Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(8):948-960. doi:10.7150/ijbs.12468 This issue Cite
Research Paper
Overexpression of FGF9 in Prostate Epithelial Cells Augments Reactive Stroma Formation and Promotes Prostate Cancer Progression
Yanqing Huang1, Chengliu Jin1#, Tomoaki Hamana1, Junchen Liu1, Cong Wang2, Lei An1, Wallace L. McKeehan1, Fen Wang1,2,3 ![]()
1. Center for Cancer and Stem Cell Biology, Institute of Biosciences and Technology, Texas A&M Health Science Center, Houston, TX, USA
2. Wenzhou Medical College, Wenzhou, Zhejiang, China
3. Department of Molecular and Cellular Medicine, College of Medicine, Texas A&M, Health Science Center, College Station, TX, USA.
# Current address: Transgenic and Gene Targeting Core, Georgia State University, Atlanta, GA, USA
Received 2015-4-21; Accepted 2015-5-15; Published 2015-6-11
Abstract

Bone metastasis is the major cause of morbidity and mortality of prostate cancer (PCa). Fibroblast growth factor 9 (FGF9) has been reported to promote PCa bone metastasis. However, the mechanism by which overexpression of FGF9 promotes PCa progression and metastasis is still unknown. Herein, we report that transgenic mice forced to express FGF9 in prostate epithelial cells (F9TG) developed high grade prostatic intraepithelial neoplasia (PIN) in an expression level- and time-dependent manner. Moreover, FGF9/TRAMP bigenic mice (F9TRAMP) grew advanced PCa earlier and had higher frequencies of metastasis than TRAMP littermates. We observed tumor microenvironmental changes including hypercellularity and hyperproliferation in the stromal compartment of F9TG and F9TRAMP mice. Expression of TGFβ1, a key signaling molecule overexpressed in reactive stroma, was increased in F9TG and F9TRAMP prostates. Both in vivo and in vitro data indicated that FGF9 promoted TGFβ1 expression via increasing cJun-mediated signaling. Moreover, in silico analyses showed that the expression level of FGF9 was positively associated with expression of TGFβ1 and its downstream signaling molecules in human prostate cancers. Collectively, our data demonstrated that overexpressing FGF9 in PCa cells augmented the formation of reactive stroma and promoted PCa initiation and progression.
Keywords: fibroblast growth factor, receptor tyrosine kinase, conditional gene knockout, prostate cancer progression, EMT, reactive stroma.
Introduction
Prostate cancer (PCa) is the most common cause of cancer death in American men over age 75. The progression of PCa is a multiple-step process of orderly events, complicated by its heterogeneity and driven by various genetic abnormalities. PCa almost invariably metastasizes to the bone, which accounts for most of the morbidity and mortality of PCa patients (1). At early stages, PCa is androgen responsive; androgen deprivation is a common treatment that generally causes PCa to regress. However, most PCa eventually recurs, becomes castration-resistant, and eventually develops metastases (2).
The prostate consists of epithelial and stromal compartments separated by basement membranes. Cells in the two compartments maintain active two-way communications through paracrine mechanisms in which ligands and receptors are partitioned between the two compartments (3, 4). These reciprocal communications are precisely balanced and are critical for preserving tissue homeostasis. It is well documented that the accumulation of somatic mutations in epithelial cells is able to cause adenocarcinoma. Moreover, PCa cells are surrounded by a complex microenvironment, called prostate reactive stroma. The reactive stroma includes extracellular matrix (ECM), diffusible growth factors and cytokines, and a variety of non-epithelial cell types. The tumor microenvironment also has a profound influence on the development and progression of carcinomas and PCa is no exception (5). Tumor-associated reactive stroma emerges as early as the formation of pre-malignant prostatic intraepithelial neoplasia (PIN), and arises due to modulation of a large spectrum of cell signaling, either due to stimuli from epithelial cells or intrinsic genetic or epigenetic changes (6-10). Prostate reactive stroma promotes PCa progression by supporting tumor cell proliferation, inducing a fibroblastic phenotype, remodeling the extracellular matrix, and augmenting metastasis (6).
Fibroblast growth factor (FGF) signaling is essential for prostate development and prostate tissue homeostasis (11, 12). The FGF family consists of 18 FGF ligands that activate FGF receptor (FGFR) tyrosine kinases encoded by four highly homologous genes (13). Abnormal FGF signaling has been implicated in developmental disorders and tumorigenesis in various tissues (14). Extensive evidence shows that aberrant expression of FGF or FGFR isoforms and activation of the FGF/FGFR signaling axis are associated with PCa development and progression (12, 15-20). Amplification of the Fgfr1 gene is frequently found in human PCa (21). The acquisition of ectopic expression of FGFR1 in tumor epithelial cells stands out as the most frequent change among FGFR isotypes (22-25). Forced expression of constitutively active FGFR1 or multiple FGF ligands has been shown to induce prostate lesions in mouse models (18, 26-33). Ablation of Fgfr1 or Frs2α that encodes FGFR substrate 2α (FRS2α), an adaptor protein for FGFR to activate multiple downstream signaling pathways, reduces development and progression of PCa induced by T antigens in mice (12, 34). However, how aberrant FGF signals contribute to PCa progression is still not fully understood.
Accumulating evidence supports a role for FGF9 in PCa progression and metastasis. Previous studies have shown that FGF9 mediates osteogenesis induced by androgen receptor-negative human PCa cells (26). In addition, FGF9-positive PCa shows a higher risk of biochemical recurrence (35). In spite of the correlation between FGF9 and progression and bone metastases of PCa, whether overexpression of FGF9 initiates prostate tumorigenesis is still elusive. To study whether FGF9 overexpression contributes to initiation and progression of PCa, transgenic mice expressing FGF9 in prostate epithelial cells were generated and crossed with the TRAMP (transgenic adenocarcinoma of the mouse prostate) mouse model. Forced expression of FGF9 in the prostate led to PIN in a time- and dosage-dependent manner. Furthermore, it augmented the formation of reactive stroma and accelerated PCa progression in TRAMP mice. Both in vivo and in vitro data showed that activation of cJun-dependent TGFβ1 expression in stromal cells of the prostate by FGF9 constituted a paracrine loop that contributed to PCa progression. Moreover, in silico analyses of the TCGA database demonstrated that expression of FGF9 was correlated with that of TGFβ1 and its downstream effectors. Together, the results support a mechanism by which FGF9 overexpression in PCa contributes to progression and metastasis of PCa.
Materials and methods
Animals
All animals were housed in the Program for Animal Resources of the Texas A&M Health Science Center, Houston Campus. The mice were maintained and handled in accordance with the principles of the Guide for the Care and Use of Laboratory Animals. All experimental procedures were approved by the Institutional Animal Care and Use Committee. Mice carrying the FGF9 and the TRAMP transgenes were bred and genotyped as described (36). The primers for genotyping are, FGF9 forward: CTTTGGCTTAGAATATCCTTA; FGF9 reverse: AGTGACCACCTGGGTCAGTCC; TRAMP forward: CCGGTCGACCGGAAGCTTCCACAAGT; TRAMP reverse: CTCCTTTCAAGACCTAGAAGGTCCA. Prostate tissues and tumors were harvested after the animals were euthanized by CO2 asphyxiation. Nude mice were purchased from Charles River Laboratory and maintained in sterile conditions according to the Institutional Guidelines.
Generation of transgenic mice
The full-length rat FGF9 cDNA including the Kozak sequence was amplified by PCR using rat FGF9 cDNA as the template. After digestion with BamHI and EcoRV, the PCR product was subcloned into the pBluescript SK vector and sequenced. The insert was excised with the two restriction enzymes and cloned into the SSI vector (27). The ARR2PB-FGF9 transgene was excised with BssHII restriction enzyme and purified for pronuclear microinjection. Fertilized eggs were collected from FVB females and pronucleus were injected with the ARR2PB-FGF9 DNA construct. Injected eggs were then transferred into pseudo-pregnant Swiss/Webster females for full-term development. Genomic DNA was purified from tails of founder mice at day 7 after birth and screened by PCR.
Histology
Prostates were dissected and sectioned for histological analyses as previously described (11, 36). Hematoxylin and Eosin staining, immunohistochemical analyses, and in situ hybridization were performed on 5-µm thick sections mounted on Superfrost/Plus slides (Fisher Scientific, Pittsburgh, PA). Antigens were retrieved by incubation in citrate buffer (10 mmol/L) for 20 minutes at 100°C or as suggested by antibody manufacturers. The sources and concentrations of primary antibodies used are: anti-α-smooth muscle actin (1:1) from Sigma (St Louis, MO); anti-Vimentin (1:200), anti-E-cadherin (1:200) from Cell Signaling Technology; anti-androgen receptor (1:200) from Santa Cruz; anti-CD31 (1:200) from Abcam; anti-Ki67 (1:500) from Novus Biologicals. For immunofluorescence, the specifically bound antibodies were detected with FITC-conjugated secondary antibodies and visualized under a Zeiss LSM 510 Confocal Microscope. For immunohistochemical staining, specifically bound antibodies were detected with biotinylated anti-Rabbit IgG or biotinylated anti-mouse IgG antibodies (Vector labs). The signal was enhanced using the VECTASTAIN ABC system and visualized with a VECTOR NovaRED Substrate kit. Prostate lesion grading was performed as described (37, 38).
Isolation of primary stromal cells
Prostates were dissected and single primary prostate cells were obtained as described (39). Briefly, prostates dissected from 4 to 8-month-old male mice were minced with steel scissors, followed by incubation with collagenase (Sigma) in 10 ml DMEM at a concentration of 1mg/ml with 10% FBS at 37°C for 90 minutes. Cells were washed with PBS and further digested with 0.25% trypsin/EDTA for 10 minutes at 37°C. After adding FBS to inactivate trypsin, cells were passed through a 40-μm cell strainer, washed with DPBS (Sigma), and seeded in 10-cm cell plates. After overnight incubation at 37oC, the unattached cells were removed and only attached stromal cells were kept in cultures. Both primary stromal cells prepared from mouse prostates and human HPS-19I prostate stromal cells were cultured as described (6).
Luciferase assay
A pGL3 plasmid containing the full-length TGFβ1 promoter region (40) was kindly provided by Dr. Zhengxin Wang, University of Texas MD Anderson Cancer Center. Luciferase assay was done following the instructions of dual-luciferase reporter assay system (Promega).
Immunoblotting
Prostate tissues or PCa cells were homogenized in RIPA buffer (50 mM Tris-HCl buffer pH 7.4, 1% NP40, 150 mM NaCl, 0.25% Na-deoxycholate, 1 mM EGTA, and 1 mM PMSF). Extracted soluble proteins were harvested by centrifugation. Samples containing 50 µg proteins were separated by SDS-PAGE and electroblotted onto PVDF membranes for Western blotting analysis with the indicated antibodies. The dilutions of the antibodies are: anti-cJun, 1:1000; anti-TGFβ1, 1:1000; anti-pSmad2/3: 1:1000; and anti-β-Actin, 1:3000. Specific bands were visualized using the ECL-Plus chemoluminescent reagents. The films were scanned with a densitometer and the bands were quantified using Image J software (NIH).
Gene expression
For in situ hybridization, paraffin-embedded tissue sections were rehydrated, followed by proteinase K (10 µg/ml) digestion for 15 minutes at room temperature. After prehybridization at 65°C for 2 hours, the hybridization was carried out by overnight incubation at 65°C with digoxigenin-labeled RNA probes (1 µg/ml) for FGF9. Nonspecifically bound probes were removed by washing three times with 0.1X DIG washing buffer at 65°C for 30 minutes. Specifically bound probes were detected by alkaline phosphatase-conjugated anti-digoxigenin antibody (Roche, Indianapolis, IN).
For real-time RT-PCR analyses, total RNA was extracted from prostates using TRIzol RNA isolation reagents (Life Technologies). The first-strand cDNAs were reverse transcribed from 1 µg RNA templates using the SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA) and random primers according to manufacturer's protocols. Real-time RT-PCR analyses were carried out using the Fast SYBR Green Master Mix (Life Technologies) as instructed by the manufacturer.
Chromatin immunoprecipitation (ChIP) assays
For the ChIP experiments, extracts were prepared from primary prostate stromal cells. ChIP assays were carried out with the EZ-Magna ChIP Kit (Millipore, Billerica, MA) according to the manufacturer's protocols. The following set of real-time PCR primers specifically for the cJun binding regions on the TGFβ1 promoter was used: TGFβ1-Forward: GGCTGCATCTCCAAGCATT and TGFβ1-Reverse: GGTTCTCATCTCCCACTCACTC.
Statistical analyses
Statistical analysis was performed using the two tailed t test, with significance set at P<0.05. Error bars indicate standard deviation.
Results
Overexpression of FGF9 disrupts tissue homeostasis of the prostate in an expression level- and time-dependent manner
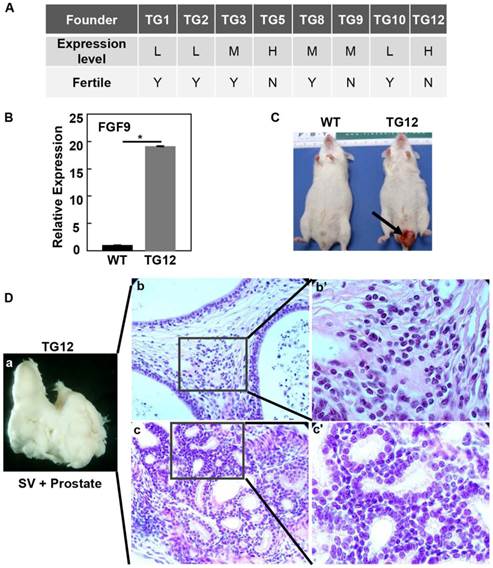
To determine whether FGF9 overexpression caused prostate lesions, we generated 8 strains of ARR2PBi-FGF9 transgenic mice, in which expression of FGF9 in prostate epithelial cells was driven by the ARR2PB composite probasin promoter as described (27). Among them, TG5 and TG12 highly expressed the FGF9 transgene; TG3, TG8, and TG9 expressed the transgene at moderate levels; and TG1, TG2, and TG10 expressed the transgene at low levels (Fig. 1A). The Fgf9 expression level in TG12 was shown in Fig. 1B. Around the age of 3-8 months, the mice suffered penis protrusion (Fig. 1C) and the prostate was fused with the seminal vesicles (Fig. 1Da). H&E staining revealed that the prostate developed severe hyperplasia in both stromal (Fig. 1Db) and epithelial compartments (Fig. 1Dc). Since both TG5 and TG12 that expressed FGF9 at high levels were infertile, the line TG8 with moderate expression levels of FGF9 was chosen for the following studies and hereafter is designated as F9TG.
Overexpression of FGF9 leads to fusion of the prostate and seminal vesicles. A. Generation of FGF9 transgenic founders. Expression of the transgene was examined with RT-PCR analysis. Note that the two high expressers were infertile. B. Real time RT-PCR analyses of FGF9 expression in TG12 founder prostate. C. TG12 founder developed a penis protrusion, indicated by the arrow. D. Fusion of the prostate with seminal vesicles in TG12 founder. Panels b&c are H&E staining showing hyperplasia in both stromal and epithelial compartments. Panels b'&c' are high magnification views of panels b and c. L, low expression level; M, moderate expression level; H, high expression level; WT, wildtype; SV, seminal vesicles.
In situ hybridization demonstrated that the Fgf9 mRNA was highly expressed in the prostate epithelium of F9TG mice (Fig 2A). In contrast, expression of endogenous Fgf9 mRNA was below the detection limit under the same condition. No apparent difference was observed in the histology and tissue size between F9TG and control prostates at young adult stages. However, mild hyperplasia was detected in the ventral prostates of 8-month-old F9TG mice (Fig. 2B). At 11 months of age, F9TG prostates displayed anomalies ranging from multifocal hyperplasia to low grade PIN, with atypical larger cells with irregular hyperchromatic nuclei. At 18 months, F9TG prostates (n=4) progressed to high-grade PIN and invasive carcinoma as shown by the loss of the boundary between epithelium and stroma compartments (Fig. 2B). Highly pleomorphic atypical cells arranged in a cribriform pattern were observed in the lumen. Extracellular matrix remodeling and stromal hypercellularity was notable adjacent to the invasive carcinoma. In contrast, WT prostates only showed a few mild hyperplasia lesions. Quantified analyses of 18-month-old mice showed that F9TG mice developed more prostate lesions, ranging from low grade PIN, high grade PIN, to invasive carcinoma, than control prostates (Fig. 2C).
Forced expression of FGF9 in prostatic epithelial cells disrupts prostate tissue homeostasis. A. In situ hybridization demonstrates expression of Fgf9 mRNA in the mouse prostate. Inserts are negative control using sense probes. B. H&E staining demonstrating prostate lesions in F9TG prostates at the indicated ages. Arrows indicate hyperplasia, LGPIN, and HGPIN. Inserts are high magnification views from the same slides. C. Quantitation of the histology of 18-month-old WT and F9TG prostates. N=4 per group. D. Immunostaining of Ki67 showing proliferating epithelial cells in 11-month-old WT and F9TG prostates. E. Quantitation of Ki67 positive cells in F9TG and control prostates at the indicated ages. F. Ki67 staining showing proliferating cells in the stromal compartment. G. Immunohistochemical or immunofluorescence analysis for androgen receptor, E-cadherin, α-smooth muscle actin, or CD3 expression in 18-month-old WT and F9TG prostates. WT, wild type; F9TG, FGF9 transgenic mice; HGPIN, high grade prostatic intraepithelial neoplasia; LGPIN, low grade prostatic intraepithelial neoplasia; F9TRAMP, FGF9TG/TRAMP bigenic mice; scale bars, 50 µm.
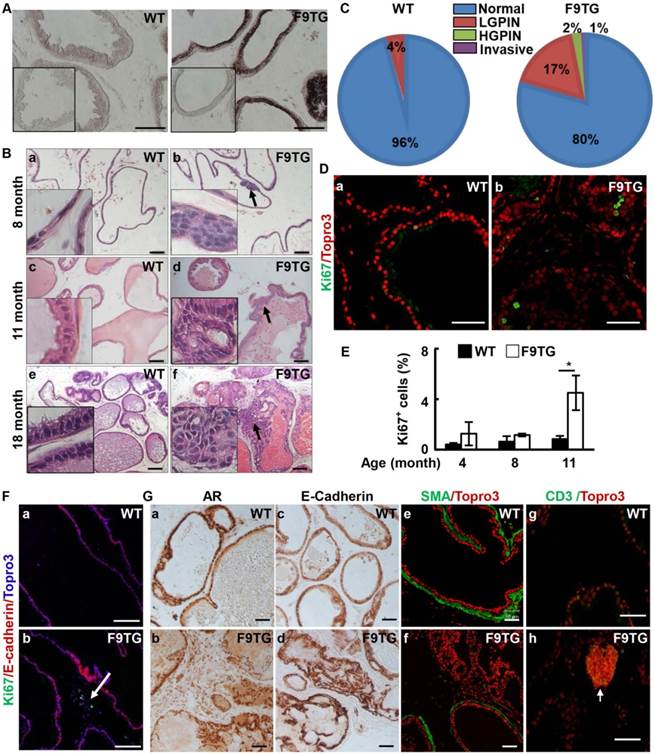
Compared with the control prostate, cells in the F9TG prostate was more proliferative (Fig. 2D&E). At 11 months of age, F9TG prostates possessed nearly four times as many proliferating epithelial cells as control prostates. Moreover, stromal proliferation became prominent as shown by the Ki67 staining of the cells that did not expressed E-cadherin (Fig. 2Fa,b). Both androgen receptor (Fig. 2Ga,b) and E-cadherin (Fig. 2Gc,d) expressions in F9TG prostates appeared to be similar to that of control prostates. However, the smooth muscle-like stromal cells were reduced adjacent to the high-grade PIN lesions (Fig. 2Ge,f). Clusters of CD3 positive cells were particularly notable in the stromal region of F9TG prostates (Fig. 2Gg,h), indicating the presence of T lymphocyte infiltrations. Together, the data indicate that overexpression of FGF9 disrupts tissue homeostasis of the prostate in both epithelial and stromal compartments and leads to invasive adenocarcinomas in a time- and dosage-dependent manner.
Overexpression of FGF9 promoted initiation and progression of TRAMP tumors
To determine whether overexpression of FGF9 contributed to initiation and progression of PCa, F9TG mice were crossed with TRAMP mice to generate FGF9/TRAMP bigenic mice (hereafter designated as F9TRAMP). In situ hybridization (Fig. 3A) and real-time RT-PCR (Fig. 3B) analyses revealed that FGF9 was highly expressed in the epithelium of the F9TRAMP prostate. F9TRAMP mice had a median survival time of 218 days, which was shorter than that of TRAMP mice that had a median survival time of 234 days. Although similar at early ages, the average weight of F9TRAMP prostates (n=13) was heavier than that of TRAMP prostates (n=10) at the age of 6 months or older (Fig. 3C).
Overexpression of FGF9 in prostatic epithelial cells promotes PCa progression in mice. A. In situ hybridization showing expression of Fgf9 mRNA in mouse prostates. B. Real-time RT-PCR analyses of Fgf9 expression in TRAMP and F9TRAMP prostates at 4 months. C. Statistical analysis of prostate/body weight ratio of TRAMP and F9TRAMP prostates at the ages of 4, 5, and 6 months. D. H&E staining of TRAMP and F9TRAMP prostates at the ages of 3, 4, and 6 months. E. Quantitative analyses of prostate lesions of TRAMP and F9TRAMP mice at the age of 3 or 6 months. N=5 per group. F. Immunostaining comparing AR, P63, and Ki67 expression in TRAMP and F9TRAMP prostates. Panel g, statistical analyses of proliferating cells in TRAMP and F9TRAMP prostates at the indicated ages. Scale bars, 50 µm
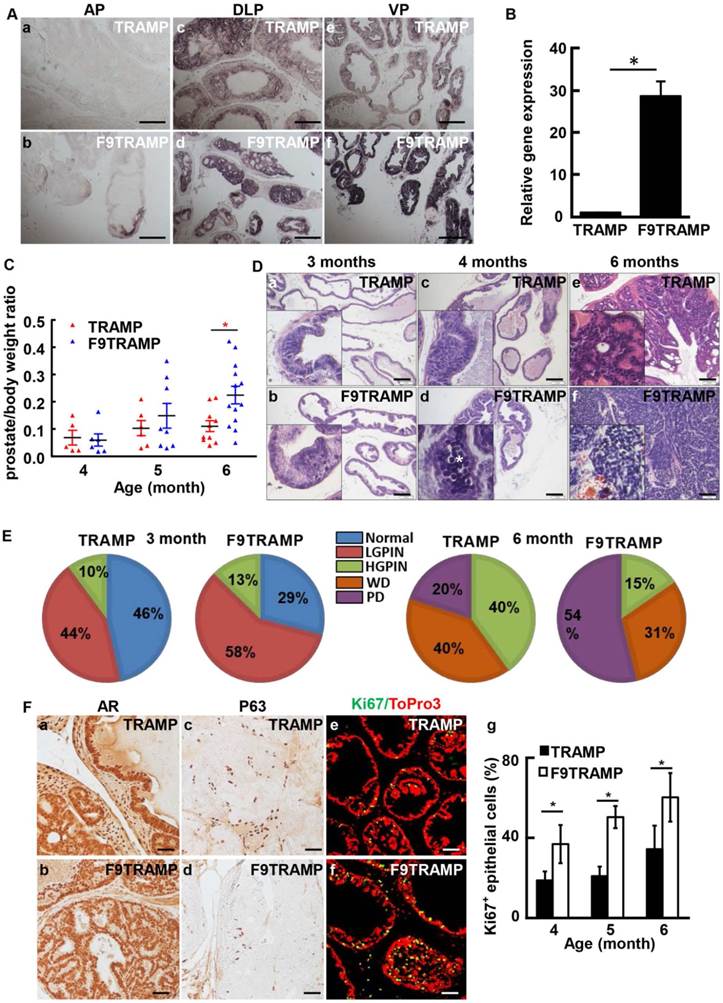
Since the majority of lesions in the TRAMP prostate were initiated in ventral prostate (37), H&E staining was carried out to analyze the lesion development in F9TRAMP ventral prostates at the ages of 3, 4, and 6 months (Fig. 3D). Apoptotic bodies were prominent in 4-month F9TRAMP ventral prostates. Cribriform architecture was seen in 6-month-old TRAMP ventral prostates, while F9TRAMP mice already developed poorly differentiated PCa, composed of sheets of cells with little or no glandular formation. To quantitate development of the lesion, the standardized prostate lesion grading system (37, 38) was employed to assess the phenotype at the ages of 3 and 6 months (Fig. 3E). The F9TRAMP prostates had more high-grade lesions in both groups than the TRAMP prostates. About 54% of the F9TRAMP prostate had poorly differentiated (PD) carcinomas, while only 20% TRAMP mice developed PD carcinomas at 6 months. No obvious defect was observed in AR expression and localization in the F9TRAMP prostate (Fig. 3Fa,b). However, the number of P63-expressing basal cells was reduced in F9TRAMP prostates (Fig. 3Fc,d), which was consistent with the notion that basal cells were diminished in PCa (41, 42). Moreover, cells in the F9TRAMP prostate were more proliferative than in the TRAMP prostate (Fig. 3Fe-g).
Overexpression of FGF9 promoted metastasis of TRAMP prostate tumors
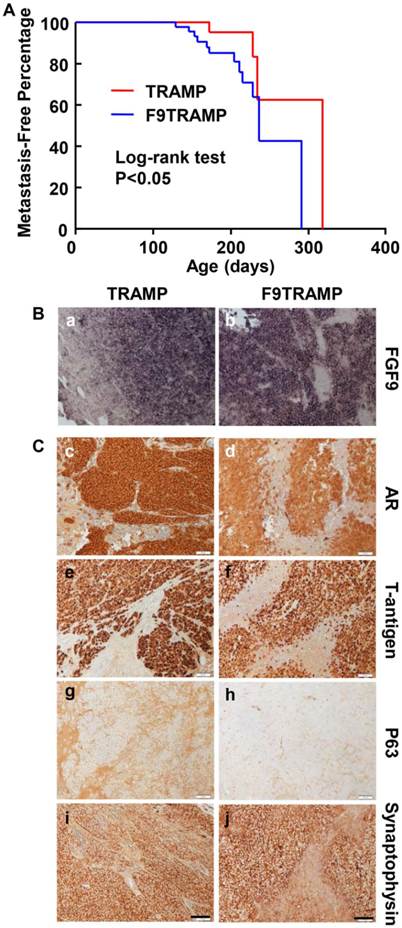
To determine whether F9TRAMP tumors were more metastatic, we then examined the metastatic tumors in F9TRAMP and TRAMP mice at the times of sacrifice. Most of the metastatic loci were located in lymph node, liver, and kidney. The metastasis-free curve and log-rank test (P<0.05) showed that F9TRAMP mice (n=54) developed metastases earlier than TRAMP mice (n=39) (Fig. 4A). Moreover, the frequency of metastasis was higher in F9TRAMP mice than in TRAMP mice at all inspection times. In situ hybridization showed that Fgf9 mRNA was expressed in metastatic PCa cells in both F9TRAMP and TRAMP mice but not adjacent non-cancerous tissues (Fig. 4Ba,b). The metastatic PCa cells in both strains expressed AR and T-antigens (Fig. 4Bc-f). Intriguingly, most of the metastatic tumors were synaptophysin positive and P63 negative (Fig. 4Bg-j). This indicates the presence of neuroendocrine differentiation in metastatic tumors, which is frequently observed in advance human PCa.
Overexpression of FGF9 promotes PCa metastasis in mice. A. Kaplan-Meier analysis of metastasis-free time in TRAMP and F9TRAMP mice. B. In situ hybridization showing Fgf9 expression in lymph node metastases of F9TRAMP and TRAMP tumors. C. Immunostaining of the indicated proteins in lymph node metastases of F9TRAMP and TRAMP tumors. Scale bars, 50 µm.
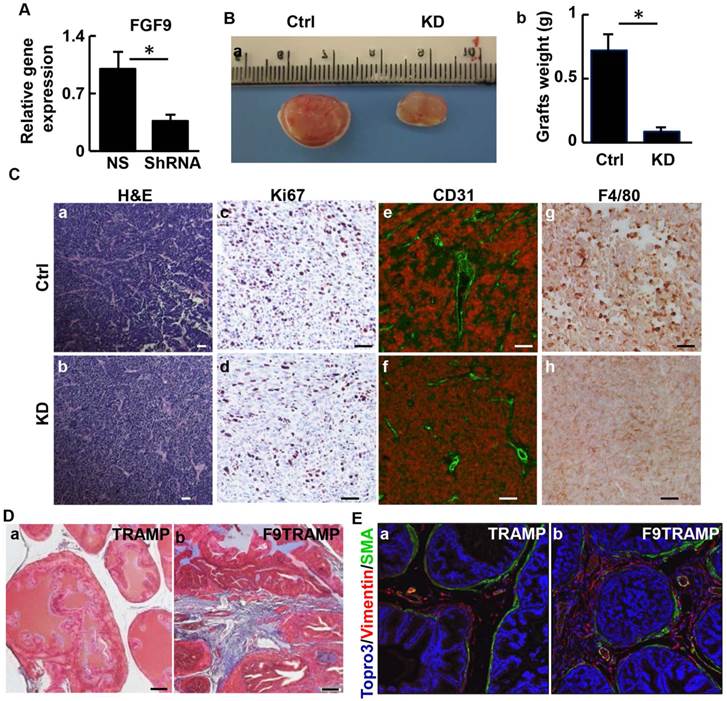
TRAMP-C2 cells are derived from TRAMP tumors and express Fgf9 at a moderate level. To further characterize the role of FGF9 in prostate tumorigenesis, expression of Fgf9 in TRAMP-C2 cells was depleted by infecting the cells with lentivirus bearing shRNA specific for Fgf9 mRNA (Fig. 5A). The cells were then mixed with mouse urogenital sinus mesenchymal cells and grafted in the flanks of nude mice. Eight weeks after the implantation, tumor grafts were excised for analysis. The graft size of the FGF9 depleted cells was smaller than that of the control cells (Fig. 5B). Although no significant difference in general histology between the two groups was detected (Fig. 5C a,b), depletion of FGF9 compromised cell proliferation in the graft (Fig. 5Cc,d). In addition, the FGF9-depleted grafts had fewer endothelial cells that are CD31 positive than the control group, indicating compromised angiogenesis in the FGF9 depleted graft (Fig. 5Ce,f). This is in agreement with a previous report that FGF9 induces VEGF-A expression in PCa (43). Interestingly, immunostaining with anti-F4/80 antibodies revealed that depletion of FGF9 reduced macrophage infiltration in TRAMP-C2 grafts (Fig. 5Cg,h), implying a role of epithelial-expressed FGF9 in reactive stroma formation and microenvironmental remodeling. Trichrome staining revealed that the stroma of F9TRAMP prostates exhibited extensive collagen deposition, which is indicative of reactive stroma (Fig. 5D). In addition, the well-organized smooth muscle layer surrounding the epithelial cells became fragmented with increased vimentin-positive fibroblast-like cells (Fig. 5E). Consistently, the F9TG prostates also exhibited a disrupted stromal compartment (Fig. 2Ff). Together, the results indicate that overexpression of FGF9 promotes growth and metastasis of PCa, in part by remodeling tumor microenvironment.
Depletion of FGF9 suppresses tumorigenicity of TRAMP tumor cells. A. Real-time RT-PCR analyses for Fgf9 expression in TRAMP-C2 cells and FGF9 depleted TRAMP-C2 cells. B. Subcutaneous grafts of TRAMP-C2/UGM cells with or without FGF9 depletion in TRAMP-C2 cells. Graft weight is mean ± sd from 3 grafts per group. C. H&E and immunostaining of graft sections with the indicated antibodies. D. Masson Trichrome staining of prostate sections of 64-month-old TRAMP or F9TRAMP mice, demonstrating excessive collagen deposition in the stromal compartment of F9TRAMP mice. E. Co-staining of α-smooth muscle actin and vimentin in 4-month-old TRAMP and F9TRAMP prostates. UGM, urogenital mesenchymal cells; Ctrl, control; KD, knockdown; scale bar, 50 µm.
FGF9 augments formation of reactive stroma by promoting TGFβ1 signaling
To investigate how overexpression of FGF9 contributes to the two-way communication between the epithelium and the stroma in human PCa, LNCaP cells were forced to express FGF9 by transfection (Fig. 6A). The transfected cells were separated into high FGF9-expressing (FGF9high) and low FGF9-expressing (FGF9low) groups by limited dilution (Fig. 6A). The migration activity of the cells were then assessed with the transwell migration assay. The cells were seeded in the upper chamber of transwell dishes with or without HPS-19I human prostate stromal cells seeded in the bottom chambers (Fig. 6B). After incubation for 24 hours, LNCaP cells that migrated through the membrane were stained and counted. Although both FGF9high and FGF9low cells exhibited a low baseline level of migration, FGF9high cells possessed a slightly higher migration rate than did FGF9low cells, which was consistent with a previous report (43). However, in the presence of HPS-19I cells, significantly more FGF9high cells migrated through the membrane than FGF9low cells (Fig. 6Be). The results indicate that FGF9 promotes HPS-19I cells to release secretory factors that promote LNCaP cell migration.
Overexpression of FGF9 promotes the two-way communication between human PCa and stromal cells. A. Real time RT-PCR analyses for Fgf9 expression in LNCaP cells. B. The indicated LNCaP cells were cultured with or without HPS-19I human prostate stromal cells in transwell assay. LNCaP cells migrating through the membrane were stained with hematoxylin (panels a-d), and the average numbers of cells from triplicate samples were calculated and presented as mean ± sd (panel e). C. Real time RT-PCR analysis of LNCaP cells with or without HPS-19I cell coculture (a), with or without FGF9 treatment (b), or HPS-19I cells cocultured with FGF9high or FGF9low cells (c). FGF9high, high FGF9-expressing LNCaP cells; FGF9low, low FGF9-expressing LNCaP cells; HPS, HPS-19I human prostate stromal cells; SMA, α-smooth muscle actin.
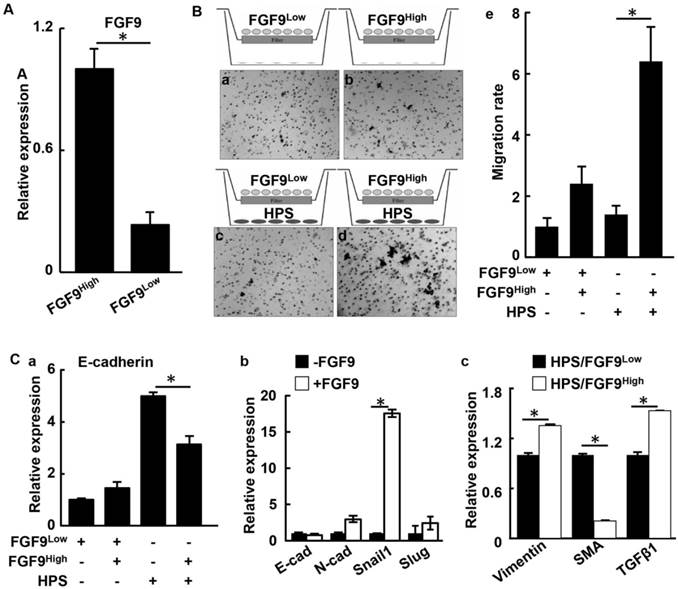
We next examined proteins that might promoted LNCaP migration. Expression of E-cadherin was equivalent between both LNCaP groups in the absence of HPS-19I cells. However, when they were cocultured with HPS-19I cells, FGF9high cells expressed E-cadherin at a lower level than did FGF9low cells (Fig. 6Ca). This suggests that overexpression of FGF9 in LNCaP cells induced HPS-19I cells to release factors that, in turn, promoted EMT in LNCaP cells. LNCaP cells treated with exogenous FGF9 showed an increase of Snail1 expression (Fig. 6Cb). Real-time RT-PCR analyses showed that coculture with LNCaP also changed the expression pattern of genes in HPS-19I cells in an FGF9 expression level-dependent manner (Fig. 6Cc). Among the upregulated genes was TGFβ1, a key signaling molecule expressed in the reactive stroma. Reactive stromal is a reservoir of multiple stromal derived factors, including FGF2, CTGF, BMP6, and IL6 (44-46).
Similarly, isolated stromal cells from the F9TG prostate expressed TGFβ1 at higher levels than stromal cells from non-transgenic mice (Fig. 7Aa). In addition, treating wildtype prostate stromal cells with FGF9 increased TGFβ1 expression at the mRNA levels within 6 hours, further demonstrating upregulation of TGFβ1 by FGF9 at the transcriptional level (Fig. 7Ab). Blocking FGFR kinase activity with FGFR inhibitors diminished the induction effect of FGF9 on TGFβ1 expression (Fig. 7Ac). To further determine whether FGF9 upregulated TGFβ1 expression, 293T cells were transfected with a luciferase reporter, the expression which was driven by the TGFβ1 promoter. Treating the cells with FGF9 increased expression of the reporter (Fig. 7B). The results further indicate upregulation of TGFβ1 by FGF9 at the transcription level.
FGF9 promotes TGFβ1 expression in prostate stromal cells via upregulating cJun. A. Real time RT-PCR analysis of TGFβ1 expression demonstrating upregulation of TGF-β1 expression by FGF signaling. Panel a, TGFβ1 in F9TG and wildtype prostates; panel b, mouse prostate stromal cells treated with 10 μg/ml FGF9 for the indicated time; panel c, mouse prostate stromal cells treated with vehicle control (Ctrl) or FGFR inhibitors (FGFRi) and then 10 μg/ml FGF9. B. Luciferase reporter assay in 293T cells showing that FGF9 enhanceed TGFβ1 promoter activity. C. ChIP analysis showing FGF9 enhanced the binding of cJun to the TGFβ1 promoter in primary prostate stromal cells. Left panel, real time PCR analysis; right panel, agarose gel electrophoresis showing specific DNA bands. D. Western blot analyses of cJun and TGFβ1 expression in primary prostate stromal cells with or without FGF9 stimulation. E. Real time RT-PCR analysis showing that cJun downregulation in HPS-19I cells suppressed TGFβ1 expression. F. Real time RT-PCR analysis showing that FGFR inhibitor abrogated the induction of cJun expression by FGF9. G. Western blot analysis demonstrating expression of the indicated proteins in WT, F9TG, TRAMP, F9TRAMP prostates at indicated ages. H. Pearson correlation analysis of the TCGA dataset (downloaded from cBioPortal database) revealed a positive correlation between FGF9 and TGFβ1 (panel a) or vimentin (panel b) mRNA expression in human PCa. I, Pearson correlation analysis of another genome-wide study (47) revealed that the expression of FGF9 is positively correlated with TGFβ1, Twist1 and Smad2 expression. Data are mean ± sd from triplicate samples. Ctrl, control; FGFRi, FGFR inhibitor; F, F9TG; W, wildtype; T, TRAMP; FT, F9TRAMP; *, P< 0.05.
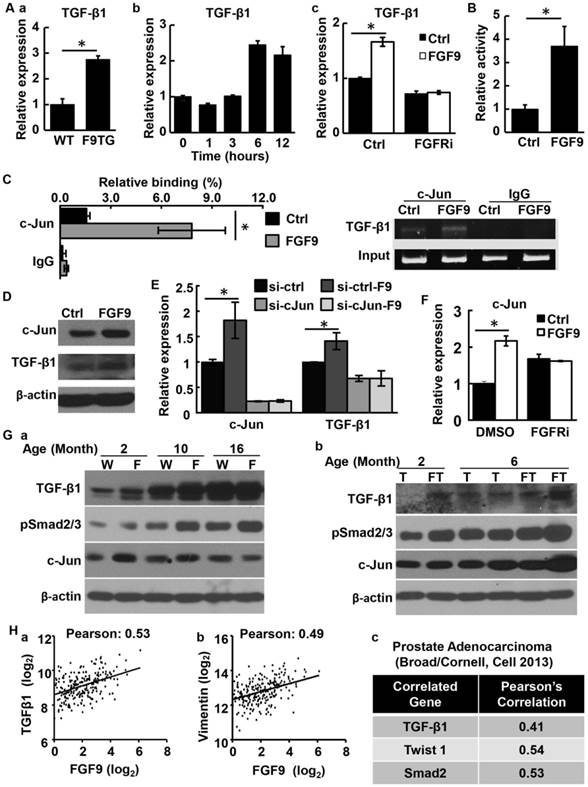
To identify the transcription factor(s) that mediated the induction of TGFβ1 expression by FGF9 signaling, TFSEARCH (http://diyhpl.us/~bryan/irc/protocol-online/protocol-cache/TFSEARCH.html) was used to predict the binding sites on the TGFβ1 promoter region for transcription factors that could be regulated by FGF signaling. Two cJun binding sites were found within a 2 kb proximal region of the TGFβ1 promoter. Treating mouse primary prostate stromal cells with FGF9 increased the binding of cJun to the TGFβ1 promoter (Fig. 7C), suggesting that cJun activation was involved in the upregulation of TGFβ1 expression by FGF9. Moreover, FGF9 promoted both cJun and TGFβ1 expression at the protein level in mouse primary prostate stromal cells (Fig. 7D). In line with this finding, depletion of cJun expression by siRNA compromised the induction of TGFβ1 expression by FGF9 (Fig. 7E). Inhibition of FGFR activity also diminished cJun expression induced by FGF9 (Fig. 7F). Consistent with the cell culture data, both F9TG and F9TRAMP prostates exhibited a higher expression level of cJun and TGFβ1, as well as higher levels of phosphorylated Smad2/3 that was an indication of TGFβ activation (Fig. 7G). Together, the study demonstrates that overexpression of FGF9 in PCa cells induces expression of TGFβ1 via cJun expression and activation in prostate stromal cells, which, in turn, promotes PCa cell migration.
To determine whether the level of FGF9 was correlated with TGFβ1 expression and stromal remoldering in human PCa, in silico analyses was carried out with data from the TCGA (cBioPortal) dataset. Expression of FGF9 in human PCa was positively correlated with TGFβ1 and vimentin expression (Fig. 7Ha,b). Furthermore, in a separate dataset including genome-wide sequencing of 57 prostate tumors and matched non-cancerous tissues (47), expression of Fgf9 was positively correlated with that of TGFβ1, Twist1, and Smad2 (Fig. 7Hc). This further indicates that FGF9 promotes EMT and reactive stroma formation in PCa.
Discussion
Aberrantly expressed FGF9 has oncogenic activity in various human cancers. Overexpression of FGF9 in human PCa predicts high bone metastasis and biochemical recurrence of the tumor (26, 35). However, how FGF9 promotes tumor epithelial-stromal interactions that contribute to PCa progression is not clear. Herein, we report that forced expression of FGF9 in prostate epithelial cells disrupts homeostasis prostate tissue of the in a time- and expression level-dependent manner. Overexpression of FGF9 promotes PCa progression and metastasis the TRAMP mouse prostate tumor model. It augments the formation of reactive stroma and increases the production of TGFβ1 in prostate stroma via increasing cJun expression and activation. Moreover, the expression level of FGF9 is positively associated with that of cJun, TGFβ1, and Smad2 in human PCa. Together, our data show that overexpression of FGF9 leads to invasive adenocarcinoma, and accelerates PCa progression in conjunction with additional genetic aberrations. Therefore, suppression of aberrant FGF signaling has the potential to constrain PCa progression.
FGF9 expression in epithelial cells of normal prostates is low. However, both human and mouse PCa frequently have a high FGF9 expression at advanced stages. In fact, our data showed that at late stages, expression of endogenous FGF9 was frequently comparable to that of the FGF9 transgene. This suggests that activation of endogenous FGF9 expression confers PCa cell growth and progression. Treating animals with neutralizing antibodies against FGF9 reduces bone metastasis in preclinical PCa xenograft model (26). Thus, although the F9TRAMP mice developed prostate lesions earlier than TRAMP mice, the difference in PCa progression in both strains at late stages was diminishing, likely due to the expression of endogenous FGF9 at high levels. Therefore, overexpression of FGF9 can serve as a biomarker for PCa diagnosis and prognosis. Inhibition of FGF9 signaling shall be of therapeutic values for the PCa with high FGF9 expression.
Bidirectional communication between epithelial cells and stromal cells is influential in carcinogenesis. Generation of reactive stroma promotes tumorigenesis in many human cancers, including PCa (6, 7). The reactive stroma serves as a reservoir of extracellular matrix, inflammatory cells, nerve cells, angiogenic cells, smooth muscle cells, fibroblasts, cytokines, and other factors that promote tumor progression (7). Elevated expression of TGFβ1 is a characteristic change in reactive stroma and is required for remodeling the tumor microenvironment (6, 48). Overexpression of FGF9 in PCa cells potentiated reactive stromal responses, including increased proliferation, inflammation, and expression of TGFβ1. It reduced smooth muscle actin (SMA)-expressing cells and increased the vimentin positive cell population in the tumor. Moreover, expression of slug was increased in F9TRAMP tumors, suggesting that overexpression of FGF9 also promoted tumor EMT. TGFβ1 is overexpressed in PCa metastases and bone marrow, which might explain why bone metastatic prostate cancer has a high TGFβ1 level. However, whether FGF9 could induce TGFβ1 in the epithelial cells is still unknown.
Rat Dunning prostate tumors have two types of prostate stromal cells: DTS1 expresses high levels of FGFR1 and no SMA and DTS2 expresses high levels of FGFR3 and SMA (49, 50). When co-implanted with PCa cells, DTS2 cells reduce tumor incidence and size relative to DTS1 cells (51). Forced expression of FGFR3 in DTS1 cells restrains PCa tumor growth. This highlights the importance of the two-way communications between stroma and epithelium mediated by signaling within the FGFR family during progression to malignancy. Although overexpression of FGF9 is associated with bone metastasis of human PCa, we did not observe any bone metastasis in the F9TRAMP mouse model albeit the mice developed more aggressive tumors than TRAMP littermates. What additional factors are required for PCa bone metastasis? In addition, a number of key questions also remain to be addressed. FGF9 binds to IIIc isoforms of FGFR1-3, and FGFR3IIIb isoform at low affinity (52, 53). How do the two FGFRs expressed in prostate stroma, FGFR1 and FGFR3, elicit distinct signals to control tissue homeostasis or induce a reactive stromal response? Are there receptor-specific pathways downstream of FGFR1 or FGFR3? How does overexpression of FGF9 diminish SMA-positive cells in TRAMP tumors (Fig. 4E)? Answers to these questions are important for translational applications of FGF pathway-related therapies since they determine whether we can treat PCa patients simply with general FGFR inhibitors or only with receptor isoform- or pathway-specific inhibitors.
In summary, our study revealed the dual roles of FGF9 in PCa progression: promoting proliferation and EMT of epithelial cell as well as augmenting reactive stroma formation. Thus targeting FGF signaling will be of therapeutic future for constraining PCa progression and metastasis.
Abbreviations
FGF, fibroblast growth factor; FGFR, FGF receptor; FRS2α, FGFR substrate 2α; F9TG, FGF9 transgenic mouse; PCa, prostate cancer; PIN, prostatic intraepithelial neoplasia.
Acknowledgements
We thank Dr. David Rowley for sharing HPS-19I human prostate stromal cells. We thank Dr. Stefan Siwko for critical reading of the manuscript. This work was supported in part by the National Institutes of Health CA96824 and DE023106 to FW, CA140388 to NN, WLM and FW, and The Cancer Prevention and Research Institution of Texas CPRIT110555 to FW and WLM, and the National Natural Science Foundation of China 81101712, 31371470, and 81270761 to CW.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Logothetis CJ, Lin SH. Osteoblasts in prostate cancer metastasis to bone. Nature reviews Cancer. 2005;5(1):21-8 doi: 10.1038/nrc1528. PubMed PMID: 15630412
2. Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes & development. 2010;24(18):1967-2000 Epub 2010/09/17. doi: 10.1101/gad.1965810. PubMed PMID: 20844012; PubMed Central PMCID: PMCPmc2939361
3. Cunha GR, Cooke PS, Kurita T. Role of stromal-epithelial interactions in hormonal responses. Arch Histol Cytol. 2004;67(5):417-34 PubMed PMID: 15781983
4. Cunha GR, Ricke W, Thomson A, Marker PC, Risbridger G, Hayward SW. et al. Hormonal, cellular, and molecular regulation of normal and neoplastic prostatic development. J Steroid Biochem Mol Biol. 2004;92(4):221-36 PubMed PMID: 15663986
5. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432(7015):332-7 Epub 2004/11/19. doi: 10.1038/nature03096. PubMed PMID: 15549095; PubMed Central PMCID: PMCPmc3050735
6. Tuxhorn JA, Ayala GE, Smith MJ, Smith VC, Dang TD, Rowley DR. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clinical cancer research: an official journal of the American Association for Cancer Research. 2002;8(9):2912-23 Epub 2002/09/17. PubMed PMID: 12231536
7. Barron DA, Rowley DR. The reactive stroma microenvironment and prostate cancer progression. Endocrine-related cancer. 2012;19(6):R187-204 Epub 2012/08/30. doi: 10.1530/ERC-12-0085. PubMed PMID: 22930558; PubMed Central PMCID: PMC3716392
8. Tuxhorn JA, McAlhany SJ, Yang F, Dang TD, Rowley DR. Inhibition of transforming growth factor-beta activity decreases angiogenesis in a human prostate cancer-reactive stroma xenograft model. Cancer research. 2002;62(21):6021-5 Epub 2002/11/05. PubMed PMID: 12414622
9. Yang F, Strand DW, Rowley DR. Fibroblast growth factor-2 mediates transforming growth factor-beta action in prostate cancer reactive stroma. Oncogene. 2008;27(4):450-9 doi: 10.1038/sj.onc.1210663. PubMed PMID: 17637743
10. Franco OE, Jiang M, Strand DW, Peacock J, Fernandez S, Jackson RS 2nd. et al. Altered TGF-beta signaling in a subpopulation of human stromal cells promotes prostatic carcinogenesis. Cancer research. 2011;71(4):1272-81 doi: 10.1158/0008-5472.CAN-10-3142. PubMed PMID: 21303979; PubMed Central PMCID: PMC3076790
11. Lin Y, Liu G, Zhang Y, Hu YP, Yu K, Lin C. et al. Fibroblast growth factor receptor 2 tyrosine kinase is required for prostatic morphogenesis and the acquisition of strict androgen dependency for adult tissue homeostasis. Development. 2007;134(4):723-34 PubMed PMID: 17215304
12. Zhang Y, Zhang J, Lin Y, Lan Y, Lin C, Xuan JW. et al. Role of epithelial cell fibroblast growth factor receptor substrate 2{alpha} in prostate development, regeneration and tumorigenesis. Development. 2008;135(4):775-84 PubMed PMID: 18184727
13. Wang F, Luo Y, McKeehan W. The FGF signaling axis in prostate tumorigenesis. In: (ed.) E. G, C. S, F. R. Molecular Oncology: Causes of Cancer and Targets for Treatment. London Cambridge University Press. 2013 p. 186-9
14. Corn PG, Wang F, McKeehan WL, Navone N. Targeting fibroblast growth factor pathways in prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19(21):5856-66 doi: 10.1158/1078-0432.CCR-13-1550. PubMed PMID: 24052019
15. Thomson AA. Role of androgens and fibroblast growth factors in prostatic development. Reproduction. 2001;121(2):187-95 PubMed PMID: 11226043
16. Gowardhan B, Douglas DA, Mathers ME, McKie AB, McCracken SR, Robson CN. et al. Evaluation of the fibroblast growth factor system as a potential target for therapy in human prostate cancer. British journal of cancer. 2005;92(2):320-7 PubMed PMID: 15655558
17. Abate-Shen C, Shen MM. FGF signaling in prostate tumorigenesis--new insights into epithelial-stromal interactions. Cancer Cell. 2007;12(6):495-7 PubMed PMID: 18068626
18. Acevedo VD, Gangula RD, Freeman KW, Li R, Zhang Y, Wang F. et al. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell. 2007;12(6):559-71 PubMed PMID: 18068632
19. Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. The Biochemical journal. 2011;437(2):199-213 doi: 10.1042/BJ20101603. PubMed PMID: 21711248
20. McKeehan WL, Wang F, Luo Y. The fibroblast growth factor (FGF) signaling complex. Handbook of Cell Signaling. 2dn ed. Bradshaw R, Dennis E, editors. New York: Academic/Elsevier Press. 2009
21. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS. et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11-22 doi: 10.1016/j.ccr.2010.05.026. PubMed PMID: 20579941; PubMed Central PMCID: PMC3198787
22. Giri D, Ropiquet F, Ittmann M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 1999;5(5):1063-71 PubMed PMID: 10353739
23. Ozen M, Giri D, Ropiquet F, Mansukhani A, Ittmann M. Role of fibroblast growth factor receptor signaling in prostate cancer cell survival. Journal of the National Cancer Institute. 2001;93(23):1783-90 PubMed PMID: 11734594
24. Devilard E, Bladou F, Ramuz O, Karsenty G, Dales JP, Gravis G. et al. FGFR1 and WT1 are markers of human prostate cancer progression. BMC cancer. 2006;6:272. PubMed PMID: 17137506
25. Wang J, Stockton DW, Ittmann M. The fibroblast growth factor receptor-4 Arg388 allele is associated with prostate cancer initiation and progression. Clinical cancer research: an official journal of the American Association for Cancer Research. 2004;10(18 Pt 1):6169-78 PubMed PMID: 15448004
26. Li ZG, Mathew P, Yang J, Starbuck MW, Zurita AJ, Liu J. et al. Androgen receptor-negative human prostate cancer cells induce osteogenesis in mice through FGF9-mediated mechanisms. The Journal of clinical investigation. 2008;118(8):2697-710 PubMed PMID: 18618013
27. Jin C, McKeehan K, Guo W, Jauma S, Ittmann MM, Foster B. et al. Cooperation between ectopic FGFR1 and depression of FGFR2 in induction of prostatic intraepithelial neoplasia in the mouse prostate. Cancer research. 2003;63(24):8784-90 PubMed PMID: 14695195
28. Wang F, McKeehan K, Yu C, Ittmann M, McKeehan WL. Chronic activity of ectopic type 1 fibroblast growth factor receptor tyrosine kinase in prostate epithelium results in hyperplasia accompanied by intraepithelial neoplasia. The Prostate. 2004;58(1):1-12 PubMed PMID: 14673947
29. Song Z, Powell WC, Kasahara N, van Bokhoven A, Miller GJ, Roy-Burman P. The effect of fibroblast growth factor 8, isoform b, on the biology of prostate carcinoma cells and their interaction with stromal cells. Cancer research. 2000;60(23):6730-6 PubMed PMID: 11118059
30. Memarzadeh S, Xin L, Mulholland DJ, Mansukhani A, Wu H, Teitell MA. et al. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell. 2007;12(6):572-85 PubMed PMID: 18068633
31. Giri D, Ropiquet F, Ittmann M. FGF9 is an autocrine and paracrine prostatic growth factor expressed by prostatic stromal cells. Journal of cellular physiology. 1999;180(1):53-60 PubMed PMID: 10362017
32. Polnaszek N, Kwabi-Addo B, Peterson LE, Ozen M, Greenberg NM, Ortega S. et al. Fibroblast growth factor 2 promotes tumor progression in an autochthonous mouse model of prostate cancer. Cancer research. 2003;63(18):5754-60 PubMed PMID: 14522896
33. Valta MP, Tuomela J, Bjartell A, Valve E, Vaananen HK, Harkonen P. FGF-8 is involved in bone metastasis of prostate cancer. International journal of cancer Journal international du cancer. 2008. PubMed. PMID: 18386787
34. Yang F, Zhang Y, Ressler SJ, Ittmann MM, Ayala GE, Dang TD. et al. FGFR1 is essential for prostate cancer progression and metastasis. Cancer research. 2013;73(12):3716-24 Epub 2013/04/12. doi: 10.1158/0008-5472.CAN-12-3274. PubMed PMID: 23576558; PubMed Central PMCID: PMC3686853
35. Teishima J, Shoji K, Hayashi T, Miyamoto K, Ohara S, Matsubara A. Relationship between the localization of fibroblast growth factor 9 in prostate cancer cells and postoperative recurrence. Prostate cancer and prostatic diseases. 2012;15(1):8-14 Epub 2011/10/19. doi: 10.1038/pcan.2011.48. PubMed PMID: 22006051
36. Zhang Y, Zhang J, Lin Y, Lan Y, Lin C, Xuan JW. et al. Role of epithelial cell fibroblast growth factor receptor substrate 2alpha in prostate development, regeneration and tumorigenesis. Development. 2008;135(4):775-84 doi: 10.1242/dev.009910. PubMed PMID: 18184727
37. Gingrich JR, Barrios RJ, Foster BA, Greenberg NM. Pathologic progression of autochthonous prostate cancer in the TRAMP model. Prostate cancer and prostatic diseases. 1999;2(2):70-5 Epub 2002/12/24. doi: 10.1038/sj.pcan.4500296. PubMed PMID: 12496841
38. Suttie A, Nyska A, Haseman JK, Moser GJ, Hackett TR, Goldsworthy TL. A grading scheme for the assessment of proliferative lesions of the mouse prostate in the TRAMP model. Toxicologic pathology. 2003;31(1):31-8 Epub 2003/02/25. PubMed PMID: 12597447
39. Xin L, Ide H, Kim Y, Dubey P, Witte ON. In vivo regeneration of murine prostate from dissociated cell populations of postnatal epithelia and urogenital sinus mesenchyme. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(Suppl 1):11896-903 PubMed PMID: 12909713
40. Qi W, Gao S, Wang Z. Transcriptional regulation of the TGF-beta1 promoter by androgen receptor. The Biochemical journal. 2008;416(3):453-62 Epub 2008/07/25. doi: 10.1042/bj20080651. PubMed PMID: 18651839
41. Grisanzio C, Signoretti S. p63 in prostate biology and pathology. Journal of cellular biochemistry. 2008;103(5):1354-68 doi: 10.1002/jcb.21555. PubMed PMID: 17879953
42. Humphrey PA. Diagnosis of adenocarcinoma in prostate needle biopsy tissue. Journal of clinical pathology. 2007;60(1):35-42 doi: 10.1136/jcp.2005.036442. PubMed PMID: 17213347; PubMed Central PMCID: PMC1860598
43. Teishima J, Yano S, Shoji K, Hayashi T, Goto K, Kitano H. et al. Accumulation of FGF9 in prostate cancer correlates with epithelial-to-mesenchymal transition and induction of VEGF-A expression. Anticancer research. 2014;34(2):695-700 Epub 2014/02/11. PubMed PMID: 24511001
44. Strand DW, Liang YY, Yang F, Barron DA, Ressler SJ, Schauer IG. et al. TGF-beta induction of FGF-2 expression in stromal cells requires integrated smad3 and MAPK pathways. American journal of clinical and experimental urology. 2014;2(3):239-48 Epub 2014/11/07. PubMed PMID: 25374926; PubMed Central PMCID: PMCPmc4219310
45. Yang F, Tuxhorn JA, Ressler SJ, McAlhany SJ, Dang TD, Rowley DR. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer research. 2005;65(19):8887-95 Epub 2005/10/06. doi: 10.1158/0008-5472.can-05-1702. PubMed PMID: 16204060
46. Yang F, Chen Y, Shen T, Guo D, Dakhova O, Ittmann MM. et al. Stromal TGF-beta signaling induces AR activation in prostate cancer. Oncotarget. 2014;5(21):10854-69 Epub 2014/10/22. PubMed PMID: 25333263; PubMed Central PMCID: PMCPmc4279415
47. Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y. et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153(3):666-77 doi: 10.1016/j.cell.2013.03.021. PubMed PMID: 23622249; PubMed Central PMCID: PMC3690918
48. Rowley DR. What might a stromal response mean to prostate cancer progression? Cancer metastasis reviews. 1998;17(4):411-9 Epub 1999/08/24. PubMed PMID: 10453285
49. Wu X, Jin C, Wang F, Yu C, McKeehan WL. Stromal cell heterogeneity in fibroblast growth factor-mediated stromal-epithelial cell cross-talk in premalignant prostate tumors. Cancer research. 2003;63(16):4936-44 PubMed PMID: 12941818
50. Jin C, Wang F, Wu X, Yu C, Luo Y, McKeehan WL. Directionally specific paracrine communication mediated by epithelial FGF9 to stromal FGFR3 in two-compartment premalignant prostate tumors. Cancer research. 2004;64(13):4555-62 PubMed PMID: 15231666
51. Jin C, Yang C, Wu X, Wang F, McKeehan WL. FGFR3-expressing smooth muscle-like stromal cells differentiate in response to FGFR2IIIb-expressing prostate tumor cells and delay tumor progression. In vitro cellular & developmental biology Animal. 2011;47(7):500-5 doi: 10.1007/s11626-011-9432-5. PubMed PMID: 21691921; PubMed Central PMCID: PMC3471788
52. Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. The Journal of biological chemistry. 2006;281(23):15694-700 PubMed PMID: 16597617
53. Santos-Ocampo S, Colvin JS, Chellaiah A, Ornitz DM. Expression and biological activity of mouse fibroblast growth factor-9. The Journal of biological chemistry. 1996;271(3):1726-31 PubMed PMID: 8576175
Author contact
![]() Corresponding author: Fen Wang, Center for Cancer and Stem Cell Biology, Institute of Biosciences and Technology, Texas A&M Health Science Center, Houston, TX 77030-3303. E-mail: fwangtamhsc.edu
Corresponding author: Fen Wang, Center for Cancer and Stem Cell Biology, Institute of Biosciences and Technology, Texas A&M Health Science Center, Houston, TX 77030-3303. E-mail: fwangtamhsc.edu