Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(11):1411-1425. doi:10.7150/ijbs.26086 This issue Cite
Research Paper
Polydatin attenuates diet-induced nonalcoholic steatohepatitis and fibrosis in mice
Rui Li1, Jingzhi Li3, Yiji Huang1, Hui Li2, Sishan Yan1, Jiaxin Lin1, Ying Chen1, Limin Wu4, Bing Liu1, Genshu Wang2 ![]() , Tian Lan1
, Tian Lan1 ![]()
1. Department of Pharmacology, School of Pharmacy, Guangdong Pharmaceutical University, Guangzhou 510006, China.
2. Department of Hepatic Surgery and Liver transplantation Center of the Third Affiliated Hospital, Sun Yat-sen University; Guangzhou 510630, China.
3. School of Nursing, Guangdong Pharmaceutical University, Guangzhou 510006, China.
4. Guangdong ShowYong Nature Medical Technology Co., Ltd., Foshan 528000, China
Received 2018-3-15; Accepted 2018-7-8; Published 2018-7-30
Abstract

Scope: Non-alcoholic steatohepatitis (NASH) is characterized by lipid accumulation in hepatocytes and inflammatory cell infiltration.
In view of the anti-oxidative and anti-inflammatory effects of polydatin, the current study aimed to investigate the pharmacological effects of polydatin on NASH and its related fibrosis.
Methods: C57BL/6 mice were fed with methionine-choline deficient (MCD) diet to induce NASH and liver fibrosis, and treated with or without polydatin (5 mg/kg, every other day, i.p) for 4 weeks. HepG2 cells induced by palmitic acid (PA) were treated with polydatin.
Results: The elevations of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST), active caspase-3, TUNEL-positive cells, and triglyceride content were decreased by polydatin treatment. In addition, administration of polydatin to MCD-fed mice reduced oxidative stress by down-regulating NOX4 enzymes. Furthermore, the reduction in inflammation and CD68 macrophage activation correlated with inhibition of toll-like receptor (TLR)-4/NF-κB p65 signaling pathway by polydatin treatment. Polydatin also attenuated lipid accumulation, inflammation and apoptosis in HepG2 cells challenged by palmitic acid (PA) combined with or without lipopolysaccharide (LPS). Finally, the reduction of hepatic fibrosis by polydatin treatment corresponded to a reduction in hepatic gene expression of fibrosis markers.
Conclusions: These results suggest that polydatin prevents NASH and fibrosis via inhibition of oxidative stress and inflammation, highlighting polydatin as a potential therapeutic agent for prevention and treatment of NASH.
Keywords: Polydatin, NASH, Inflammation, Oxidative stress, Apoptosis, Liver fibrosis.
Introduction
With the increasing incidence of obesity, diabetes, and metabolic syndrome in the general population, nonalcoholic fatty liver diseases (NAFLD) is emerging as one of the most common causes of chronic liver disease. NAFLD encompasses a wide spectrum of liver damage ranging from benign lipid accumulation in the liver (steatosis) to nonalcoholic steatohepatitis (NASH) characterized by fat vacuoles, lobular inflammation and hepatocyte damage in the form of ballooning and apoptosis [1]. As an advanced stage of NAFLD, NASH patients are at risk of disease progression that leads to liver fibrosis, cirrhosis and hepatocellular carcinoma (HCC). The causes of progression from steatosis to NASH is involved in lipotoxicity, oxidative stress and inflammation. Although many attempts were made to find an appropriate molecular target and related chemical drugs to treat NAFLD, there is no effective therapy approved for NAFLD yet. Thus, novel and effective therapies to improve the prognosis of NASH is urgently needed. It has been demonstrated that necroinflammatory process is attributed to an initial metabolic disturbance that increases the influx of free fatty acids and lipid synthesis, followed by inflammation, oxidative stress and lipid peroxidation [2, 3]. Oxidative stress reflects an imbalance between the production and clearance of reactive oxygen species (ROS) which leads to deviant tissue repair in the liver. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidative (NOX) is an enzyme system that catalyzes the overproduction of ROS under pathological conditions. Among the NOX family, abnormal expression of NOX4 has been implicated in oxidative stress in patients with NASH and mice with diet-induced steatohepatitis [3]. Thus, treatment of targeting NOX may be a potential therapy for NASH.
Apart from oxidative stress, inflammation also plays a prominent role in NASH. Increased levels of blood lipopolysaccharide (LPS), a well-known inducer of inflammation, are often observed in patients with NASH [4, 5]. Toll-like receptor 4 (TLR4) is a pattern-recognition receptor for LPS and induces the activation of innate immune signaling through adaptor proteins MyD88 and TIR-domain containing adaptor-inducing interferon-b (TRIF) [6]. Previous studies have clearly verified the critical roles of TLR4 and MyD88 in promoting NASH and its related fibrosis [7, 8]. These results well demonstrated the relevant roles of TLR4 and ROS signaling in promoting the progression of NASH.
Polydatin, a resveratrol glucoside (resveratrol-3-O-β-mono-D-glucoside), is an active component isolated from the roots of Polygonum with a cuspidatum Sieb. et Zucc. Unlike resveratrol, which passively penetrates cells, polydatin enters cells via an active mechanism using glucose carrier [9]. Additionally, polydatin is more resistant to enzymatic oxidation than resveratrol and possesses much better solubility in water.
We previously found that polydatin protects against liver fibrosis in mice through inhibition of oxidative stress and inflammation. Recently, One group suggested that polydatin may ameliorate diet-induced NAFLD in rat through inhibition of insulin resistance and lipid metabolism [10, 11]. However, the therapeutic role of polydatin in methionine- and choline-deficient (MCD)-induced hepatic steatohepatitis and fibrosis, as well as its underlying mechanism, especially it's relating to anti-oxidant and anti-inflammatory function, has not been clearly defined. This study therefore evaluates the effects of polydatin on steatohepatitis and fibrosis in mice induced by MCD diet and HepG2 cells induced by palmitic acid (PA).
Materials and methods
Materials and reagents
Polydatin, Oil red O, lipopolysaccharide (LPS) and dimethyl sulfoxide (DMSO) were purchased from Sigma (St. Louis, CA). Dulbecco's modified Eagle's medium (DMEM) were from Invitrogen Life Technologies (Carlsbad, CA, USA). Dihydroethidium (DHE) Fetal bovine serum (FBS), penicillin/streptomycin, solution 0.25% Trypsin-EDTA, phosphate-buffered-saline (PBS) were from Invitrogen Life Technologies (Carlsbad, CA, USA). TRIzol reagent were from TransGen Biotech (Beijing, China). One Step TUNEL Apoptosis Assay Kit was from Beyotime biotechnology. Anti-α-SMA, anti-CD68, anti-β-actin, anti-α-tubulin and anti-GAPDH antibodies were from Boster Biological Technology Co, Ltd. (Wuhan, China); Anti-caspase3 was from Cell Signaling Technology, Inc. (Beverly, MA, USA); Anti-4HNE was from Abcam Inc. (Cambridge, MA); Anti-TLR4 antibody was from Proteintech Group, Inc. (Proteintech, Rosemont, USA). ROS Fluorescent Probe-DHE, Alexa Fluor 488 and 594-conjugated secondary antibodies were from Invitrogen Life Technologies (Carlsbad, CA, USA). Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG and anti-rabbit IgG were purchased from Zhongshan Golden Bridge Biotechnology Co, Ltd. (Beijing, China). Annexin V-FITC kit and caspase-3 activity kit were from BestBio (Shanghai, China). Enhanced chemiluminescence (ECL) substrate for detection of HRP was obtained from Bio-Rad Laboratories. Protease Inhibitor Cocktail Kit was obtained from Pierce Thermo Scientific (Rockford, USA).
Animals and treatment
All animal procedures were conducted in accordance with the China Animal Welfare Legislation and were approved by the Ethics Committee on the Care and Use of Laboratory Animals in Guangdong Pharmaceutical University (Guangzhou, China). C57BL/6 mice (28 male, 10-week-old, 25-27 g) were purchased from Experimental Animals Center of Guangdong Province, China. The mice were housed in a temperature-controlled environment (22±2 °C) under standard 12h light/dark conditions and received food and water ad libitum. Mice were fed with methionine-choline deficient diet (MCD) for 4 weeks to induce NASH (n=10). Mice were fed with methionine and choline supplemented (MCS) as control (n=8). In treatment group (n=10), mice were intraperitoneally injected with polydatin (5 mg/kg, dissolved in saline) following each MCD diet induced. Mice in untreated groups were administrated with the same volume of saline as vehicle control.
Serum Biochemistry
Serum levels of alanine aminotransferase (ALT), triglycerides (TG) and aspartate aminotransferase (AST) were measured using standard enzymatic procedures according to the manufacture's instruction (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Hydroxyproline assay
Hepatic hydroxyproline content was measured using commercial hydroxyproline assay kit according to the manufacturer's instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Briefly, liver samples were hydrolyzed at 95 ℃ for 20 min, then adjusted to pH 6.5 and filtered through activated charcoal. After centrifugation, the supernatant was mixed with detecting liquid and incubated at 60 ℃ for 15 min. Last, samples were measured using a microplate reader at 550 nm.
Caspase3 activity analysis
Hepatic caspase3 activity was measured using caspase3 activity assay kit according to the manufacturer's instructions (Bestbio, China). Briefly, liver tissues were grinded and centrifuged in 12000 rpm, the supernatant was detected at 405nm.
Cell culture and treatment
Human hepatocellular carcinoma cell line HepG2 (Shanghai Institute of Biochemistry and Cell Biology, Shanghai, China) was cultured in Cells DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin in a 5% CO2 incubator at 37°C. Stock solutions of 5 mM palmatic acid (PA)/5% BSA were heated for 15 min at 55°C and cooled to room temperature to obtain the PA-BSA complex. The PA-BSA mixture was added to serum-containing cell culture medium to final concentration of 250 μM. Cells were starvation in serum-free DMEM for 12 h followed by PA induction for additional 24 h in the absence or presence of polydatin (5, 10, 20 µM)). HepG2 cells maintained in culture medium containing 5% BSA served as a control. Lactate dehydrogenase (LDH) and triglycerides (TG) were measured using standard enzymatic procedures according to the manufacture's instruction (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Western blot analysis
Liver tissues or cells were lysed in RIPA buffer. Lysates were centrifuged (12,000 x g for 20 min at 4˚C) and the supernatant was collected. Equal amounts of total proteins (30 µg) were fractionated by SDS-PAGE and then transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore Corp, Bedford, MA, USA). After blocked with 5% non-fat milk in Tris-buffered saline with Tween-20 for 1 h at room temperature, the membranes were incubated with primary antibodies at 4 ˚C overnight, followed by incubation with secondary antibodies (1:5000 dilutions) for 1 h at room temperature. Bands were detected by enhanced chemiluminescent (ECL) method (Bio-Rad, USA) and captured by chemiluminescence system (New Life Science Products, Boston, MA, USA).
Histopathology
Liver tissue was fixed in 10% formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) according to a standard procedure. Histological scoring of the liver lesions were assessed histologically by NASH activity score (NAS), as described [26]. NAS included assessments of three histological features: steatosis (0-3), hepatocellular ballooning (0-2) and lobular inflammation (0-3). Samples with scores >5 were designated as “NASH”, and samples with scores <3 were designated “not NASH”. For Oil red O staining, OCT-embedded frozen livers were sectioned at 10μm with a cryostat, slides or cells were fixed in 10% (v/v) formalin for 10 min, and stained with Oil Red O working solution (Sigma Aldrich, St. Louis, MO, USA).
Immunohistochemistry and immunofluorescent staining
Liver specimens fixed in 10% buffered formalin were embedded in paraffin blocks. Liver sections (4μm thick) were processed using a standard immunostaining protocol. For immunohistochemical analyses, liver sections were separated, rehydrated and sequentially incubated with primary antibodies and secondary antibody. The area of positive staining was measured in high-power (x20) fields on each slide and quantified using Image J software. For immunofluorescent staining, the livers were collected and fixed in 10% PBS buffered formalin for 24 h. The fixed samples were sequentially exposed to 10% and 30% sucrose in PBS for 10 h each and then embedded in Tissue Tek OTC compound (Sakura Finetek, Torance, CA). The liver sections were permeabilized by 0.25% Triton X-100 and incubated with primary antibody (1:100) overnight at 4˚C. Then, the liver sections were incubated with corresponding Alexa Fluor 488 and 594-conjugated secondary antibodies (1:500) for 1 h at room temperature and stained with DAPI (1 µg/mL) for 10 min. Finally, the sections were captured by Olympus BX51 microscope (Olympus Co, Tokyo, Japan).
Liver oxidative stress in mice
Superoxide generation was assessed by quantifying the conversion of dihydroethidium to ethidium. Images were obtained with Olympus BX51 microscope (Olympus Co, Tokyo, Japan). Fluorescence was detected with a 594-nm long-pass filter. Laser settings were identical for acquisition of images from all specimens.
Quantitative Real-time RT-PCR
Total RNA was extracted from mouse liver tissues or HepG2 cells with TRIzol reagent followed by treatment with RNase-free DNase (Takara, Dalian, China) for 30 min at 37°C. Total RNA was reverse transcribed to cDNA using a first-strand cDNA kit (Takara, Dalian, China) according to the manufacturers' instructions. Real-time RT-PCR was conducted using the Prime Script RT-PCR Kit (Takara, Dalian, China) according to the manufacturers' instructions. The PCR was run on a Step-One Plus Real-time PCR System (Applied Biosystems, Foster City, CA, USA). The PCR reactions were carried out at: 95°C for 30 s; and 40 cycles of 95°C/5 s, 60°C/30 s. Relative difference in fold change between an experimental and calibrator sample were calculated by using the comparative cycle threshold (2-ΔΔCt) method. GAPDH was used as internal control. Sequences of PCR primers were summarized in Table 1.
Primer sequences for real-time PCR
| Genes | Forward primer (5'-3') | Reverse primer (5'-3') |
|---|---|---|
| Mouse | ||
| IL-1β | TGCCACCTTTTGACAGTGATG | ATGTGCTGCTGCGAGATTTG |
| IL-6 | ACCAGAGGAAATTTTCAATAGGC | TGATGCACTTGCAGAAAACA |
| Col1α1 | TAGGCCATTGTGTATGCAGC | ACATGTTCAGCTTTGTGGACC |
| TGF-β1 | GTGGAAATCAACGGGATCAG | ACTTCCAACCCAGGTCCTTC |
| TIMP1 | AGGTGGTCTCGTTGATTTCT | GTAAGGCCTGTAGCTGTGCC |
| SREBP-1c | TAGAGCATATCCCCCAGGTG | GGTACGGGCCACAAGAAGTA |
| FAS | TCCTGGAACGAGAACACGATCT | GAGACGTGTCACTCCTGGACTTG |
| GAPDH | AGGAGTAAGAAACCCTGGAC | CTGGGATGGAATTGTGAG |
| Human | ||
| IL-1β | TTCGACACATGGGATAACGAGG | TTTTTGCTGTGAGTCCCGGAG |
| IL-6 | ACTCACCTCTTCAGAACGAATTG | CCATCTTTGGAAGGTTCAGGTTG |
| TNF-α | CCTGTAGCCCATGTTGTAGCAA | CCTTGAAGAGGACCTGGGAGTAG |
| SREBP-1c | GCCCCTGTAACGACCACTG | CAGCGAGTCTGCCTTGATG |
| FAS | AGATTGTGTGATGAAGGACATGG | TGTTGCTGGTGAGTGTGCATT |
| GAPDH | ACAACTTTGGTATCGTGGAAGG | GCCATCACGCCACAGTTTC |
Flow cytometric analysis of apoptosis
A fluorescein isothiocyanate (FITC) Annexin-V Apoptosis Detection Kit (Keygentec, China) was used according to the manufacturers' instruction to evaluate apoptosis. HepG2 cells were cultured in 6-well plates for 24 h, followed by incubation with vehicle, PA or polydatin. After 24 h, the HepG2 cells were harvested and washed with cold PBS. The cells were resuspended in 1ml of 1 x binding buffer. The resuspended cells (100 μl) were transferred to a 5ml culture tube, and Annexin V-FITC (5 μl) and propidium iodide (PI, 5 μl) were added. The cells were vortexed and incubated for 15 min in the dark. Binding buffer (400 μl) was added to each tube. Flow cytometric analysis was performed immediately after staining. Data acquisition and analysis were performed by a fluorescence-activated cell scanner (FACS) flow cytometer (Becton Dickinson, San Jose, CA, USA). Cells in the early stages of apoptosis were Annexin V-positive and PI-negative, whereas cells in the late stages of apoptosis were positive for both Annexin V and PI. PI stained the necrotic cells.
Statistical Analysis
All experiments were performed in at least triplicate and the results are expressed as mean ± standard deviation (SD). Statistical differences between two groups were analyzed by the unpaired student's t test and differences between multiple groups of data were analyzed by one-way ANOVA with Bonferroni correction (GraphPad Prism 5.0). P < 0.05 was considered statistically significant.
Completed tissue sections was scanned ten photos in sequence with 10x microscopic scale of each tissue and the positive area was analyzed by Image J software. Again, three random fields of each tissue section were selected for quantification and statistical analysis with GraphPad Prism 5.0.
Results
Polydatin attenuated liver steatosis and injury induced by MCD diet in mice
To investigate the hepatoprotective activity of polydatin in NASH, mice were induced by MCD diet for 4 weeks. Male age-matched mice fed a control (chow) diet, were included as a reference group in most assessments. As expected, a significant reduction in body weight by MCD diet feeding was observed (Fig. 1F).
Liver steatosis and injury were attenuated by polydatin in mice fed with MCD diet. Mice were fed with MCD diet or control diet for 4 weeks. Mice fed with MCD diet were intraperitoneal injected with 5 mg/kg polydatin daily in the period of experiment. (A-B) Representative histology of HE and Oil red O staining. (C) Histological analysis of steatosis, ballooning, inflammation and overall NAFLD activity scores in MCD-fed and polydatin-treated mice. Mean SE of results from controls (n=5), MCD diet-fed group (n=7) and PD treatment group (n=9); (D) Serum levels of ALT, AST and T. (E) The mRNA levels of FAS, SREBP-1c were measured by q-PCR. (F) The curve of body weight of mice in the period of diet feeding. Mean SE of results from controls (n=5), MCD diet-fed group (n=7) and PD treatment group (n=9). ## p < 0.01, ###p < 0.001, ####p < 0.0001 vs control mice; *p < 0.05, **p < 0.01, ****p < 0.0001 vs mice induced by MCD.
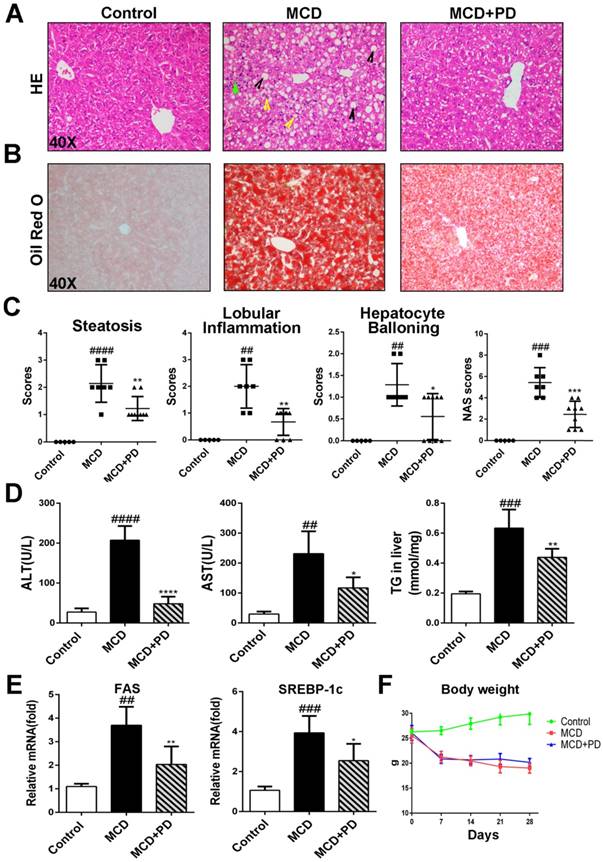
Histological examination of H&E and Oil Red O staining results revealed that MCD diet triggered marked macrovesicular steatosis in mice; however, the hepatic lipid deposition in polydatin-treated mice significantly decreased (Fig. 1A, B and D).
Treatment with polydatin ameliorated liver damage with NAS score being similar to that in the control group. Notably, morphometric analysis further showed that the lipid droplet density was significantly higher in MCD-fed mice but decreased in polydatin-treated mice (Fig. 1B). Moreover, there was a shift in the lipid droplet size distribution: the number of large droplets was significantly higher and lower in the livers of MCD-fed mice and polydatin-treated mice, respectively, compared with livers from control mice (Fig. 1A and 1B).
We next studied the protective role of polydatin in the hepatic injury induced by MCD. Numbers of evidence suggests that increased hepatocyte apoptosis is a critical mechanism contributing to oxidative stress, inflammation and fibrogenesis in NASH [12]. We found that the number of apoptotic hepatocytes, as shown by TUNEL and caspase-3 assays, was marked increased in MCD-fed mice, while significantly decreased in polydatin (Fig. 2). Furthermore, these features were less prominent in polydatin-treated mice fed an MCD diet. Consistent with these histologic findings, MCD diet induced hepatocyte damage represented by serum transaminases ALT and AST as well. However, polydatin treatment significantly reduced the levels of ALT and AST in mice fed by MCD diet (Fig. 1D). Furthermore, q-PCR assay showed that hepatic mRNA levels of lipid synthesis genes (FAS and SREBP-1c) were increased in MCD-fed mice, whereas significantly decreased by polydatin treatment (Fig. 1E).
Polydatin attenuated liver injury and hepatocyte apoptosis in mice induced by MCD diet. Mice were fed with MCD diet or control diet for 4 weeks. Mice fed with MCD diet were intraperitoneal injected with 5 mg/kg polydatin daily in the period of experiment. Apoptotic cells of liver were detected by means of TUNEL assay (n=5/group). (B) Representative immunofluorescent staining of cleaved-caspase3 (n=5/group). (C-D) Quantification of positive staining areas was measured by Image J software. (E) Caspase3 activity assay of mice liver tissue. Mean SE of results from controls (n=5), MCD diet-fed group (n=7) and PD treatment group (n=8); (F) The protein expression of cleaved-caspase3, caspase3 was examined by Western blot and analysis. # p < 0.05, ###p < 0.001, ####p < 0.0001 vs control mice; *p < 0.05, **p < 0.01 vs mice induced by MCD.
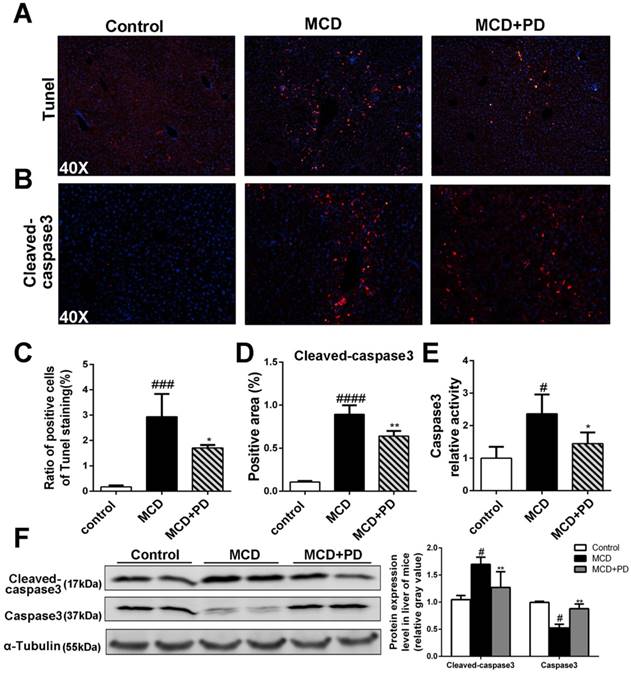
Polydatin relieves hepatocyte apoptosis in mice induced by MCD diet
In order to discover the mechanism of polydatin alleviating liver injury, apoptotic cells of liver as determined by the method of TdT mediated dUTP nick end labeling (TUNEL) and immunochemical staining of cleaved-caspase3 were significantly decreased in mice treated by polydatin (Fig. 2A, B, C and D). The cleaved-caspase 3 expression and caspase 3 activity were also markedly increased in MCD-induced mice, but significantly decreased in mice treated by polydatin (Fig. 2D, E and F).
Polydatin attenuates oxidative stress induced by MCD diet in mice
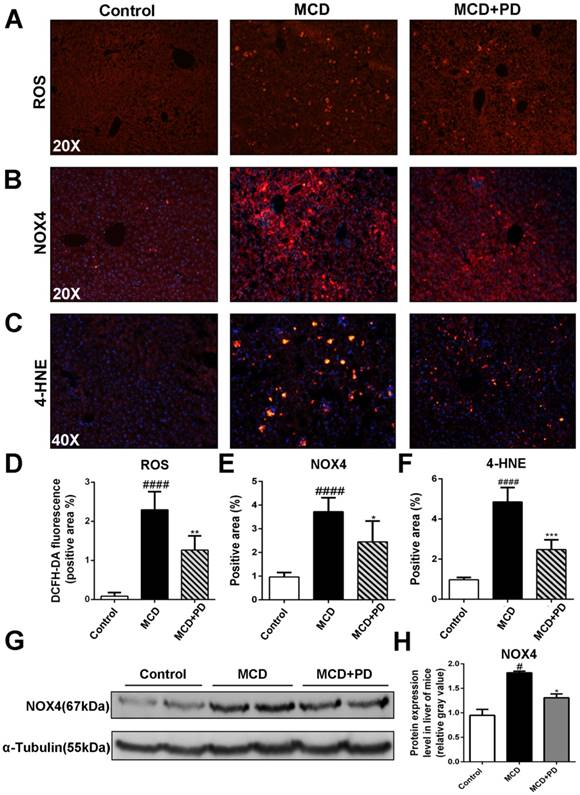
Human NASH and MCD-diet-induced experimental steatohepatitis in rodents are associated with increased oxidative stress [13]. We found that ROS production, represented by fluorescence probe DCFH-DA and its key enzyme, NOX4 and 4-HNE, were significantly increased in MCD diet-fed mice. Conversely, ROS, NOX4 and 4-HNE were markedly decreased in the liver of MCD-fed mice treated with polydatin (Fig. 3).
Polydatin reduced NASH in MCD-fed mice through inhibition of oxidative stress. Mice were fed with MCD diet or control diet for 4 weeks. Mice fed with MCD diet were intraperitoneal injected with 5 mg/kg polydatin daily in the period of experiment (n=5/group). (A) The Representative fluorescent detection of DCFH-DA. (B) Representative immunofluorescent staining of NOX4. (C) Representative immunofluorescent staining of 4-HNE. (D-F) Quantification of positive staining areas was measured by Image J software. (G) The protein expression of NOX4 was examined by Western blot and analysis. #p < 0.05, ####p < 0.0001 vs control mice; *p < 0.05, **p < 0.01, ***p < 0.001 vs mice induced by MCD.
Polydatin blocks inflammation in mice induced by MCD diet
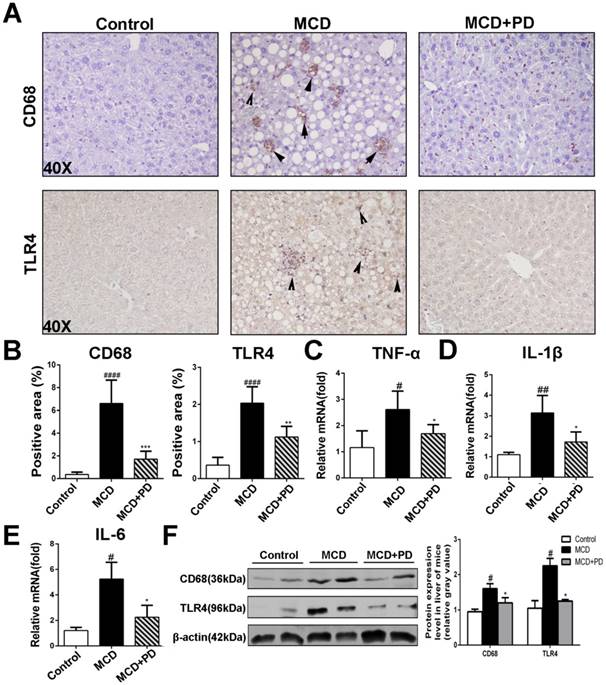
Analysis of H&E staining of liver sections revealed that polydatin treatment protected MCD-fed mice from hepatic inflammation. Hepatic macrophage, central inflammatory mediators in the pathogenesis of NASH, can arise either from circulating monocytes or from self-renewing embryo-derived local macrophages, named Kupffer cells [14]. As shown in Fig. 4, the expression of CD68 (macrophage marker), was markedly increased in vehicle-treated mice fed by MCD diet. However, the expression of CD68 was significantly decreased in polydatin-treated mice fed by MCD diet. Given TLR4/NF-κB signaling also plays vital roles in the inflammatory response in NASH [15], we presumed that polydatin inhibits pro-inflammatory cytokines synthesis by downregulation of TLR4. As expected, TLR4 expression was significantly downregulated by polydatin treatment in mice fed by MCD diet (Fig. 4A). Consistent with the change of TLR4, the hepatic mRNA levels of pro-inflammatory cytokines such as TNF-α, IL-1β and IL-6 were increased in mice fed by MCD diet, but decreased by polydatin treatment (Fig. 4C, D and E). The downregulation of CD68 and TLR4 by polydatin treatment was also confirmed by western blotting assay (Fig. 4F).
Polydatin blocks inflammation in mice induced by MCD. Mice were fed with MCD diet or control diet for 4 weeks. Mice fed with MCD diet were intraperitoneal injected with 5 mg/kg polydatin daily in the period of experiment. Polydatin suppressed inflammatory reaction and the expression of immunity correlating cytokines induced by MCD diet. (A) Representative histology of immunohistochemical staining of CD68 and TLR4 (n=5/group). (B) Quantification of positive staining areas was measured by Image J software. (C-E) The mRNA levels of TNF-α, IL-1β and IL-6 were measured by q-PCR. Mean SE of results from controls (n=5), MCD diet-fed group (n=7) and PD treatment group (n=9). (F) The protein expression of CD68 and TLR4 was examined by Western blot and analysis. #p < 0.05, ##p < 0.01, ####p < 0.0001 vs control mice(n=5-9/group); *p < 0.05, **p < 0.01, ***p < 0.001 vs mice induced by MCD.
Polydatin represses steatosis, hepatocellular lipotoxicity and apoptosis induced by palmitate
To investigate the hepatoprotective roles of polydatin in NASH, we next examined the effects of polydatin on hepatocellular damage and steatosis in HepG2 cells incubated with PA, replicates the early features of NASH in vivo Oil red O staining showed that lipid droplet accumulated in cells cultured with PA. However, polydatin treatment significantly reduced the accumulation of lipid droplet (Fig. 5A). Consistently, polydatin significantly reduced cellular TG content in response to PA induction (Fig. 5B). Furthermore, q-PCR assay showed that hepatic mRNA levels of lipid synthesis genes (FAS and SREBP-1c) were markedly increased in PA-induced HepG2 cells, whereas significantly decreased by polydatin treatment (Fig. 5 C).
Polydatin suppressed hepatocellular lipotoxicity and hepatocyte apoptosis. HepG2 cells were induced by 0.25 mM palmitic acid (PA) in the absence or presence of polydatin (5, 10, 20 μM) for 24 h. (A) Lipid droplets in HepG2 cells were measured by Oil red O staining assay. (B) The contents of triglycerides in HepG2 cells. (C) The mRNA levels of FAS, SREBP-1c were measured by q-PCR. (D) The proportion of LDH release from HepG2 cells. (E) The protein expression of cleaved-caspase3, caspase3 was examined by Western blot and analysis. (F) The apoptotic cells were determined by flow cytometric analysis. #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001 vs Control; *p < 0.05, **p < 0.01, ***p < 0.001 vs HepG2 induced by PA.
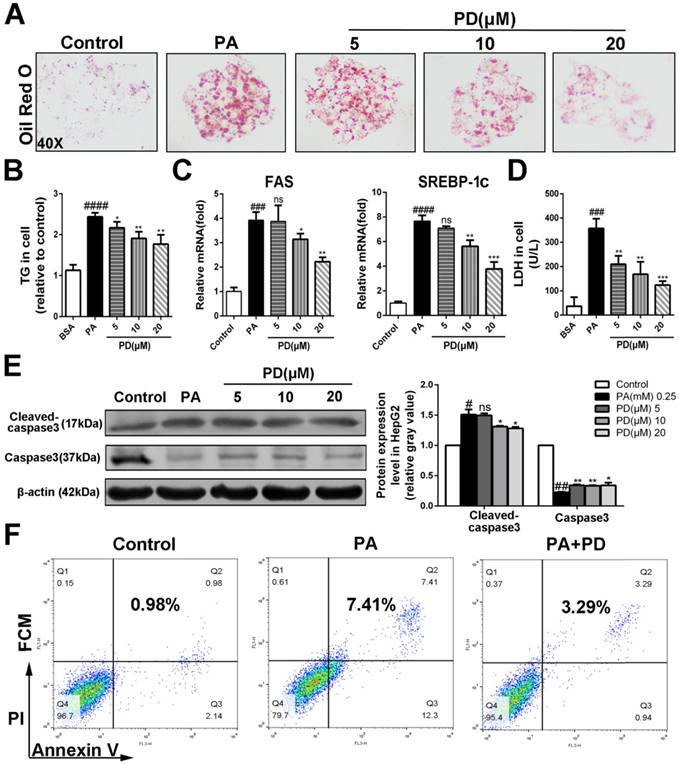
As expected, LDH release into the medium, and hepatocellular apoptosis as measured by enhanced levels of cleaved-caspase3 and Annexin V were markedly increased in PA-cultured HepG2 cells. In contrast, the levels of LDH, cleaved-caspase3 and Annexin V were significantly decreased in polydatin-treated cells (Fig. 5D, E and F).
Polydatin blocks TLR4/NF-κB p65 inflammatory signaling pathway in hepatocyte challenged by LPS
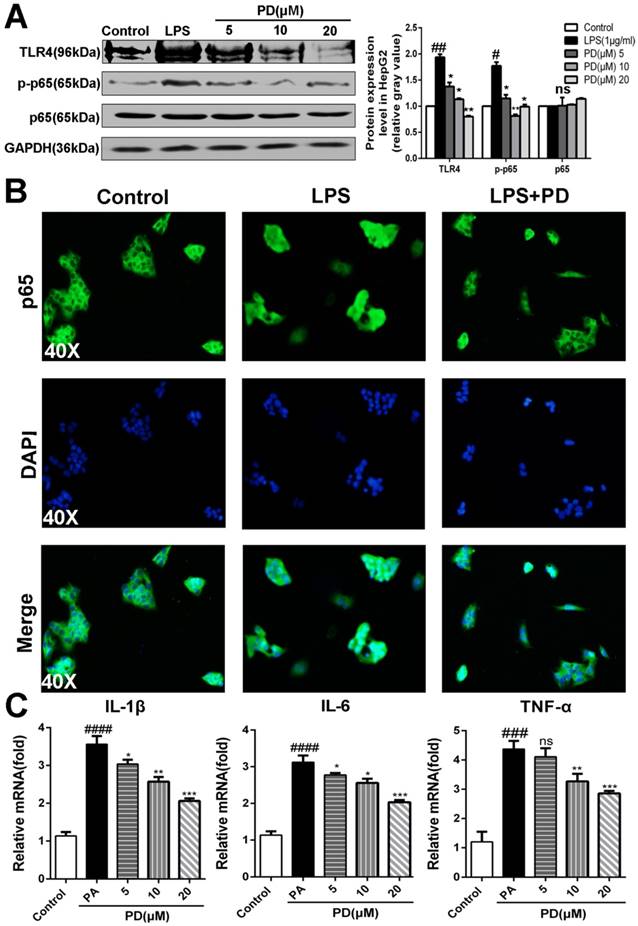
Toll-like receptor 4 (TLR4) is a pattern-recognition receptor for LPS and induces activation of innate immune signaling. Previous studies clearly have shown the critical roles of TLR4 in promoting NASH and its related fibrosis [13-15]. NF-κB p65 is the downstream of TLR4 and involved in the NASH [5]. Considering the important relationship between TLR4/NF-κB signaling pathway and pro-inflammatory, we presumed that polydatin attenuates pro-inflammatory cytokines by inhibition of TLR4/NF-κB signaling pathway. As shown in Fig. 6A and B, polydatin treatment significantly reduced TLR4 expression and p65 nuclear translocation in HepG2 cells induced by PA. Furthermore, q-PCR assay also showed that the mRNA levels of proinflammatory cytokines, such as IL-1β, IL-6 and TNF-α were increased after 24 h 0.25mM PA incubation, whereas decreased by polydatin treatment (Fig. 6C). These data demonstrated that polydatin attenuated NASH-related inflammation by inhibition of TLR4/NF-κB p65 signaling pathway.
Polydatin blocks TLR4/NF-κB p65 inflammatory signaling pathway in hepatocyte challenged by LPS. HepG2 cells were induced by 1 μg/ml LPS in the absence or presence of polydatin for 24 h. (A) The protein expression of TLR4, p-p65 and p65 was examined by Western blot and analysis. (B) Nuclear translocation of NF-kB p65 in HepG2 was assayed by immunofluorescence assay. (C) The mRNA levels of IL-1β, IL-6 and TNF-α were measured by q-PCR assay. #p <0.05, ##p <0.01, ###p <0.001, ####p <0.0001 vs Control. *p <0.05, **p < 0.01, ***p <0.001 vs HepG2 induced by PA.
Polydatin attenuates hepatic steatosis and apoptosis induced by LPS and PA
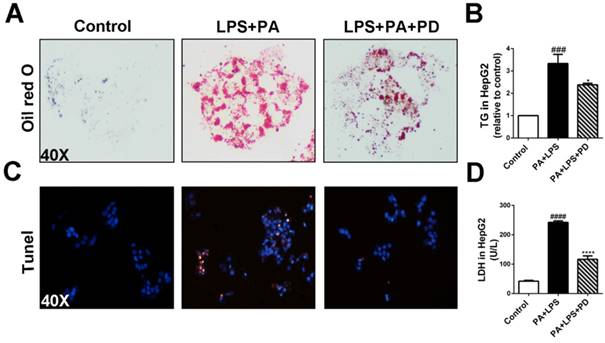
It has been demonstrated that LPS alone did not increase lipid accumulation and TLR4 signaling pathway promotes free fatty acid-induced lipid accumulation and apoptosis in hepatocytes co-cultured with PA [5, 16]. Given that systemic LPS levels are elevated in human NASH, we next examine the effects of polydatin on hepatocellular steatosis and injury in response to combination of LPS and PA stimulation [5, 17]. HepG2 cells cultured in medium containing LPS and PA showed severe lipid accumulation as assayed by Oil red O staining, whereas polydatin treatment significantly reduced the hepatocellular lipid accumulation (Fig. 7A). Consistently, hepatocellular TG levels were increased dramatically by co-stimulated with LPS and PA, but reduced significantly by polydatin treatment (Fig. 7B). We next investigated the hepatocellular protection of polydatin against the hepatocyte injury challenged by inflammation and lipotoxicity. After 24 h of LPS and PA stimulation, hepatocyte apoptosis and LDH release were remarkably increased. However, polydatin treatment significantly reduced apoptosis and LDH release to the similar levels of control (Fig. 7C and D). These data suggest that polydatin plays protective roles in the hepatic steatosis and injury induced by lipotoxic and inflammatory stimulation replicating NASH in vitro.
Polydatin attenuates hepatic steatosis and apoptosis induced by LPS and PA. HepG2 cells were induced by the combination of 1 μg/ml LPS and 0.25 mM PA and treated with or without polydatin for 24 h. (A) Oil red O staining in HepG2 cells. (B) The contents of triglyceride in HepG2 cells. (C) Apoptotic cells were measured by means of TUNEL assay. (D) The proportion of LDH release from HepG2 cells. ###p < 0.001, ####p < 0.0001 vs Control. *p < 0.05, ****p < 0.0001 vs HepG2 induced by PA.
Polydatin retards development of liver fibrosis in mice fed by MCD diet
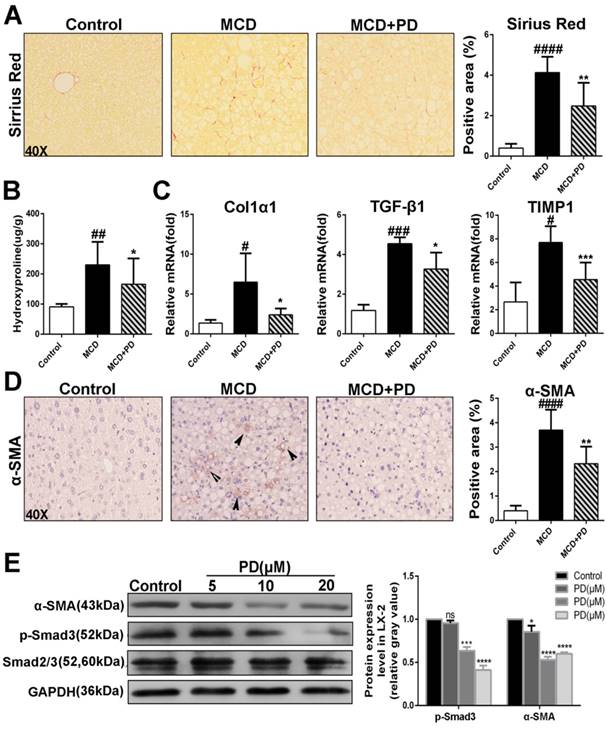
The presence of activated hepatic stellate cells (HSCs) and fibrosis is the major differences that distinguish NASH from simple steatosis. As expected, MCD diet-fed mice showed collagen deposition by Sirius red staining (Fig. 8A), with corresponding increases in hydroxyproline content, a marker of collagen deposition (Fig. 8B). However, polydatin treatment significantly diminished collagen deposition (Fig. 8A and B). Consistently, pro-fibrotic markers such as Col1α1, TGF-β1 and tissue inhibitor of matrix metalloproteinase (TIMP-1) transcripts were increased in MCD diet-fed mice and decreased by polydatin treatment (Fig. 8C). Furthermore, immunostaining for α-SMA, a marker for activated HSCs, was attenuated by polydatin treatment in mice fed by MCD diet (Fig. 8D). Because of a key role of activated HSCs in hepatic fibrosis, this finding suggests that polydatin treatment may suppress hepatic fibrosis by blocking HSC activation. Finally, LX-2 cells, (a human HSC line with key features of activated HSCs despite limitations due to immortalization) were treated by polydatin (5, 10, 20 μM). Strikingly, Smad3 phosphorylation and α-SMA expression in LX-2 cells were significantly attenuated by polydatin treatment in a dose-dependent manner (Fig. 8E). These data suggest that polydatin attenuates liver fibrosis in mice induced by MCD diet at least in part through inhibition Smad3 pathway and HSC activation.
Development of liver fibrosis was suppressed after treatement of Polydatin in mice fed by MCD diet. Mice were fed with MCD diet or control diet for 4 weeks. Mice fed with MCD diet were intraperitoneal injected with 5 mg/kg polydatin daily in the period of experiment. (A) Representative histology of immunohistochemical staining of α-SMA and Sirius red staining from controls (n=5), MCD-fed group (n=7) and PD treatment group (n=9). (B) Quantification of positive staining areas was measured by Image J software. (C) The hepatic mRNA levels of Col1α1, TGF-β1 and TIMP1 were measured by q-PCR assay. (D) Hepatic hydroxyproline content. (E) The hepatic expression of α-SMA in mice was examined by Western blot and analysis. # p < 0.05, ## p < 0.01, ###p < 0.001, ####p < 0.0001 vs control mice. *p <0.05, **p < 0.01, ***p <0.001 vs mice induced by MCD; *p < 0.05, ***p <0.001, ****p < 0.0001 vs Control.
Discussion
In the present study, we found that polydatin treatment in mice fed an MCD diet results in dramatic attenuation of integral features of steatohepatitis compared with mice fed control diet, including repressed liver steatosis and hepatocellular damage, decreased plasma ALT and AST, suppression of hepatic oxidative stress, inflammation, collagen deposition, HSC activation and nutritional fibrosis. NASH pathohistological features were blocked by polydatin treatment in mice fed by MCD diet and human liver cells cultured with PA.
Feeding mice with MCD diet is a well-established nutritional model of NASH with serum aminotransferase elevation, and liver histological changes similar to human NASH, including hepatic steatosis, lobular inflammation and pericelluar fibrosis [2]. In the MCD model, the “first hit” is the impaired mitochondrial β-oxidation, and the “second hit” is increased oxidative stress and lipid peroxidation [18], which induce proinflammatory gene expression and HSC activation [19]. Hepatocellular apoptosis is considered a critical factor that contributes to liver inflammation and a characteristic marker for the progression of steatosis to steatohepatitis [12]. In current study, we found that polydatin protected MCD diet-fed mice from hepatocellular apoptosis and liver injury, suggesting polydatin is a promising hepatoprotective agent for NASH therapy.
Excessive hepatic triglyceride accumulation in steatohepatitis can be caused by enhanced synthesis of fatty acids and reduced fatty acid oxidation. Our current study demonstrated that polydatin regulates hepatic fatty acids synthesis by suppression of fatty acid synthesis regulators such as FAS and SREBP-1c. Notably, SREBP-1c is a transcription factor for TG synthesis through activating genes required for fatty acid synthesis and storage of TG such as FAS [20].
The severity of hepatic steatosis correlates with the stage of liver injury and fibrosis, and liver lipid accumulation is considered a main risk factor for development of NASH [21]. However, many obese humans with type 2 diabetes, and mice with diet-induced obesity, readily develop hepatic steatosis, but seldom experience progressive liver damage or fibrosis [22]. Thus, only regulation of liver lipid deposition may not be sufficient to induce progression toward steatohepatitis. The multiple-hit hypothesis of NASH pathogenesis suggests that lipid accumulation in hepatocytes increases liver sensitivity to a second injury to cause NASH lesions. Oxidative stress has been proposed as one of these cofactors, and hepatic oxidative stress markers correlate with the severity of necroinflammation and fibrosis [23]. Furthermore, oxidative stress can stimulate hepatic inflammation in NASH through adaptive immune responses [13]. Mechanistically, NADPH oxidases (NOXs) received a considerable attention as major producers of ROS, especially, NOX4 induction in hepatocytes leads to their apoptosis, further triggering the cascade of events leading to NASH and fibrosis [3]. It is well-known that antioxidant properties of polydatin have great potential for use as an alternative to more toxic synthetic antioxidants as an additive in food, cosmetics and pharmaceutical preparations for the treatment of oxidative diseases [24]. Additionally, polydatin promotes Nrf2-ARE anti-oxidative pathway to prevent diabetic kidney fibrosis [25, 26]. Consistently, our present data showed that polydatin attenuates oxidative stress in mouse liver induced by MCD through downregulation of NOX4.
The contribution of TLR4/NF-κB signaling and its activation by translocated gut-derived LPS to chronic liver diseases including NASH have been well demonstrated [27]. Regarding hepatic inflammation, the number of CD68 positive cells, which reflect the recruitment of macrophages; the hepatic inflammation score; and the IL-1β, IL-6 and TNF-α mRNA levels were obviously increased by MCD diet induction. NF-κB is an important regulator of liver injury and hepatic inflammation in NASH, suggesting NF-κB activation promotes NASH development [28]. TLR4 is capable of triggering the rapid activation of its downstream signaling, NF-κB, which up-regulates the production of proinflammatory cytokines such as IL-1β and IL-6 [29]. The deletion of TLR4 prevents liver injury, inflammation and fibrosis [16]. Increasing evidences showed that polydatin effectively attenuates inflammatory reaction in various diseases of lung, kidney and colon[30, 31]. In the current study, polydatin is capable of suppressing inflammatory response both in the HepG2 cell line and MCD-fed mouse with NASH features. Polydatin counteracted the MCD-induced increase in hepatic inflammation score and the number of macrophages. Our data showed that polydatin alleviates the expression of pro-inflammatory factors in livers from MCD diet-fed mice and PA-cultured HepG2 cells at least in part through inhibition of LPS/NF-κB signaling pathway. Recent studies showed that although patients with NASH are much more likely to develop cirrhosis than patients with simple hepatic steatosis, cirrhosis is not an inevitable consequence of NASH. Thus, considerable efforts have been devoted to identify and treat individuals with NASH who are at great risk for fibrosis progression [32]. Hepatic fibrosis is one of the key histopathological features in NASH patients, suggesting a more severe and progressive liver injury [33]. HSC activation is the central component for hepatic fibrosis. Our recent study found that polydatin protected against liver fibrosis through inhibition of HSC activation in mice induced by CCl4 [34]. Our results consistently indicate that polydatin attenuated collagen deposition and hepatic expression of profibrotic growth factors (Col1α1, TGF-β1, TIMP-1 and α-SMA) in mice fed by MCD diet, suggesting that polydatin protects against NASH-related fibrosis through suppression of HSC activation.
The MCD diet feeding used in the current study is considered as the best established rodent NASH models, which mimics classic histopathological features of liver steatosis, apoptosis, oxidative stress, inflammation and fibrosis, as similar as seen in human NASH [35]. However, the model dose not fully manifest all human NASH features. Mice fed with MCD diet lose weight instead of being obese and lack insulin resistance which are normally seen in patients with NASH [36, 37]. Thus, in future, high-fat and high-fructose model, which may result in an obese animal with several steatosis, oxidative stress, inflammation and insulin resistance at 16 weeks [36, 38], addressing the protective roles of polydatin in the obesity-induced NASH.
In summary, the present study presents compelling evidence that polydatin attenuates lipid accumulation, oxidative stress, inflammation and fibrosis in NASH, suggesting that polydatin may be useful for the treatment of NASH.
Abbreviations
LPS, lipopolysaccharide; MCD, methionine and choline deficiency; MCS, methionine and choline supplemented; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; NF-κB, nuclear factor kappa-B; ROS, reactive oxygen species; α-SMA, α-smooth muscle actin; TLR, Toll-like receptor; PA, palmitic acid; LPS, lipopolysaccharide. PD, polydatin; LDH, lactate dehydrogenase; TG, triacylglyceride.
Acknowledgements
This work was supported by the research grants from the; the Science and Technology Planning Project of Guangdong Province (2017A020211007), China; the Key Project of Natural Science Foundation of Guangdong Province (2016A030311014), China; National Natural Science Foundation of China (81170422); and Science and Technology Planning Project of Guangdong Province (2017A020215178). We also thank the support from the Guangzhou Key Laboratory of Construction and Application of New Drug Screening Model Systems.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Marra F, Gastaldelli A, Svegliati Baroni G. et al. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends in molecular medicine. 2008;14:72-81
2. Wang X, Hausding M, Weng SY. et al. Gliptins Suppress Inflammatory Macrophage Activation to Mitigate Inflammation, Fibrosis, Oxidative Stress, and Vascular Dysfunction in Models of Nonalcoholic Steatohepatitis and Liver Fibrosis. Antioxidants & redox signaling. 2018;28:87-109
3. Bettaieb A, Jiang JX, Sasaki Y, Chao TI. et al. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity During Development of Steatohepatitis in Mice. Gastroenterology. 2015;149:468-480
4. Alisi A, Manco M, Devito R, Piemonte F. et al. Endotoxin and plasminogen activator inhibitor-1 serum levels associated with nonalcoholic steatohepatitis in children. Journal of pediatric gastroenterology and nutrition. 2010;50:645-649
5. Farhadi A, Gundlapalli S, Shaikh M, Frantzides C. et al. Susceptibility to gut leakiness: a possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver international official journal of the International Association for the Study of the Liver. 2008;28:1026-1033
6. Takaki A, Kawai D, Yamamoto K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). International journal of molecular sciences. 2013;14:20704-20728
7. Wenzel P, Kossmann S, Munzel T, Daiber A. Redox regulation of cardiovascular inflammation- Immunomodulatory function of mitochondrial and Nox-derived reactive oxygen and nitrogen species. Free radical biology & medicine. 2017;109:48-60
8. Tanaka N, Takahashi S, Fang Z, Matsubara T. et al. Role of white adipose lipolysis in the development of NASH induced by methionine- and choline-deficient diet. Biochimica et biophysica acta. 2014;1841:1596-1607
9. Mikulski D, Molski M. Quantitative structure-antioxidant activity relationship of trans-resveratrol oligomers, trans-4,4'-dihydroxystilbene dimer, trans-resveratrol-3-O-glucuronide, glucosides: trans-piceid, cis-piceid, trans-astringin and trans-resveratrol-4'-O-beta-D-glucopyranoside. European journal of medicinal chemistry. 2010;45:2366-2380
10. Zhang Q, Tan Y, Zhang N, Yao F. Polydatin supplementation ameliorates diet-induced development of insulin resistance and hepatic steatosis in rats. Molecular medicine reports. 2015;11:603-610
11. Zhang J, Tan Y, Yao F, Zhang Q. Polydatin alleviates non-alcoholic fatty liver disease in rats by inhibiting the expression of TNF-alpha and SREBP-1c. Molecular medicine reports. 2012;6:815-820
12. Hatting M, Zhao G, Schumacher F, Sellge G. et al. Hepatocyte caspase-8 is an essential modulator of steatohepatitis in rodents. Hepatology. 2013;57:2189-2201
13. Sutti S, Jindal A, Locatelli I, Vacchiano M. et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. 2014;59:886-897
14. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. Journal of hepatology. 2014;60:1090-1096
15. Song CY, Zeng X, Wang Y, Shi J. et al. Sophocarpine attenuates toll-like receptor 4 in steatotic hepatocytes to suppress pro-inflammatory cytokines synthesis. Journal of gastroenterology and hepatology. 2015;30:405-412
16. Yang L, Miura K, Zhang B, Matsushita H. et al. TRIF Differentially Regulates Hepatic Steatosis and Inflammation/Fibrosis in Mice. Cellular and molecular gastroenterology and hepatology. 2017;3:469-483
17. Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology. 2014;146:1513-1524
18. Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. International journal of experimental pathology. 2006;87:1-16
19. George J, Pera N, Phung N, Leclercq I. et al. Lipid peroxidation, stellate cell activation and hepatic fibrogenesis in a rat model of chronic steatohepatitis. Journal of hepatology. 2003;39:756-764
20. Zhang X, Han J, Man K, Li X. et al. CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophages and autophagy. Journal of hepatology. 2016;64:160-170
21. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836-1846
22. Begriche K, Massart J, Robin MA, Bonnet F. et al. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology. 2013;58:1497-1507
23. Seki S, Kitada T, Yamada T, Sakaguchi H. et al. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. Journal of hepatology. 2002;37:56-62
24. Jin J, Li Y, Zhang X, Chen T. et al. Evaluation of Both Free Radical Scavenging Capacity and Antioxidative Damage Effect of Polydatin. Advances in experimental medicine and biology. 2016;923:57-62
25. Gong W, Li J, Chen Z, Huang J. et al. Polydatin promotes Nrf2-ARE anti-oxidative pathway through activating CKIP-1 to resist HG-induced up-regulation of FN and ICAM-1 in GMCs and diabetic mice kidneys. Free radical biology & medicine. 2017;106:393-405
26. Huang K, Chen C, Hao J, Huang J. et al. Polydatin promotes Nrf2-ARE anti-oxidative pathway through activating Sirt1 to resist AGEs-induced upregulation of fibronetin and transforming growth factor-beta1 in rat glomerular messangial cells. Molecular and cellular endocrinology. 2015;399:178-189
27. Petrasek J, Csak T, Szabo G. Toll-like receptors in liver disease. Advances in clinical chemistry. 2013;59:155-201
28. Dela Pena, A Leclercq I, Field J, George J. et al. NF-kappaB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology. 2005;129:1663-1674
29. Luo M C, Zhou SY, Feng DY, Xiao J. et al. Runt-related Transcription Factor 1 (RUNX1) Binds to p50 in Macrophages and Enhances TLR4-triggered Inflammation and Septic Shock. The Journal of biological chemistry. 2016;291:22011-22020
30. Liu HB, Meng QH, Huang C, Wang JB. et al. Nephroprotective Effects of Polydatin against Ischemia/Reperfusion Injury: A Role for the PI3K/Akt Signal Pathway. Oxidative medicine and cellular longevity. 2015;2015:362158
31. Shiyu S, Zhiyu L, Mao Y, Lin B. et al. Polydatin up-regulates Clara cell secretory protein to suppress phospholipase A2 of lung induced by LPS in vivo and in vitro. BMC cell biology. 2011;12:31
32. Witek RP, Stone WC, Karaca FG, Syn WK. et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology. 2009;50:1421-1430
33. Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99-S112
34. Zhao X, Li R, Liu Y, Zhang X. et al. Polydatin protects against carbon tetrachloride-induced liver fibrosis in mice. Archives of biochemistry and biophysics. 2017;629:1-7
35. Coulon S, Legry V, Heindryckx F, Van Steenkiste C. et al. Role of vascular endothelial growth factor in the pathophysiology of nonalcoholic steatohepatitis in two rodent models. Hepatology. 2013;57:1793-1805
36. Kohli R, Feldstein AE. NASH animal models: are we there yet? Journal of hepatology. 2011;55:941-943
37. Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. Journal of hepatology. 2004;40:47-51
38. Tetri LH. Basaranoglu M, Brunt E, M Yerian, et al. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. American journal of physiology Gastrointestinal and liver physiology. 2008;295:G987-995
Author contact
![]() Corresponding author: Tian Lan, 280 Wai Huan Dong Road, Department of Pharmacology, School of Pharmacy, Guangdong Pharmaceutical University, Guangzhou Higher Education Mega Center, Guangzhou 510006, China. Email: lantian012345com and Genshu Wang, 600 Tianhe Road, Tianhe District, Department of Hepatic Surgery and Liver transplantation Center of the Third Affiliated Hospital, Sun Yat-sen University; Guangzhou 510630, China. Email: wgsh168com
Corresponding author: Tian Lan, 280 Wai Huan Dong Road, Department of Pharmacology, School of Pharmacy, Guangdong Pharmaceutical University, Guangzhou Higher Education Mega Center, Guangzhou 510006, China. Email: lantian012345com and Genshu Wang, 600 Tianhe Road, Tianhe District, Department of Hepatic Surgery and Liver transplantation Center of the Third Affiliated Hospital, Sun Yat-sen University; Guangzhou 510630, China. Email: wgsh168com