. DRB1*15-DQA1*0602-DQB1*0102 is a protective haplotype to reduce T1D risk in most populations.
Individuals with only one susceptibility haplotype have an increased but modest T1DM risk, whereas others with two high risk DRB1-DQA1-DQB1 haplotypes show a significantly higher T1D risk than those with one or no high risk haplotype. The estimate of relative risk ranges from 10-45 and 3-7, respectively, for these groups, relying on race or ethnicity [15]. In terms of absolute risk, 6% of Caucasian with two susceptibility haplotypes will develop T1DM by 35 years of age. In fact, this figure is much lower in populations where T1D is rare, such as < 1% among Asians.
Insulin gene (INS). The insulin gene is located in the insulin-linked polymorphic region (ILPR, also known as IDDM2) on chromosome 11p15.5. A variable number tandem repeat (VNTR) region consisting of a 14 to 15 bp consensus sequence upstream of the INS gene, in the INS promoter, is comprised of three classes of alleles: there is a higher frequency of class I alleles (26-63 repeats) with shorter repeat sequences in individuals with T1DM while individuals with longer class III alleles (141-209 repeats) are relatively protected from T1DM [38, 39]. The biological plausibility of these associations may be due to the insulin mRNA expression in the thymus. Compared to class I variants, class III variants can generate higher levels of insulin mRNA, contributing to the establishment of a state of immune tolerance with the increased negative selection of autoreactive T-cell clones. The effect of insulin gene varies among different ethnicity groups, with lesser effects in non-Caucasian populations [40].
CTLA-4 (cytotoxic T lymphocyte antigen 4). The CTLA-4 gene is located on chromosome 2q31-q33 [41], in which multiple T1D genes are located. Evidence from combined linkage and association analyses indicates that CTLA-4 gene and T1DM are linkage disequilibrium. It is demonstrated that the impaired activity is associated with a Thr17Ala variant; this may contribute to the increase of T1D risk. On the whole, the relative increase in the risk for the CTLA-4 Ala17 variant is estimated as 1-2.
PTPN22 lyp. Lymphoid protein tyrosine phosphatase (Lyp) encoded by the PTPN22 locus on chromosome 1p13.3-13.1 [30] has the relation to T1DM. Lyp, a suppressor of T-cell activation, is an∼105 kDa Class I protein tyrosine phosphatase (PTP) consisting of an N-terminal PTP domain and a long noncatalytic C terminus with proline-rich motifs [36]. The variants encoded by the two alleles, 1858C and 1858T, are different in a crucial amino acid residue which is involved in the association of LYP with the negative regulatory kinase Csk (C-terminal Src kinase). The variant associated with T1D does not bind Csk, and the PTPN22 allele 1858T has higher frequency in individuals with T1D than those in healthy individuals: 30.6% of individuals with T1D are heterozygous with respect to 1858C, whereas 21.3% are heterozygous in healthy controls, and 3.7% of the patients with T1D are homozygous, although only 1.0% are homozygous in healthy controls (χ2 = 14.84 with 2 degrees of freedom, P< 0.001) [42]. As the cohorts were matched for age and race, these results demonstrate that the PTPN22 allele 1858T predisposes individuals to the development of T1D.
Epigenetics and T1DM
Disease concordance rates of monozygotic twins range from 12.0% to 67.7% [43-45]. The low disease concordance rates observed in adult-onset T1DM (<20%) indicate that epigenetic changes may have a predominant effect on the onset of T1DM in adults, compared to young patients. It is thus necessary to look further into the status of DNA methylation and histone modifications caused by external factors in patients with T1DM, because these modifications are related to altered gene expressions [46].
DNA methylation in T1DM. Comprehensive DNA methylation profiling suggests that a total of 276 CpG loci can be affiliated with promoters of 254 genes, displaying significantly different DNA methylation patterns in diabetic islets [47]. Because increased body weight and insulin resistance may be closely related to T1D in adults, the epigenetic dysregulation such as DNA methylation is critically involved in the onset of the disease. Therefore, impaired β-cell functions can be driven by epigenetic changes in patients with less HLA genetic susceptibility such as those affected by LADA (latent autoimmune diabetes of the adult) [48].
Rakyan and his collegues [49] conducted a comparative study on the epigenome-wide association in CD14+ monocytes from T1D-discordant monozygotic twin pairs. They identified 132 different CpG sites significantly linked with diabetic condition and discovered that some of the genes were hypomethylated or hypermethylated (e.g. GAD2 and HLA-DQB1), which are known to be correlated with T1DM. In addition, T1D-associated methylation in variable positions arises early in the progression of the disease, as they are found in islet autoantibody positive individuals many years before clinical diagnosis. The difference in the methylation patterns between T1DM patients and non-diabetic controls was reported by Bougnères's group [50]. In addition, a 3-CpG-hypomethylation pattern that seemed to be present only in T1D patients was identified. As these three CpG sites are proximal to the transcription initiation site in the insulin promoter gene, they could be a marker for the prediction of T1DM. Furthermore, 19 CpG sites were linked to the onset time of a dominating T1DM complication of nephropathy, of which one CpG site was found to be hypermethylated. The UNC13B gene is shown to be associated with the risk of diabetic nephropathy [51]. Akirav et al. [52] demonstrated that hypomethylated insulin DNA could be detected in the blood of newly-diagnosed type 1 diabetic patients and this finding might lead to the development of an effective means for the detection of β-cell death.
Histone modification in T1DM. Gray and DeMeyts [53] demonstrated that histone modification was correlated with diabetes. Natarajan's group [54] performed ChIP-chip for the comparison of genome-wide histone H3K9me2 patterns in peripheral lymphocytes and monocytes between T1D patients and non-diabetes controls. In lymphocytes, they found a notable increase in H3K9me2 in some T1D high risk genes such as CTLA4 gene. Curcumin treatment, which provides protection against T1D nephropathy, increased acetylation of histone H3 [55]. Moreover, hyperglycemia affects histone methylation [56]. The upregulation of the NF-κB-p65 gene due to the histone methylation of the gene promoter region can be caused by prior hyperglycemia.
MicroRNA regulation in T1DM. Dotta's group [57] measured miR-326 expression levels in peripheral lymphocytes from T1D patients who possessed autoantibodies to glutamic acid decarboxylase and insulinoma antigen-2. A high level expression of miR-326 was observed among T1D patients with autoantibodies, compared to antibody-negative T1D patients. The miRNAs are reported to be related to β-cell death. Michalek's group [58] analyzed the miRNAs expression in regulatory T cells derived from T1D patients, and found that miR-342 and miR-191 were downregulated, while miR-510 upregulated. As reviewed by Fernandez-Valverde [59], a large quantity of other miRNAs may be involved in the T1D pathogenesis.
Environmental factors
Based on comparative studies to determine the effect of genetic and environmental factors on the onset of diabetes, 88% of phenotypic variances are ascribable to genetic factors, and the rest to unshared environmental factors. Environmental risk factors are thought to be 'initiators' or 'accelerators' of β-cell autoimmunity, or 'precipitators' of overt symptoms in individuals suffering from β-cell destruction. The factors may act directly on the pancreas, or provoke abnormal immune responses to proteins normally expressed in the cells.
Monozygous and dizygous twins. Twin studies suggest an important role of environmental risk factors in the development of T1DM. The concordance of T1DM development differs much more dramatically between monozygotic and dizygotic twins [60]. Although the probability of monozygotic twins who live under different environment to develop T1DM decreases with the duration of discordance, concordance can happen in twins more than 40 years after the development of diabetes in their twin sibling. In some degree, the risk for diabetes of a dizygotic twin is similar to the risk of a twin of a patient with diabetes (5%). Therefore, the development of diabetes is not substantially enhanced in dizygotic twins under the shared environment. Anti-islet autoantibodies are found more frequently in monozygotic twins, compared to dizygotic twins, and most of the monozygotic twins of T1DM patients expressing anti-islet autoantibodies progress to diabetes [61].
In most studies, anti-islet cell autoantibodies are frequently observed in nondiabetic monozygotic twin siblings of patients with T1DM, ranging from 42% to 76% [62, 63], which is in concordance with their high progression to diabetes. Radioassays show that autoantibodies are consistently expressed prior to the development of diabetes, and most monozygotic twin siblings with multiple autoantibodies develop diabetes in the long period. Studies indicate a low concordance rate for diabetes in dizygotic twins between 0% [63] and 13% [64], while, in monozygotic twins, the concordance rate ranges from 21% to 70% [63, 64]. Life table analysis and long term follow-up studies show the highest rate for the progression of diabetes in monozygotic twin siblings [62].
Viral infections. Viral infections have been implicated in the T1D etiology for more than 100 years. The epidemiological data show that some viruses such as enteroviruses, coxsackie virus B (CVB), mumps, rubella, cytomegalovirus, parvovirus, rotaviruses, and encephalomyocarditis virus might contribute to T1D pathogenesis [65, 66]. On the basis of seroepidemiological human studies, enteroviruses, in particular, may induce T1D [67, 68], and enteroviral infections occurring early in utero may increase a child's subsequent risk to develop the disease [69]. Coxsackie viruses, which contain a peptide homologous to glutamic acid decarboxylase 65 (GAD65), are often observed in childhood and are known to have effects on the pancreas.
Recently, Mycobacterium avium subsp. paratuberculosis (MAP), the etiological agent of paratuberculosis [70], has been proposed as a new environmental factor [71] that might play a role in the pathogenesis of T1D [72]. This pathogen is widely spread and can be detected in milk and dairy products derived from infected ruminants that are asymptomatic reservoir [73], owing to its ability to survive pasteurization and chlorination. The prevalence of MAP infection is high in T1D patients in Sardinia [74-77], one of the regions with the highest T1D incidence all over the world. As a matter of fact, MAP DNA was detected in 63% of Sardinian T1D patients, but 16% of healthy individuals [78]. Similarly, the MAP envelope protein MptD was detected in 47% Sardinian T1D patients, but only 13% in healthy individuals [72]. MAP protein, named MAP3865c, has a sequence homology with the β-cell antigen zinc transporter 8 (ZnT8) [79] targeted by Abs in T1D patients [80].
Two possible mechanisms may be involved in the virus infection-mediated development of T1D: one is via a direct cytolytic effect, and the other through triggering autoimmune responses gradually leading to β-cell destruction. In addition, the study of structural homology between viral structures and β-cell antigens suggests that molecular mimicry may play an essential role in diabetes-associated autoimmune responses. Furthermore, persistent or slow virus infections may also be essential for the development of autoimmunity.
Other related genes
PD-1. Programmed cell death 1 (PD-1), an immunoinhibitory receptor which belongs to the CD28/CTLA-4 family, is expressed on activated T cells. PD-1 inhibits T cell activation and provides negative costimulation with the recruitment of the protein tyrosine phosphatase SHP-2 (src homology 2 domain-containing tyrosine phosphatase 2), upon binding to its ligands, PD-L1 and PD-L2 [81-83]. Because PD-1 plays an important role in the regulation of peripheral tolerance, PD-1-deficiency may lead to various autoimmune diseases [84]. The onset and frequency of T1DM in NOD mice are specifically accelerated under the condition of PD-1 deficiency, with strong T helper 1 polarization of T cells infiltrating into islets, and this is more pronounced in male animals. The diabetic incidence of NOD-Pdcd1-/- mice was controlled by five genetic loci, including Idd (insulin-dependent diabetes) 1, Idd17, and Idd20, in which recessive loci are included. Ansari et al. [85] demonstrated that antibodies specific to PD-1 or PD-L1, but not PD-L2, would contribute to the acceleration of insulitis and subsequent development of diabetes in NOD mice. Based on these findings, PD-1/PD-L1 pathway plays a crucial role in the diabetic incidence in NOD mice.
Recently, Lillevang's group [86] showed for the first time that the A allele of PD-1 7146G/A SNP (single nucleotide polymorphism) had significant association with susceptibility to T1DM in Caucasians, which was confirmed in two separate populations of T1D patients from different regions in Denmark. Testing the pooled material further confirmed this finding.
PD-1 can induce immune tolerance to pancreatic islet cells in animal models. Roles of PD-1 in T1DM were examined with the use of PD-1 transgenic mice (Tg). Multiple low doses of streptozotocin (STZ) were injected into mice to achieve T cell-mediated destruction of β-cells [87]. Insulitis and hyperglycemia appeared in male mice 7 days after the treatment of low doses of STZ [88]. Although the development of autoimmune diabetes was not completely prevented by PD-1 transgene expression, the severity of the disease in PD-1 Tg mice was significantly reduced. On the contrary, PD-1 deficiency accelerated T1DM in NOD mice, demonstrating that PD-1 deficiency would accelerate the development of autoimmune responses [89]. Accumulating evidence demonstrates that PD-1 delays the incidence of diabetes and it may play an essential role in the induction of immune tolerance in the pancreas.
PD-Ls expressed on non-lymphoid organs can prevent tissue destruction through the suppression of effector functions of autoreactive lymphocytes. In NOD mice, PD-L1, but not PD-L2, is highly expressed on β-cells in pancreatic islets of patients with insulitis [90]. It is intriguing that the islets are surrounded by infiltrating lymphocytes which form a cluster but are rarely invaded. PD-L1 on β-cells might thus serve as a barrier to suppress the effector function of diabetogenic T cells. In NOD-Pdcd1 K/K mice, this barrier is missing and the islets are deeply invaded by lymphocytes in spite of augmented PD-L1 expression on β-cells. As a consequence, NOD-Pdcd1 K/K mice develop T1DM much faster than PD-1-sufficient NOD mice, with the islets being extensively destructed [91]. As T cells are much more activated in the islets than in draining lymph nodes, PD-1/PD-L1 interaction can also inhibit the in situ activation of T cells. Blockade of the PD-1-PD-L pathway by antibodies in prediabetic NOD mice induces T1DM within 10 days [92]. Taken together, the PD-1/PD-L pathway plays a pivotal role in the maintenance of peripheral tolerance at the frontline of the immune response.
c-kit. c-kit, a receptor tyrosine kinase, and its ligand, stem cell factor, dominate various cellular events, such as pancreatic β-cell survival and differentiation as revealed in c-kit Wv mice. The c-kit Wv mice, which have a point mutation in the c-kit allele, resulting in the loss of function of this kinase, develop diabetes. The hematopoietic stem cell marker c-kit plays quite important roles in the development and function of islets of Langerhans, particularly in β-cell proliferation, maturation, and survival [93].
Li et al. [94] demonstrated that c-kit was expressed during the development of human fetal pancreas in early and mid-gestation in a dynamic, temporally-regulated fashion. Their findings are consisting with previous investigations [95-98] showing that c-kit is a marker for β-cell progenitors. In addition, they have also shown that pancreatic duodenal homeobox-1 (PDX-1) and insulin expression at both mRNA and protein levels increased or reduced by the enhancement or downregulation of c-kit receptor tyrosine kinase activity in separated human fetal islet-epithelial cell clusters. This indicates that the c-kit receptor tyrosine kinase has crucial effects on the modulation in various aspects of islet biology during the development of human fetal pancreas. On the basis of this result, c-kit is considered as a marker for β-cell progenitors in humans. It is essential to identify such factors to establish new islet cell-based therapies for β-cell destruction in insulin-dependent diabetes.
Feng et al. [99] examined whether c-kit overexpression could prevent β-cell defects in c-kit Wv mice. The c-kitβTg Wv mice not only showed normal fasting glycaemia and glucose tolerance, but also enhanced glucose-induced insulin secretion. They also demonstrated that c-kit overexpression in β-cells could improve β-cell proliferation and function, and protect mice from developing HFD-induced diabetes. Moreover, the c-kit overexpression on specific β-cells had the ability to prevent β-cell dysfunction in c-kitWv mice. Thus, c-kit plays a primary physiological role in β-cells, and may be a target for the development of gene and cell therapeutic schemes for diabetes patients.
Treatment and prevention trials
Today, diabetic patients can have a near-normal life relying on the administration of exogenous insulin by daily injections, continual pump therapy, or islet transplantions and adhering to various self-care tasks. Through these methods, diabetics can optimize the glycemic control and decrease the incidence of diabetes complications, such as cardiovascular disease, kidney failure, neuropathy, and retinopathy. However, currently available therapies fail to quell the risks for long-term hypoglycemia and microvascular damage and the treatments are quite costly [100].
In order to optimize the treatment for T1DM, large multi-national investigations have been designed and conducted to evaluate primary and secondary prevention trials [101].
Primary prevention trials. Primary prevention is treatment in infants with increased genetic risk. The primary prevention studies include several dietary manipulations, such as infant formulas free of either cow's milk or bovine insulin, delayed exposure of gluten-containing foods, and vitamin D supplementation. Because primary prevention is directed at individuals who have no clinical signs of autoimmune diseases or metabolic impairment, and because it is uncertain whether they will develop T1DM, the designed interventions must be effective, safe, and free of side effects. To date, all primary prevention trials have been dietary interventions designed to interrupt putative environmental factors of T1DM. So far, none of the specific dietary factors has been shown to be an unequivocal risk factor for β-cell-targeted autoimmune diseases or T1DM, and their effects have still been contradictory [102]. It is thus difficult to evaluate the efficacy of the primary interventions.
Dietary interventions. Cow's milk: Epidemiological studies and meta-analyses indicated that early introduction of cow's milk may be a risk factor for the development of T1DM [103, 104]. In 1995, a pilot study was initiated to evaluate in babies, who have high genetic risk markers of T1DM, about whether weaning replacement with a formula based on casein hydrolysate instead of cow's milk might decrease the development of T1DM [105]. Infants were enrolled in the study when they had HLA-conferred susceptibility to T1DM and one or more family members develop T1DM. In the trial, 230 infants were randomly assigned to receive either a casein hydrolysate formula or a conventional cow's-milk formula whenever breast milk was not sufficient during the first 6-8 months of life. Children were followed up for 10 years to monitor diabetes-related autoantibodies and T1DM. The study suggested that the risk of the development of autoimmune responses to β-cells was reduced among the group with casein hydrolysate formula (appearance of at least one autoantibody) [106].
Bovine insulin: Vaarala and his colleagues demonstrated that cow's-milk formula containing bovine insulin induced autoimmune responses to insulin [107]. Then, a pilot study named the Finnish Dietary Intervention Trial for the Prevention of T1DM [FINDIA] was initiated, whose purpose was to confirm whether a formula without bovine insulin might reduce diabetes. Compared to the cow's-milk formula group, the FINDIA formula group showed a reduced risk of the development of autoimmune responses to β-cells (appearance of one or more antibodies) [108]. In the meantime, it might be prudent to encourage breastfeeding for as long as possible whatever the outcome of the study.
Gluten: It has been suggested by prospective observational studies that the age for the introduction of solid food, for example, gluten-containing foods or cereals, would have an effect on the development of anti-islet autoimmune responses in children who are genetically susceptible to T1DM [109]. A pilot study showed that the β-cell function of islet autoantibody-positive children can be improved by gluten deprivation for 6 months [110]. Therefore, it might be safe to delay the exposure of gluten till 12-month of age. It is, however, likely that the delay of the introduction of solid food fails to achieve substantial reduction in the risk of anti-islet autoimmune responses in genetically susceptible children.
Vitamin D: It has been shown that vitamin D plays a role in the prevention of insulitis and T1DM in mouse disease models and vitamin D supplementation in early childhood may reduce the risk of the development of T1DM. Retrospective studies also suggest that regular vitamin D supplementation in early life shows benefits in reducing the risk of T1DM in the later lifetime [111]. However, the prospective Diabetes Autoimmunity Study in the Young (DAISY) had revealed that the intake of vitamin D during childhood was not correlated with the risk of anti-islet autoimmune responses or T1DM [112]. More recently, a meta-analysis of data indicated that the risk of T1DM was strikingly decreased by 29% in infants supplemented with vitamin D, compared to those who were not supplemented [113]. Controlled studies with vitamin D performed in new-onset T1DM have shown mixed outcomes, with one showing benefits [114] and two others not [115, 116]. A nationwide study has been proposed in Canada to confirm the hypothesis that vitamin D supplementation can reduce the risk of anti-islet autoimmune responses and the development of T1DM.
Secondary prevention trials. Secondary prevention is targeted at individuals with persistent islet autoantibodies. Ongoing trials involve the use of nicotinamide or antigen-specific therapies, including parenteral insulin, oral and nasal insulin or the intradermal administration of proinsulin peptides, and a vaccine with Glutamic acid decarboxylase (GAD).
Nicotinamide: Nicotinamide, a water-soluble vitamin (B6) isolated from nicotinic acid, has been shown to increase insulin synthesis and inhibit the development of diabetes if administered prior to the onset of the disease. Early in 1947, nicotinamide was found to be effective to prevent the development of diabetes in alloxan-treated rats. Subsequently, it was indicated that the compound was effective in the prevention of streptozotocin-induced diabetes and in the spontaneous development of diabetes in the NOD mouse [117].
Furthermore, The European Nicotinamide Diabetes Intervention Trial (ENDIT) [118] evaluated the effects of nicotinamide in at-risk relatives of individuals with Type 1 diabetes. ENDIT recruited islet cell antibody (ICA)-positive individuals aged 5-40 years old with T1DM for less than 20 years. The study randomized 552 participants either to nicotinamide (1.2 g m-2 day-1) or placebo groups. 35 000 first-degree relatives were screened to identify eligible subjects. After following up for about 4 years, it was shown that the rates of T1DM development in nicotinamide and placebo groups were essentially the same [119]. Nicotinamide thus had no effect on the prevention or delay of T1DM development in at-risk relatives.
Antigen-specific therapy: Antigen-specific therapy, a kind of immunotherapy to prevent T1DM [120], is based on the concept that the appropriate administration of a diabetes autoantigen has a potential to control the autoimmune responses by diverting the immune system to a protective rather than destructive response, and potentially to induce or restore tolerance. Antigens used for the treatment are safe, as they are specific for T1DM and are not expected to change generalized immune responses. Mucosal administration of autoantigens, such as oral or intranasal immunization, was expected to yield protective immunity, and thus has been the route used in some studies.
Because insulin is a β-cell-specific antigen, multiple approaches have been conducted for the interventions using insulin. It is quite advantageous to employ the insulin therapy in individuals with anti-islet autoimmune responses [121]. Firstly, the β-cell load will be reduced in the state of subclinical T1DM. Secondly, immunological tolerance is expected to be induced. In fact, delayed disease progression was observed in pilot studies of parenteral insulin (subcutaneous or intravenous administration) as prophylaxis among first-degree relatives of T1DM patients with anti-islet cell autoantibodies [122].
Parenteral insulin: In the Diabetes Prevention Trial Type 1 (DPT-1) trial, more than 80,000 first-degree relatives of T1DM patients were screened for anti-islet cell autoantibodies [123]. The intervention included low-dose subcutaneous ultralente insulin twice every day with a total dose of 0.25 units per kg body weight per day. The result failed to demonstrate the delay or prevention in T1DM. As only one dose of insulin was tested and the subjects already showed reduced β-cell function in randomization, it was impossible to evaluate the effect of insulin in the protection of the β-cells and the induction of immunomodulation.
Oral insulin: DPT-1 subjects' positive for anti-islet cell autoantibodies and anti-insulin autoantibodies without impaired glucose tolerance were randomly allocated to receive oral insulin 7.5 mg per day or placebo [124]. The original study demonstrated that there was no delay in the clinical onset of T1DM. A post hoc analysis indicated that a considerable delay in the clinical onset of T1DM was achieved in a subgroup of individuals with high-titer anti-insulin autoantibodies. A 13-year follow-up also revealed that the β-cell function was preserved for so long as the oral insulin was taken [125]. Currently, TrialNet, an international network seeking ways for the prevention, delay or reverse of T1DM progression, is recruiting subjects in an attempt to test whether oral insulin has effect on the prevention of T1DM in individuals with T1DM relatives.
Nasal insulin: Nasal insulin has also been tested for the induction of immune tolerance. In the Intranasal Insulin Trial (INIT), in phase I and II stages, a double-blind, crossover design was used to examine Australian individuals with anti-insulin autoantibodies and first-degree relatives with T1DM. INIT-I showed that there were no significant effects on β-cell function, but the immune tolerance to insulin was improved [126]. INIT-II is an ongoing randomized, placebo-controlled trial with nasal insulin at either 1.6 mg or 16 mg, whose purpose is to evaluate whether nasal insulin is effective on anti-islet autoimmune responses. The Diabetes Prediction and Prevention (DIPP) trial in Finland was a double-blind trial using nasal insulin in children with genetic risk of T1DM who were positive for islet cells and anti-insulin autoantibodies. The trial showed that the nasal insulin had no effect on the protection of the disease [127] and the modulation of the anti-insulin autoantibodies, indicating that the anti-insulin autoimmunity was already mature at the start of the intervention [128]. The ancillary or mechanistic studies, however, showed signs of immune tolerance to insulin after administration of nasal insulin, and the INIT and DIPP trials demonstrated the safety of nasal insulin. Future studies should include broader dose-response analyses to determine the association between the immune responses to autoantigens and the HLA-DQ genotype of the individuals, because the analysis of insulin alone might not be sufficient to obtain conclusive results.
Proinsulin peptide(s): The intradermal administration or a cocktail of proinsulin peptides is an alternative antigen-based therapy which could be used for the prevention of T1DM. A pilot safety study with a single proinsulin peptide administration has been performed in individuals with established T1DM [129]. The peptides chosen were those with epitopes which were recognized by HLA-DR4. Recently, several trials with various proinsulin peptides are underway.
Glutamic acid decarboxylase: Glutamic acid decarboxylase (GAD) is another antigen used for the antigen-specific therapy. Whether a vaccine using GAD combined with an aluminum hydroxide (alum) adjuvant can promote the prevention of T1DM is being assessed by the Diabetes Prevention-Immune Tolerance (DIA-PREV-IT) Study in Southern Sweden. This 50-subject double-masked randomized controlled clinical study is fully enrolled. Eligible children are at least 4 years old, have positive anti-GAD antibodies and one or more additional autoantibodies, and have not yet developed T1DM [119].
Immunomodulation. A French pilot trial was conducted focusing on whether immunosuppression with low-dose cyclosporine in first-degree relatives of T1DM patients with anti-islet cell autoantibodies reduced first-phase insulin responses and impaired glucose tolerance [130]. Cyclosporine was given at an initial dose of 7.5 mg kg-1day-1 and tapered the first year later. The subjects in the study included six cyclosporine-treated individuals and nine historical controls. Results showed that all of the controls developed T1DM within 12 months, while two of the cyclosporine-treated individuals did not develop T1DM until 47 and 57 months after the initiation of cyclosporine therapy. This study indicated that immunomodulation may delay the development of T1DM.
Based on the current concept of the immunopathogenesis of the disease, it seems to be possible to delay or prevent T1DM [131]. There has not been, however, overt evidence of clinical benefits from any intervention tested for either primary or secondary prevention studies so far. For primary prevention studies, infant formula without cow's milk proteins in the Finish TRIGR pilot study and infant formula without bovine insulin in the FINDIA study appeared to contribute to the reduction of anti-islet autoantibodies. For secondary prevention studies, a subgroup identified by post-hoc analyses of the DPT-1 oral insulin trial [124, 132] provided the only evidence of the delay of the T1DM onset.
Challenges
The most critical step is to identify the process that initiates the immune system to attack self-pancreatic β-cells. Without this knowledge, it is difficult to design a novel strategy for an effective prevention or an ultimate cure. Even though we have the ability of producing β-cells ex vivo with the help of regenerative medicine or transplanting a large number of islets, the autoimmune responses may obliterate the transplanted β-cells.
It is also necessary to design a safe treatment because the current intervention may elicit both acute and long-term adverse effects. Recently, JDRF Continuous Glucose Monitoring Study Group [133] demonstrated that patients, especially children, are prone to develop commonly prolonged nocturnal hypoglycemia and that the alternative route for insulin administration may reduce its frequency. Improved algorithms and mathematical models combined with glucose sensors and insulin pump technology would provide a reasonable opportunity to improve life quality and decrease the incidence of complications. However, the problem of non-physiologically high insulin levels in the periphery rather than in the portal vein is not solved even if a well-functioning artificial pancreas appears.
Future directions for treatment of T1DM
For primary prevention of T1DM, it is prerequisite to elucidate the mechanism underlying the development of anti-islet autoantibodies in infancy, in which the studies should be focused on individuals who have high-risk genetic markers without manifestation of T1DM. Although most of the studies so far conducted are dietary manipulation or supplementation, conclusive evidence has not been obtained. It is thus important to perform further studies for the identification of the effective ingredients for the primary prevention of T1DM. In addition, it should be clarified which vaccine is the most effective for the primary prevention-either antigen-specific vaccines, or vaccines against putative viral or other infectious factors.
Although a variety of interventions can be designed for the primary and secondary prevention of T1DM, most of them raise concerns about patient safety. We have to seek an appropriate balance among safety, potential efficacy, and the impact of T1DM. It is likely that antigen-specific vaccines or vaccines against putative infectious agents can be used for primary prevention. For secondary prevention, immunomodulatory agents may be used for the reduction of the projected risk of T1DM.
At present, the monotherapy for T1DM is not satisfactory as a means for a cure or prevention of the disease: on one hand, the therapy fails to achieve immune tolerance and preservation of C-peptide production; on the other hand, patients will eventually lose the ability to produce endogenous insulin. It is thus necessary to employ combination therapies or to explore new treatment strategies, because various pathways and arms of the immune system can be targeted [134]. Recently, a research group in Brazil conducted autologous nonmyeloablative hematopoietic stem cell transplantation to T1D patients [135], and the results showed that C-peptide levels (an indirect measure of viable beta-cell function) increased significantly and the majority of patients achieved insulin independence with good glycemic control. Other alternatives include the combined use of immune suppressive agents such as anti-CD3 monoclonal antibody and an anti-inflammatory agent, and antigen specific agents such as GAD-Alum and BHT DNA proinsulin vaccine. The potential mechanism underlying the tolerance induction might be the development of Tregs, leading to an antigen specific therapy with immune suppression or diminished inflammation.
In addition, targeted and specific immune therapies should be considered for the prevention and ultimate cure of T1D. The trimolecular complexes comprised of class II major histocompatability (MHC II) molecules, peptides of autoantigens, and specific T cell receptors (TCR) are crucial for the tissue specific targeting therapy for autoimmune diseases. So far, the trimolecular complex for insulin has been well characterized in NOD mice [136]. MHC class II molecules serve as a genetic determinant for T1DM [137, 138]. About 90% of T1D patients have HLA-DQ8 or DQ2 alleles. Insulin has been considered as a T1D autoantigen for decades since the discovery of autoantibodies to this molecule in T1D patients [139]. The autoantibodies specific to insulin are often detected prior to the appearance of anti-islet autoantibodies [140]. Three dimensional structures of the anti-insulin trimolecular complex have been solved in the last decade. Cocrystals of an MHC class II molecule plus peptide have also been determined, including DQ8 with an insulin B chain peptide, 9 to 23 [141]. Recent studies suggest that the 9-23 amino acid region of the insulin B-chain is recognized in an unexpected, low affinity register [142]. With the understanding of the structural determinants for autoantigen recognition, novel therapies can be designed to specifically target insulin-MHC complexes and the TCRs that recognize them. Autoantigen presentation or TCR recognition of autoantigen-MHC complexes might be blocked by small molecules. Another strategy includes designing monoclonal antibodies specific to the autoantigen peptide-MHC complexes in a defined register recognized by autoreactive T cell receptors. These novel approaches based on structural information may provide a means to achieve a complete cure or prevention of T1DM.
Abbreviations
T1DM: type 1 diabetes mellitus; IDDM: insulin-dependent diabetes mellitus; GADA: glutamic acid decarboxylase autoantibody; IAA: insulin autoantibody; ZnT8A: zinc transporter 8 autoantibody; HLA: human leukocyte antigen; MHC: major histocompatibility complex; NOD: nonobese diabetic; ILPR: insulin-linked polymorphic region; VNTR: variable number of tandem repeats; CTLA-4: cytotoxic T lymphocyte antigen 4; Lyp: lymphoid protein tyrosine phosphatase; PTP: Protein tyrosine phosphatase; PTPN22: Protein tyrosine phosphatase non-receptor type 22 (lymphoid); Csk: C-terminal src kinase; IGH: immunoglobulin heavy chain; LOD: logarithm of the odds; LADA: latent autoimmune diabetes of the adult; GAD65: glutamic acid decarboxylase 65; PD-1: programmed cell death 1; SHP-2: src homology 2 domain-containing tyrosine phosphatase 2; Idd: insulin-dependent diabetes; SNP: single nucleotide polymorphism; STZ: streptozotocin; PDX-1: pancreatic duodenal homeobox-1; HFD: high-fat diet; DAISY: Diabetes Autoimmunity Study in the Young; GAD: glutamic acid decarboxylase; ENDIT: European Nicotinamide Diabetes Intervention Trial; ICA: islet cell antibody; DPT-1: Diabetes Prevention Trial Type 1; INIT: Intranasal Insulin Trial; DIPP: Diabetes Prediction and Prevention; DIA-PREV-IT: Diabetes Prevention-Immune Tolerance; TCR: T cell receptors; G-CSF: granulocyte-colony stimulating factor.
Acknowledgements
We gratefully acknowledge the financial support from Zhejiang Provincial Natural Science Foundation of China (LY12B02019), the Qianjiang Talents Program of Zhejiang Province (2009R10002), the Major Projects on Science and Technology of Zhejiang Province (2013C13G1360034) and the Program for Zhejiang Leading Team of Science and Technology Innovation (2011R50021).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464:1293-1300
2. Jönsson L, Hallström I, Lundqvist A. "The logic of care" - parents' perceptions of the educational process when a child is newly diagnosed with type 1 diabetes. BMC Pediatr. 2012;12:165
3. Rankin D, Cooke DD, Elliott J. et al. Supporting self-management after attending a structured education programme: a qualitative longitudinal investigation of type 1 diabetes patients' experiences and views. BMC Public Health. 2012;12:652
4. Alderson P, Sutcliffe K, Curtis K. Children as partners with adults in their medical care. Arch Dis Child. 2006;91:300-303
5. Liese AD, D'Agostino RB Jr, Hamman RF. et al. The burden of diabetes mellitus among US youth: Prevalence estimates from the SEARCH for diabetes in youth study. Pediatrics. 2006;118:1510-1518
6. Hawa MI, Beyan H, Buckley LR. et al. Impact of genetic and non-genetic factors in type 1diabetes. Am J Med Genet. 2002;115:8-17
7. Westerberg DP. Diabetic ketoacidosis: evaluation and treatment. Am Fam Physician. 2013;87(5):337-346
8. Barker JM, Barriga KJ, Yu L. et al. Diabetes Autoimmunity Study in the Young. Prediction of autoantibody positivity and progression to type 1 diabetes: Diabetes Autoimmunity Study in the Young (DAISY). J Clin Endocrinol Metab. 2004;89:3896-3902
9. Cucca F, Lampis R, Congia M. et al. A correlation between the relative predisposition of MHC class II alleles to type 1 diabetes and the structure of their proteins. Hum Mol Genet. 2001;10(19):2025-2037
10. Kudva YC, Rajagopalan G, Raju R. et al. Modulation of insulitis and type 1 diabetes by transgenic HLA-DR3 and DQ8 in NOD mice lacking endogenous MHC class II. Hum Immunol. 2002;63(11):987-999
11. Hanan FA, Ameer MM, Zaidan MA. Genetics of Type 1 Diabetes Mellitus. Kuwait Medical Journal. 2007;39(2):107-115
12. Genetic Aspects of Diabetes mellitus. Elizabeth T. http://www.ifcc.org/ejifcc/ejifcc/voll3no5/1305/2002/os
13. Pociot F, McDermott MF. Genetics of type 1diabetes mellitus. Genes Immun. 2002;3:235-249
14. Jones EY, Fugger L, Strominger JL. et al. MHC class II proteins and disease: a structural perspective. Nat Rev Immunol. 2006;6(4):271-282
15. Dorman JS, Bunker CH. HLA-DQ locus of the human leukocyte antigen complex and type 1 diabetes mellitus: a HuGE review. Epidemiol Rev. 2000;22:218-227
16. Noble JA, Valdes AM. Genetics of the HLA region in the prediction of type 1 diabetes. Curr Diab Rep. 2011;11(6):533-542
17. Bingley PJ, Gale EA. Progression to type 1 diabetes in islet cell antibody-positive relatives in the European Nicotinamide Diabetes Intervention Trial: the role of additional immune, genetic and metabolic markers of risk. Diabetologia. 2006;49:881-890
18. Valdes AM, Thomson G. Several loci in the HLA class III region are associated with T1D risk after adjusting for DRB1-DQB1. Diabetes Obes Metab. 2009;11(suppl 1):46-52
19. Awata T, Kawasaki E, Ikegami H. et al. Insulin gene/IDDM2 locus in Japanese type 1 diabetes: contribution of class I alleles and influence of class I subdivision in susceptibility to type 1 diabetes. J Clin Endocrinol Metab. 2007;92(5):1791-1795
20. Nisticò L, Buzzetti R, Pritchard LE. et al. The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian Diabetes Registry. Hum Mol Genet. 1996;5:1075-1080
21. Bottini N, Musumeci L, Alonso A. et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. 2004;36:337-338
22. Bottini N, Vang T, Cucca F. et al. Role of PTPN22 in type 1 diabetes and other autoimmune diseases. SeminImmunol. 2006;18:207-213
23. Zamani M, Pociot F, Raeymaekers P. et al. Linkage of type I diabetes to 15q26 (IDDM3) in the Danish population. Hum Genet. 1996;98(4):491-496
24. Nakagawa Y, Kawaguchi Y, Twells RC. et al. Fine mapping of the diabetes-susceptibility locus, IDDM4, on chromosome 11q13. Am J Hum Genet. 1998;63(2):547-556
25. Luo DF, Buzzetti R, Rotter JI. et al. Confirmation of three susceptibility genes to insulin-dependent diabetes mellitus: IDDM4, IDDM5 and IDDM8. Hum Mol Genet. 1996;5(5):693-698
26. Laine AP, Nejentsev S, Veijola R. et al. A linkage study of 12 IDDM susceptibility loci in the Finnish population. Diabetes Metab Res Rev. 2004;20(2):144-149
27. Kristiansen OP, Pociot F, Bennett EP. et al. IDDM7 links to insulin-dependent diabetes mellitus in Danish multiplex families but linkage is not explained by novel polymorphisms in the candidate gene GALNT3. Hum Mutat. 2000;15(3):295-296
28. Laine AP, Turpeinen H, Veijola R. et al. Evidence for linkage to and association with type 1 diabetes at the 3q21 region in the Finnish population. Genes Immun. 2006;7(1):69-72
29. Field LL, Tobias R, Thomson G. et al. Susceptibility to insulin-dependent diabetes mellitus maps to a locus (IDDM11) on human chromosome 14q24.3-q31. Genomics. 1996;33(1):1-8
30. Burn GL, Svensson L, Sanchez-Blanco C. et al. Why is PTPN22 a good candidate susceptibility gene for autoimmune disease? FEBS Lett. 2011;585:3689-3698
31. Larsen ZM, Kristiansen OP, Mato E. et al. IDDM12 (CTLA4) on 2q33 and IDDM13 on 2q34 in genetic susceptibility to type 1 diabetes (insulin-dependent). Autoimmunity. 1999;31(1):35-42
32. Field LL, Larsen Z, Pociot F. et al. Evidence for a locus (IDDM16) in the immunoglobulin heavy chain region on chromosome 14q32.3 producing susceptibility to type 1 diabetes. Genes Immun. 2002;3(6):338-344
33. Babu SR, Conant GC, Eller E. et al. A second-generation genome screen for linkage to type 1 diabetes in a Bedouin Arab family. Ann N Y Acad Sci. 2004;1037:157-160
34. Morahan G, Huang D, Ymer SI. et al. Linkage disequilibrium of a type 1 diabetes susceptibility locus with a regulatory IL12B allele. Nat Genet. 2001;27(2):218-221
35. Sale MM, FitzGerald LM, Charlesworth JC. et al. Evidence for a novel type 1 diabetes susceptibility locus on chromosome 8. Diabetes. 2002;51(suppl 3):S316-319
36. Liu Y, Stanford SM, Jog SP. et al. Regulation of lymphoid tyrosine phosphatase activity: inhibition of the catalytic domain by the proximal interdomain. Biochemistry. 2009;48(31):7525-7532
37. Hirschhorn JN. Genetic epidemiology of type 1 diabetes. Pediatr Diabetes. 2003;4:87-100
38. Ikegami H, Noso S, Babaya N. et al. Genetic Basis of Type 1 Diabetes: Similarities and Differences between East and West. Rev Diabet Stud. 2008;5(2):64-72
39. Bennett ST, Lucassen AM, Gough SCL. et al. Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nature Genetics. 1995;9:284-292
40. Undlien DE, Hamaguchi K, Kimura A. Type 1 diabetes susceptibility associated with polymorphism in the insulin gene region: a study of blacks, Caucasians, and orientals. Diabetologia. 1994;37:745-749
41. Kantárová D, Buc M. Genetic susceptibility to type 1 diabetes mellitus in humans. Physiol Res. 2007;56:255-266
42. Cox NJ, Wapelhorst B, Morrison VA. et al. Seven regions of the genome show evidence of linkage to type 1 diabetes in a consensus analysis of 767 multiplex families. Am J Hum Genet. 2001;69(4):820-830
43. Huber A, Menconi F, Corathers S. et al. Joint genetic susceptibility to type 1 diabetes and autoimmune thyroiditis: from epidemiology to mechanisms. Endocr Rev. 2008;29:697-725
44. Hewagama A, Richardson B. The genetics and epigenetics of autoimmune diseases. J Autoimmun. 2009;33:3-11
45. Weetman AP. Determinants of autoimmune thyroid disease. Nat Immunol. 2001;2:769-770
46. Dang MN, Buzzetti R, Pozzilli P. Epigenetics in autoimmune diseases with focus on type 1 diabetes. Diabetes Metab Res Rev. 2013;29:8-18
47. Volkmar M, Dedeurwaerder S, Cunha DA. et al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J. 2012;31:1405-1426
48. Buzzetti R, Di Pietro S, Giaccari A. et al. High titer of autoantibodies to GAD identifies a specific phenotype of adult-onset autoimmune diabetes. Diabetes Care. 2007;30:932-938
49. Rakyan VK, Beyan H, Down TA. et al. Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet. 2011;7:e1002300
50. Fradin D, Le Fur S, Mille C. et al. Association of the CpG methylation pattern of the proximal insulin gene promoter with type 1 diabetes. PLoS One. 2012;7:e36278
51. Bell CG, Teschendorff AE, Rakyan VK. et al. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genomics. 2010;3:33
52. Akirav EM, Lebastchi J, Galvan EM. et al. Detection of β cell death in diabetes using differentially methylated circulating DNA. Proc Natl Acad Sci USA. 2011;108:19018-19023
53. Gray SG, De Meyts P. Role of histone and transcription factor acetylation in diabetes pathogenesis. Diabetes Metab Res Rev. 2005;21:416-433
54. Miao F, Smit h DD, Zhang L. et al. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes. 2008;57:3189-3198
55. Tikoo K, Meena RL, Kabra DG. et al. Change in post-translational modifications of histone H3, heat-shock protein-27 and MAP kinase p38 expression by curcumin in streptozotocin-induced type I diabetic nephropathy. Br J Pharmacol. 2008;153:1225-1231
56. Brasacchio D, Okabe J, Tikellis C. et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes. 2009;58:1229-1236
57. Sebastiani G, Grieco FA, Spagnuolo I. et al. Increased expression of microRNA miR-326 in type 1 diabetic patients with ongoing islet autoimmunity. Diabetes Metab Res Rev. 2011;27:862-866
58. Hezova R, Slaby O, Faltejskova P. et al. MicroRNA-342, microRNA-191 and microRNA-510 are differentially expressed in T regulatory cells of type 1 diabetic patients. Cell Immunol. 2010;260:70-74
59. Fernandez-Valverde SL, Taft RJ, Mattick JS. Micro RNAs in β-cell biology, insulin resistance, diabetes and its complications. Diabetes. 2011;60:1825-1831
60. Hirschhorn JN. Genetic epidemiology of type 1 diabetes. Pediatr Diabetes. 2003;4:87-100
61. Redondo MJ, Rewers M, Yu L. et al. Genetic determination of islet cell autoimmunity in monozygotic twin, dizygotic twin, and non-twin siblings of patients with type 1 diabetes: prospective twin study. BMJ. 1999;318:698-702
62. Petersen JS, Kyvik KO, Bingley PJ. et al. Population based study of prevalence of islet cell autoantibodies in monozygotic and dizygotic Danish twin pairs with insulin dependent diabetes mellitus. BMJ. 1997;314:1575-1579
63. Hawa M, Rowe R, Lan MS. et al. Value of antibodies to islet protein tyrosine phosphatase-like molecule in predicting type 1 diabetes. Diabetes. 1997;46:1270-1275
64. Kumar D, Gemayel NS, Deapen D. et al. North-American twins with IDDM. Genetic, etiological, and clinical significance of disease concordance according to age, zygosity, and the interval after diagnosis in first twin. Diabetes. 1993;42:1351-1363
65. van der Werf N, Kroese FG, Rozing J. et al. Viral infections as potential triggers of type 1 diabetes. Diabetes Metab Res Rev. 2007;23(3):169-183
66. Van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev. 2011;91:79-118
67. Hyoty H, Taylor KW. The role of viruses in human diabetes. Diabetologia. 2002;45:1353-1361
68. Yeung WC, Rawlinson WD, Craig M. Enterovirus infection and type 1 diabetes mellitus: Systematic review and meta-analysis of observational molecular studies. BMJ. 2011;342:335
69. Hyöty H, Hiltunen M, Knip M. et al. A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Diabetes. 1995;44:652-657
70. Chiodini RJ, Van Kruiningen HJ, Merkal RS. Ruminant paratuberculosis (Johne's disease): the current status and future prospects. Cornell Vet. 1984;74:218-262
71. Di Sabatino A, Paccagnini D, Vidali F. et al. Detection of Mycobacterium avium subsp. paratuberculosis (MAP)-specific IS900 DNA and antibodies against MAP peptides and lysate in the blood of Crohn's disease patients. Inflamm Bowel Dis. 2011;17:1254-1255
72. Rosu V, Ahmed N, Paccagnini D. et al. Specific immunoassays confirm association of Mycobacterium avium Subsp. paratuberculosis with type-1 but not type-2 diabetes mellitus. PLoS One. 2009;4:e4386
73. Ellingson JL, Anderson JL, Koziczkowski JJ. et al. Detection of viable Mycobacterium avium subsp. paratuberculosis in retail pasteurized whole milk by two culture methods and PCR. J Food Prot. 2005;68:966-972
74. Cossu A, Rosu V, Paccagnini D. et al. MAP3738c and MptD are specific tags of Mycobacterium avium subsp. paratuberculosis infection in type I diabetes mellitus. Clin Immunol. 2011;141:49-57
75. Sechi LA, Rosu V, Pacifico A. et al. Humoral immune responses of type 1 diabetes patients to Mycobacterium avium subsp. paratuberculosis lend support to the infectious trigger hypothesis. Clin Vaccine Immunol. 2008;15:320-326
76. Rani PS, Sechi LA, Ahmed N. Mycobacterium avium subsp. paratuberculosis as a trigger of type-1 diabetes: destination Sardinia, or beyond? Gut Pathog. 2010;2:1
77. Paccagnini D, Sieswerda L, Rosu V. et al. Linking chronic infection and autoimmune diseases: Mycobacterium avium subspecies paratuberculosis, SLC11A1 polymorphisms and type-1 diabetes mellitus. PLoS One. 2009;4:e7109
78. Sechi LA, Paccagnini D, Salza S. et al. Mycobacterium avium subspecies paratuberculosis bacteremia in type 1 diabetes mellitus: an infectious trigger? Clin Infect Dis. 2008;46:148-149
79. Masala S, Paccagnini D, Cossu D. et al. Antibodies recognizing mycobacterium avium paratuberculosis epitopes cross-react with the beta-cell antigen ZnT8 in Sardinian type 1 diabetic patients. PLoS One. 2011;6:e26931
80. Wenzlau JM, Juhl K, Yu L. et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci USA. 2007;104:17040-17045
81. Okazaki T, Maeda A, Nishimura H. et al. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci USA. 2001;98:13866-13871
82. Carreno BM, Collins M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu Rev Immunol. 2002;20:29-53
83. Okazaki T, Iwai Y, Honjo T. New regulatory co-receptors: inducible co-stimulator and PD-1. Curr Opin Immunol. 2002;14:779-782
84. Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116-126
85. Ansari MJ, Salama AD, Chitnis T. et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med. 2003;198:63-69
86. Nielsen C, Hansen D, Husby S. et al. Association of a putative regulatory polymorphism in the PD-1 gene with susceptibility to type 1 diabetes. Tissue Antigens. 2003;62:492-497
87. Kwon NS, Lee SH, Choi CS. et al. Nitric oxide generation from streptozotocin. FASEB J. 1994;8:529-533
88. Won TJ, Jung YJ, Kwon SJ. et al. Forced expression of programmed death-1 gene on T cell decreased the incidence of type 1 diabetes. Arch Pharm Res. 2010;33:1825-1833
89. Yoshida T, Jiang F, Honjo T. et al. PD-1 deficiency reveals various tissue-specific autoimmunity by H-2b and dose-dependent requirement of H-2g7 for diabetes in NOD mice. Proc Natl Acad Sci USA. 2008;105:3533-3538
90. Liang SC, Latchman YE, Buhlmann JE. et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33:2706-2716
91. Wang J, Yoshida T, Nakaki F. et al. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci USA. 2005;102:11823-11828
92. Ansari MJ, Salama AD, Chitnis T. et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med. 2003;198:63-69
93. Yashpal NK, Li J, Wang R. Characterization of c-Kit and nestin expression during islet cell development in the prenatal and postnatal rat pancreas. Dev Dyn. 2004;229:813-825
94. Li J, Quirt J, Do HQ. et al. Expression of c-Kit receptor tyrosine kinase and effect on beta-cell development in the human fetal pancreas. Am J Physiol Endocrinol Metab. 2007;293:E475-483
95. Rachdi L, Ghazi LE, Bemex F. et al. Expression of the receptor tyrosine kinase KIT in mature β-cells and in the pancreas in development. Diabetes. 2001;50:2021-2028
96. Wang R, Li J, Yashpal NK. Phenotypic analysis of c-kit expression in epithelial monolayers derived from postnatal rat pancreatic islets. J Endocrinol. 2004;182:113-122
97. Welsh M, Anneren C, Lindholm C. et al. Role of tyrosine kinase signaling for beta-cell replication and survival. Ups J Med Sci. 2000;105:7-15
98. Yashpal NK, Li J, Wang R. Characterization of c-kit and nestin expression during islet cell development in the prenatal and postnatal rat pancreas. Dev Dyn. 2004;229:813-825
99. Feng ZC, Li J, Turco BA. et al. Critical role of c-Kit in beta cell function: increased insulin secretion and protection against diabetes in a mouse model. Diabetologia. 2012;55:2214-2225
100. DeWitt DE, Hirsch IB. Outpatient insulin therapy in type 1 and type 2 diabetes mellitus: scientific review. JAMA. 2003;289:2254-2264
101. Devendra D, Liu E, Eisenbarth GS. Type 1 diabetes: recent developments. BMJ. 2004;328(7442):750-754
102. Knip M, Virtanen SM, Akerblom HK. Infant feeding and the risk of type 1 diabetes. Am J Clin Nutr. 2010;91:1506S-1513S
103. Akerblom HK, Savilahti E, Saukkonen TT. et al. The case for elimination of cow's milk in early infancy in the prevention of type 1 diabetes: the Finnish experience. Diabetes Metab Rev. 1993;9:269-278
104. Gerstein HC. Cow's milk exposure and type I diabetes mellitus. A critical overview of the clinical literature. Diabetes Care. 1994;17:13-19
105. Knip M, Virtanen SM, Seppä K. et al. Dietary intervention in infancy and later signs of beta-cell autoimmunity. N Engl J Med. 2010;363:1900-1908
106. TRIGR Study Group. Study design of the Trial to Reduce IDDM in the Genetically at Risk (TRIGR). Pediatr Diabetes. 2007;8:117-137
107. Vaarala O, Ilonen J, Ruohtula T. et al. Removal of bovine insulin from cow's milk formula and early initiation of beta-cell autoimmunity in the FINDIA pilot study. Arch Pediatr Adolesc Med. 2012;166:608-614
108. Ziegler AG, Schmid S, Huber D. et al. Early infant feeding and risk of developing type 1 diabetes-associated autoantibodies. J Am Med Assoc. 2003;290:1721-1728
109. Norris JM, Barriga K, Klingensmith G. et al. Timing of initial cereal exposure in infancy and risk of islet autoimmunity. JAMA. 2003;290:1713-1720
110. Pastore MR, Bazzigaluppi E, Belloni C. et al. Six months of gluten-free diet do not influence autoantibody titers, but improve insulin secretion in subjects at high risk for type 1 diabetes. J Clin Endocrinol Metab. 2003;88:162-165
111. Takiishi T, Van Belle T, Gysemans C. et al. Effects of vitamin D on antigen-specific and non-antigen-specific immune modulation: relevance for type 1 diabetes. Pediatr Diabetes. 2013;14:81-89
112. Simpson M, Brady H, Yin X. et al. No association of vitamin D intake or 25-hydroxyvitamin D levels in childhood with risk of islet autoimmunity and type 1 diabetes: the Diabetes Autoimmunity Study in the Young (DAISY). Diabetologia. 2011;54:2779-2788
113. Zipitis CS, Akobeng AK. Vitamin D supplementation in early childhood and risk of type 1 diabetes: a systematic review and meta-analysis. Arch Dis Child. 2008;93:512-517
114. Gabbay MA, Sato MN, Finazzo C. et al. Effect of cholecalciferol as adjunctive therapy with insulin on protective immunologic profile and decline of residual β-cell function in new-onset type 1 diabetes mellitus. Arch Pediatr Adolesc Med. 2012;166:601-607
115. Walter M, Kaupper T, Adler K. et al. No effect of the 1alpha, 25-dihydroxy vitamin D3 on β-cell residual function and insulin requirement in adults with new-onset type 1 diabetes. Diabetes Care. 2010;33:1443-1448
116. Bizzarri C, Pitocco D, Napoli N. et al. No protective effect of calcitriol on beta-cell function in recent-onset type 1 diabetes: the IMDIAB XIII trial. Diabetes Care. 2010;33(9):1962-1963
117. Yamada K, Nonaka K, Hanafusa T. et al. Preventive and therapeutic aspects of large dose nicotinamide injections on diabetes associated with insulitis: an observation in non-obese diabetic (NOD) mice. Diabetes. 1982;31:749-753
118. European Nicotinamide Diabetes Intervention Trial (ENDIT) Group. European Nicotinamide Diabetes Intervention Trial (EN-DIT): a randomized controlled trial of intervention before the onset of type 1 diabetes. Lancet. 2004;363:925-931
119. Skyler JS. Primary and secondary prevention of Type 1 diabetes. Diabet Med. 2013;30:161-169
120. Peakman M, von Herrath M. Antigen-specific immunotherapy for type 1 diabetes: maximizing the potential. Diabetes. 2010;59:2087-2093
121. Lernmark A, Larsson HE. Immune therapy in type 1 diabetes mellitus. Nat Rev Endocrinol. 2013;9:92-103
122. Keller RJ, Eisenbarth GS, Jackson RA. Insulin prophylaxis in individuals at high risk of type I diabetes. Lancet. 1993;341:927-928
123. Diabetes Prevention Trial-Type 1 Diabetes Study Group (Skyler JS, Brown D, Chase HP, et al). Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med. 2002;346:1685-1691
124. Skyler JS, Krischer JP, Wolfsdorf J. et al. Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial-Type 1. Diabetes Care. 2005;28:1068-1076
125. Vehik K, Cuthbertson D, Ruhlig H. et al. Long-term outcome of individuals treated with oral insulin: diabetes prevention trial-type 1 (DPT-1) oral insulin trial. Diabetes Care. 2011;34:1585-1590
126. Harrison LC, Honeyman MC, Steele CE. et al. Pancreatic beta-cell function and immune responses to insulin after administration of intranasal insulin to humans at risk for type 1 diabetes. Diabetes Care. 2004;27:2348-2355
127. Näntö-Salonen K, Kupila A, Simell S. et al. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial. Lancet. 2008;372:1746-1755
128. Ryhänen SJ, Härkönen T, Siljander H. et al. Impact of intranasal insulin on insulin antibody affinity and isotypes in young children with HLA-conferred susceptibility to type 1 diabetes. Diabetes Care. 2011;34:1383-1388
129. Thrower SL, James L, Hall W. et al. Proinsulin peptide immunotherapy in type 1 diabetes: report of a first-in-man Phase I safety study. Clin Exp Immunol. 2009;155:156-165
130. Carel JC, Boitard C, Eisenbarth G. et al. Cyclosporine delays but does not prevent clinical onset in glucose intolerant pre-type 1 diabetic children. J Autoimmun. 1996;9:739-745
131. Eisenbarth GS. Banting Lecture 2009. An unfinished journey: molecular pathogenesis to prevention of type 1A diabetes. Diabetes. 2010;59:759-774
132. Skyler JS and the Type 1 Diabetes TrialNet Study Group. Update on worldwide efforts to prevent type 1 diabetes. Ann N Y Acad Sci. 2008;1150:190-196
133. Juvenile Diabetes Research Foundation Continuous Glucose Monitoring Study Group. Prolonged nocturnal hypoglycemia is common during 12 months of continuous glucose monitoring in children and adults with type 1 diabetes. Diabites Care. 2010;33:1004-1008
134. Matthews JB, Staeva TP, Bernstein PL. et al. Developing combination immunotherapies for type 1 diabetes: recommendations from the ITN-JDRF Type 1 Diabetes Combination Therapy Assessment Group. Clin Exp Immunol. 2010;160:176-184
135. Couri CE, Oliveira MC, Stracieri AB. et al. C-peptide levels and insulin independence following autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA. 2009;301:1573-1579
136. Michels AW, Nakayama M. The anti-insulin trimolecular complex in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. 2010;17:329-334
137. Erlich H, Valdes AM, Noble J. et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes. 2008;57:1084-1092
138. Steenkiste A, Valdes AM, Feolo M. et al. 14th International HLA and Immunogenetics Workshop: report on the HLA component of type 1 diabetes. Tissue Antigens. 2007;69:214-225
139. Palmer JP, Asplin CM, Clemons P. et al. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science. 1983;222:1337-1339
140. Achenbach P, Koczwara K, Knopff A. et al. Mature high-affinity immune responses to (pro)insulin anticipate the autoimmune cascade that leads to type 1 diabetes. J Clin Invest. 2004;114:589-597
141. Lee KH, Wucherpfennig KW, Wiley DC. Structure of a human insulin peptide/HLA-DQ8 complex and susceptibility to type 1 diabetes. Nature Immunology. 2001;2:501-507
142. Stadinski BD, Zhang L, Crawford F. et al. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci USA. 2010;107:10978-10983
Author biography
 Dr. Yan-Ling Wu is a professor in Molecular Immunology and now heads the Cellular and Molecular Immunology Research Group. She received Master and Doctoral degrees in Applied Life Science in 2003 and in Medicine Science in 2006, respectively, from Tohoku University, Japan. After that, she entered to Professor Minato's group of School of Medicine, Kyoto University, Japan, as a senior researcher working in the field of molecular immunology. Her current researches focus on understanding the molecular mechanisms of gene regulation related to diseases by immune inhibitory receptors. Dr. Wu have given oral presentations in international conferences and published related papers.
Dr. Yan-Ling Wu is a professor in Molecular Immunology and now heads the Cellular and Molecular Immunology Research Group. She received Master and Doctoral degrees in Applied Life Science in 2003 and in Medicine Science in 2006, respectively, from Tohoku University, Japan. After that, she entered to Professor Minato's group of School of Medicine, Kyoto University, Japan, as a senior researcher working in the field of molecular immunology. Her current researches focus on understanding the molecular mechanisms of gene regulation related to diseases by immune inhibitory receptors. Dr. Wu have given oral presentations in international conferences and published related papers.
 Yan-Ping Ding received her bachelor's degree in biological sciences from Zhejiang University of Chinese Traditional Medicine. Since graduation, she has been working in Zhejiang Center of Disease Control and Prevention. Her research under the guidance of Prof. Yanling Wu mainly focuses on the field of Molecular Immunology.
Yan-Ping Ding received her bachelor's degree in biological sciences from Zhejiang University of Chinese Traditional Medicine. Since graduation, she has been working in Zhejiang Center of Disease Control and Prevention. Her research under the guidance of Prof. Yanling Wu mainly focuses on the field of Molecular Immunology.
 Jian Gao received his Bachelor of Medicine degree from College of Public Heath, Shandong University in 2004. Then, he entered Chinese Centre for Disease Control and Prevention to earn his Master degree of Medicine. Since 2008, He has been working in Zhejiang Provincial Centre for Disease Control and Prevention, and now his research focuses on Molecular Virology in the Wu's group.
Jian Gao received his Bachelor of Medicine degree from College of Public Heath, Shandong University in 2004. Then, he entered Chinese Centre for Disease Control and Prevention to earn his Master degree of Medicine. Since 2008, He has been working in Zhejiang Provincial Centre for Disease Control and Prevention, and now his research focuses on Molecular Virology in the Wu's group.
 Yoshimasa Tanaka received his Ph.D. in Hokkaido University Graduate School of Agriculture with a specialization in Enzymology and Biochemistry. After graduation, he continued his research in the field of Immunobiology. Since 2008, he is an associate professor and works in the Center for Innovation in Immunoregulative Immunology and Therapeutics that belongs to the Kyoto University Graduate School of Medicine.
Yoshimasa Tanaka received his Ph.D. in Hokkaido University Graduate School of Agriculture with a specialization in Enzymology and Biochemistry. After graduation, he continued his research in the field of Immunobiology. Since 2008, he is an associate professor and works in the Center for Innovation in Immunoregulative Immunology and Therapeutics that belongs to the Kyoto University Graduate School of Medicine.
 Dr. Wen Zhang is a full professor with 25 years of research and teaching experience in Bioorganic Chemistry and Chemical Biology. Dr. Zhang earned his doctorate degree in Bioorganic Chemistry from East China University of Science and Technology, China. Then, he entered Professor Ohrui's Lab of Tohoku University, Japan, working in the field of molecular recognition as a JSPS postdoctoral fellow. After that, he joined Professor Sugiyama's Chemical Biology group of Kyoto University, Japan, as a COE and JST research fellow working on biology and chemistry of polyamide-nucleic acids interaction. Now, Dr. Zhang has a special interest in elucidating the gene regulation mechanisms with small organic molecules and the development of gene-targeted drug. His group formed in 2008 and established an extremely fruitful collaboration with Prof. Sugiyama's Group in order to better pursue aspects of gene-targeted drug research. To date, Dr. Zhang has published better papers as the first/corresponding author in excellent Journals including JACS, ChemBioChem, Int J Biol Sci etc.
Dr. Wen Zhang is a full professor with 25 years of research and teaching experience in Bioorganic Chemistry and Chemical Biology. Dr. Zhang earned his doctorate degree in Bioorganic Chemistry from East China University of Science and Technology, China. Then, he entered Professor Ohrui's Lab of Tohoku University, Japan, working in the field of molecular recognition as a JSPS postdoctoral fellow. After that, he joined Professor Sugiyama's Chemical Biology group of Kyoto University, Japan, as a COE and JST research fellow working on biology and chemistry of polyamide-nucleic acids interaction. Now, Dr. Zhang has a special interest in elucidating the gene regulation mechanisms with small organic molecules and the development of gene-targeted drug. His group formed in 2008 and established an extremely fruitful collaboration with Prof. Sugiyama's Group in order to better pursue aspects of gene-targeted drug research. To date, Dr. Zhang has published better papers as the first/corresponding author in excellent Journals including JACS, ChemBioChem, Int J Biol Sci etc.
![]() Corresponding author: Yanling Wu, Virus Inspection Department of Zhejiang Provincial Center for Disease Control and Prevention, 630 Xincheng Road, Hangzhou, 310051, PR China; Tel: +86-571-87115282; Fax: +86-571-87115282; e-mail: ylwuzj.cn. Wen Zhang, Department of Biopharmaceutical Science, College of Pharmaceutical Science, Zhejiang University of Technology, 18 Chaowang Road, Hangzhou, 310014, PR China; Tel: +86-571-88871507; Fax: +86-571-88871507; e-mail: wzhang63edu.cn.
Corresponding author: Yanling Wu, Virus Inspection Department of Zhejiang Provincial Center for Disease Control and Prevention, 630 Xincheng Road, Hangzhou, 310051, PR China; Tel: +86-571-87115282; Fax: +86-571-87115282; e-mail: ylwuzj.cn. Wen Zhang, Department of Biopharmaceutical Science, College of Pharmaceutical Science, Zhejiang University of Technology, 18 Chaowang Road, Hangzhou, 310014, PR China; Tel: +86-571-88871507; Fax: +86-571-88871507; e-mail: wzhang63edu.cn.