ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2018; 14(13):1892-1900. doi:10.7150/ijbs.28620 This issue Cite
Research Paper
DJ-1 Deficiency Protects Hepatic Steatosis by Enhancing Fatty Acid Oxidation in Mice
Min Xu1*, Hailong Wu2*, Meng Li1*, Yankai Wen1, Chang Yu1, Lei Xia1 ![]() , Qiang Xia1
, Qiang Xia1 ![]() , Xiaoni Kong1
, Xiaoni Kong1 ![]()
1. Department of Liver Surgery, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China.
2. Shanghai Key Laboratory for Molecular Imaging, Collaborative Research Center, Shanghai University of Medicine & Health Science, Shanghai, China.
*These authors contribute equally to this work.
Abstract

Our previous studies have shown that DJ-1 play important roles in progression of liver diseases through modulating hepatic ROS production and immune response, but its role in hepatic steatosis remains obscure. In the present study, by adopting a high-fat-diet (HFD) induced mice model, we found that DJ-1 knockout (DJ-1-/-) mice showing decreased HFD-induced obesity and visceral adipose accumulation. In line with these changes, there were also reduced liver weight and ameliorated hepatic triglyceride (TG) accumulation in DJ-1-/- mice compared to wild-type (WT) mice. And there were also decreased blood glucose levels and insulin resistance and reduced glucose metabolic disorder in DJ-1-/- mice, whereas there were no significant differences in total cholesterol (TC) and serum lipid in two groups of mice. Mechanistically, we found that there were no differences in food intake in these two genotypes of mice. Furthermore, there were no significant differences in fatty acid synthesis and glycolysis, but the expression of key enzymes in fatty acid oxidation and the tricarboxylic acid (TCA) cycle, such as Cpt1α, Pparα, Acox1, Cs, Idh1 and Idh2, was increased in DJ-1-/- mice liver, suggesting that there was enhanced fatty acids oxidation and TCA cycle in DJ-1-/- mice. Our data indicate that deletion of DJ-1 enhancing fatty acids oxidation resulting in lower hepatic TG accumulation in mice, which protecting mice hepatic steatosis.
Keywords: DJ-1, hepatic steatosis, fatty acid oxidation
Introduction
As a vital metabolic organ in human body, liver plays a central role in maintaining energy balance. When lipid synthesis exceeds lipolysis or export caused unbalanced lipid synthesis and hepatic lipid accumulation which finally lead to hepatic steatosis[1] . The lipid synthesis and catabolism were regulated by complicated networks which controlled by numerous transcriptional factors, including transcriptional factor like: SREBP-1[2], transcriptional coregulators, such as PGC-1, Sirt-1 [3].
DJ-1, also known as Parkinson disease 7 (Park7), is a ubiquitously expressed, multifunctional protein [4]. We have established that DJ-1 has a critical role in initiating an inflammatory response through modulating ROS generation [5]. Later, we have shown that DJ-1 has important roles in progression of liver diseases through modulating ROS and immune response[6]. We have shown that DJ-1 deficiency negatively regulates hepatic progenitor cells (HPC) proliferation by impairing the formation of HPC-associated fibrosis and inflammatory niches[7]; Deficiency of DJ-1 ameliorates liver fibrosis through inhibition of hepatic ROS production and inflammation[8]; moreover, DJ-1 ameliorates tumorigenesis and hepatocellular carcinoma cell (HCC) proliferation through regulating hepatic inflammation and decreasing interleukin 6/signal transducer and activator of transcription 3 (IL-6/ STAT3) signaling pathway in a classic diethylnitrosamine (DEN)-induced HCC mice model [9].Recently, studies have showed that DJ-1 controlled type 2 diabetes mellitus (T2DM) and energy metabolism in various tissues [10]. It was reported that DJ-1 inhibited energy metabolism through PTEN/PI3K/ AKT/FoxO1/Ucp1 pathway and promoted lipid formation and obesity-mediated inflammation in adipose tissue [11, 12]. DJ-1 deficiency accelerated glycolysis through ROS-mediated MAPK and mitochondrial uncoupling pathway in skeletal muscle [13]. Although DJ-1 has been shown to regulate energy metabolism in adipose tissue and skeletal muscle, evidence for a role of DJ-1 in liver steatosis is lacking. In this study, we characterized the function and potential mechanism of DJ-1 in liver steatosis.
In the present study, we have investigated the role of DJ-1 on hepatic lipid metabolism in an HFD-induced mice model. We found HFD-induced adipose accumulation, insulin resistance and glucose metabolic disorder were significantly inhibited in DJ-1-/- mice. Furthermore, we found there were increased hepatic fatty acids oxidation and lower hepatic TG accumulation in DJ-1-/- mice, which may protect mice form hepatic steatosis.
Materials and Methods
Mice and animal models
DJ-1 knockout (DJ-1-/-) mice on a C57BL/6 background (stock#006577) were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA). Wild-type (WT) mice were purchased from SLAC laboratory (Shanghai, China). 4- to 6-week-old male mice (14-16g) were housed in a standard environment at 22 to 24℃ with a 12:12 h light -dark cycle and ad libitum access to food and water. The WT mice and DJ-1-/- mice were fed with HFD (18.1% protein, 61.6% fat and 20.3% carbohydrates; D12492, Research Diets, New Brunswick, NJ) for 24 weeks to establish a mouse model of NAFLD. Mice fed with normal chow diet (NCD) were served as controls. All animal protocols were approved by the criteria outlined in Guide for the Care and Use of Laboratory Animals, which was approved by Bioethics Committee School of Medicine, Shanghai Jiao Tong University.
Mouse hepatic and serum lipid contents analysis
Liver and blood specimens were collected from mice after fed NCD or HFD for 24 weeks, and stored at -80℃ until use. The lipid contents of liver and serum were measured by using commercial kits (E1003 for serum TG, E1013 for liver TG, E1005 for serum TC, E1015 for liver TC; Applagin Technologies Inc., Beijing, China; A113-1 for serum low-density lipoprotein cholesterol (LDL-C), A112-1 for serum high-density lipoprotein cholesterol (HDL-C); Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer's instructions.
Histopathologic analysis
The liver sections were embedded in paraffin and cut into 5-µm-thick sections. Liver tissue sections were stained with hematoxylin-eosin (H&E) and periodic acid-Schiff (PAS) using standard procedures. Oil red O (ORO)(O0625; Sigma-Aldrich, St Louis, MO) staining of frozen liver sections were performed according to previously described protocol [14].
Glycogen contents assay
The levels of glycogen in the liver were measured using a commercially available glycogen Assay Kit (MAK016, Sigma-Aldrich, St Louis, MO) according to the manufacturer's instructions.
qPCR and western blot analysis
Total RNA from liver tissue was isolated with the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Reverse transcription was performed using random primers using the PrimeScript RT reagent Kit (Takara). SYBR-green based Quantitative real time-PCR (Takara) was applied to detect the relative expression levels of genes. The mRNA expression levels of target genes were normalized to β-actin. Primer sequences are shown in Table S1. Liver tissues or cell samples were homogenized with RIPA buffer (Thermo Scientific, Rockford, IL) containing a protease inhibitor cocktail (Caliche, Raleigh, NC) and phosphatase inhibitor cocktail (HY-K0021; Med Chen Express). The Pierce BCA Protein Assay Kit (#23227, Thermo Fisher Scientific) was used to measure total protein concentrations. Protein samples (40 µg) were separated on a 10% SDS-PAGE gel. The gel was transferred to a nitrocellulose membrane. The NC membrane was blocked with 5% non-fat milk for one hour at room temperature, and then incubated with primary antibodies against indicated antigens at 4 °C overnight under shaking conditions. The membrane incubation with the corresponding secondary antibodies coupled with horseradish peroxidase for one hour at room temperature then. The Cheviot XRS+ System with Image Lath Software (Bio-Rad) was used for blotting signal detection and quantification. Primary antibodies were rabbit anti-DJ-1 (Abcam, Cambridge, MA), rabbit anti-SREBP-1 (Santa Cruz, CA), rabbit anit-ACLY (Proteintech, Wuhan, China), IRS1(Proteintech, Wuhan, China), p-IRS1(Tyr608)(Millipore, Billerica, MA), p-AKT (Ser473) (Cell Signaling Technology, Beverly, MA), AKT (Cell Signaling Technology, Beverly, MA), anti-β-actin (Sigma-Aldrich, St Louis, MO).
Glucose tolerance tests and Insulin tolerance tests
The mice used for the glucose tolerance tests had been fasted for 16 h (17:00-09:00) with ad libitum access to water. The fasting blood glucose levels were measured, and then each mouse was intraperitoneally injected with 2.0 g per kg body weight (g kg-1) glucose (Sigma-Aldrich, St Louis, MO, USA). We measured the levels of blood glucose 15, 30, 60 and 120 min after glucose injection using a glucometer. For the insulin tolerance tests, mice were fasted for 4 h (09:00-13:00) and then intraperitoneally injected 0.75U kg-1 human insulin (Novolin R, Novo Nordisk, Bisgaard, Denmark). Blood glucose levels were measured 0,15,30,60 and 120 min after insulin injection.
In vitro lipid synthesis assays
Mouse primary hepatocyte were isolated from 6- to 8-weeks WT or DJ-1-/- mice similarly to a previously published procedure[8]. Isolated primary hepatocytes were seed into 6-well plates with DMEM medium treated with 0.25 mM palmitate (P9767, Sigma-Aldrich) for 48h, as previously reported [15]. The intracellular TG levels were measured using commercial kits (E1013, Applagin Technologies Inc.) according to the manufacturer's instructions.
Statistical analysis
All results were presented as mean ± SEM from at least 3 independent biological replicated experiments. Unpaired or paired Student's t-tests were used in this study. P<0.05 was considered statistically significant (*P<0.05; **P<0.01; ***P<0.001).
Results
Ablation of DJ-1 prevents HFD-induced obesity and visceral adipose accumulation in mice
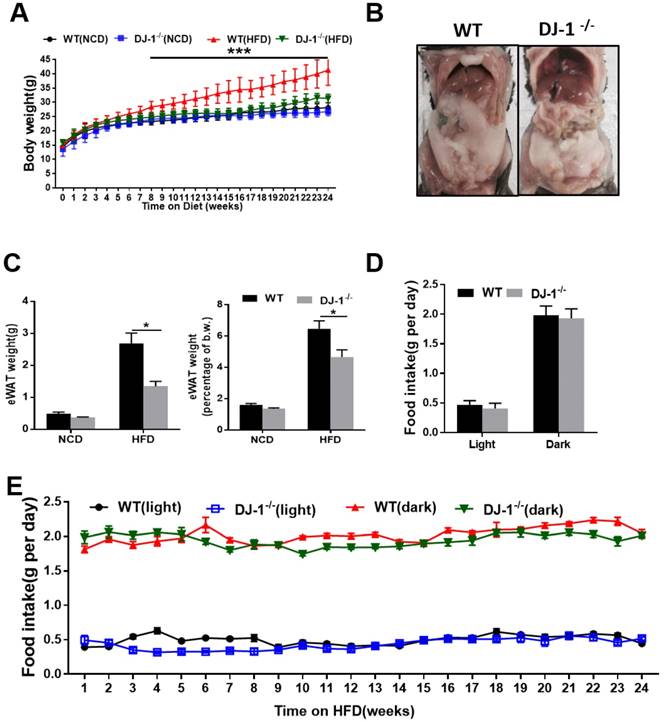
To study the role of DJ-1 in hepatic steatosis, we fed WT and DJ-1-/- mice with an NCD and HFD for different time periods. There was no significant difference in their body weight among the genotypes when fed NCD for 24 weeks and even 40 weeks (figure 1A and figure S1A). However, the body weight was significantly lower in HFD-fed DJ-1-/- mice compared to WT mice (figure 1A). The visceral adipose tissues in DJ-1-/- mice were also significantly less than those of WT mice at the end of 24th week after HFD feeding (figure 1B). The mass of epididymal white adipose tissue (eWAT) and the ratio of eWAT weight to body weight were also markedly decreased in DJ-1-/- mice after HFD feeding for 24 weeks (figure 1C). Because higher food intake could induce obesity, so we first measured the food intake amount during the day and night, and found that there was no significant difference in the food intake between DJ-1-/- mice and WT mice (figure 1D and 1E, figure S1B and S1C), therefore, the difference between DJ-1-/- mice and WT mice was not due to the food intake appetite.
DJ-1 deficiency ameliorates HFD-induced insulin resistance and glucose metabolic disorder
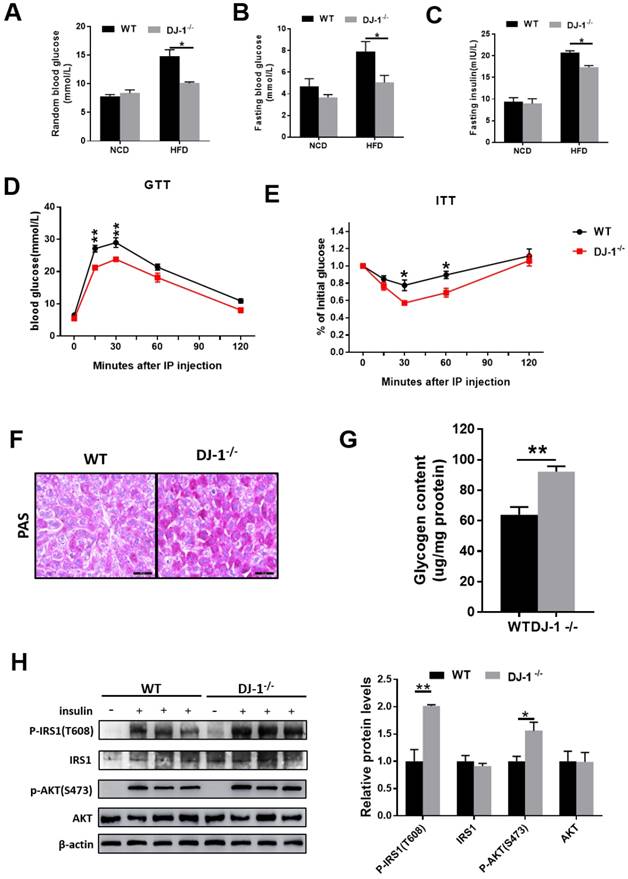
Next, we explored whether DJ-1 regulate insulin sensitivity. As show in figure 2A and 2B, both random blood glucose and fasting blood glucose levels were lower in HFD-fed DJ-1-/- mice compared to WT mice and there was no significant difference between DJ-1-/- and WT mice with NCD. In addition, fasting serum insulin levels were also lower in HFD-fed DJ-1-/- mice compared to WT mice (figure 2C). Glucose tolerance tests (GTT) was significantly improved in DJ-1-/- mice compared to WT mice at the end of 24th week of HFD feeding (figure 2D). Insulin tolerance tests (ITT) results were consistent with GTT (figure 2E). As show in figure 2F, we observed that the liver glycogen contents in DJ-1-/- mice were higher than those of WT mice by PAS staining. The liver glycogen contents measured by commercial kit were consistent with PAS staining (figure 2G). Study has shown that the phosphorylation levels of IRS1 and AKT were related to insulin sensitivity[16]. We also measured the levels of phosphorylation-IRS1 and -AKT and found that they were higher in HFD-fed DJ-1-/- mice compared to WT mice (figure 2H). It suggested that the HFD induced impairment of hepatic insulin signaling was alleviated in DJ-1-/- mice. All these results above showed that ablation of DJ-1 ameliorated HFD-induced insulin resistance and glucose metabolic disorder.
Dj-1 deficiency ameliorates HFD-induced hepatic TG accumulation
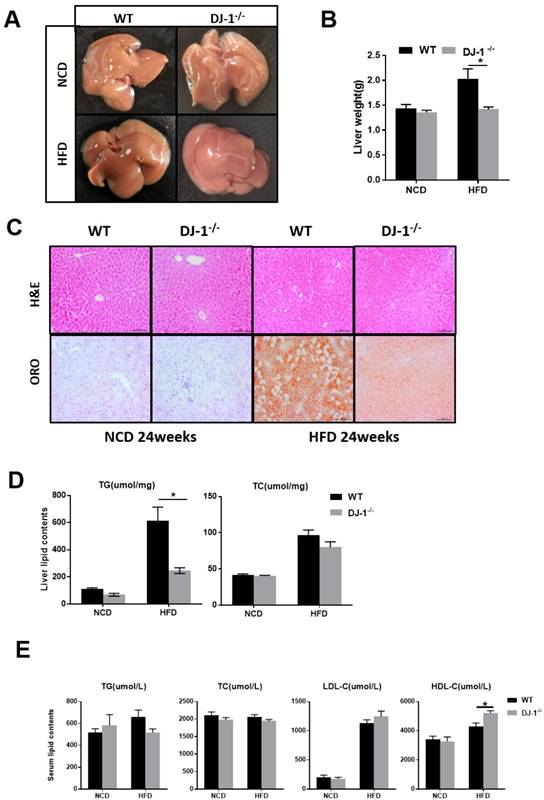
It is well known that fatty liver is closely related to obesity and insulin resistance [17]. Next, we explored whether deletion of DJ-1 inhibits hepatic lipid accumulation. In the general form, we can clearly see there were more white spots on the surface of liver in WT mice than that in DJ-1-/- mice after HFD feeding for 24 weeks (figure 3A). The liver weight was significantly lower in DJ-1-/- mice than WT mice at the end of 24th week of HFD feeding, and deletion of DJ-1 did not affect liver weights in NCD-fed group (figure 3B). There are more vacuoles in mice liver after HFD feeding as shown by H&E staining, the number and size of vacuoles in DJ-1-/- mice were much lower than those of WT mice after HFD feeding, and we also found the hepatic lipid accumulations were also markedly decreased in DJ-1-/- mice compared to WT mice as determined by ORO staining (figure 3C right). There was no significant difference between both genotypes in NCD-fed group (figure 3C left). Consistently, the hepatic TG levels in DJ-1-/- mice were lower than that in WT mice after HFD feeding (figure 3D left). But there was no significant difference in hepatic TC levels between WT and DJ-1-/- mice (figure 3D right). Except the serum HDL-C levels in DJ-1-/- mice were higher than that of WT mice in the HFD-group, all other serum lipid contents had no differences between WT and DJ-1-/- mice (figure 3E). Lipid accumulation can cause hepatic-lipotoxicity, so we measured the levels of serum ALT/AST to check the liver function, the serum ALT levels in DJ-1-/- mice were lower than that of WT mice in the HFD-group, and there was no significant difference was observed in the level of AST in two groups (figure S2). There were no significant differences in liver weight and hepatic lipid contents after fed NCD for 40 weeks by the H&E and ORO staining (figure S3A and S3B). Our results suggested that deletion of DJ-1 prevented diet-mediated hepatic TG accumulation in vivo. However, HFD did not increase the mRNA and protein expressions of DJ-1 in the liver tissues (figure S4).
Ablation of DJ-1 prevents HFD-induced obesity and visceral adipose accumulation in mice. (A)Body weight of WT and DJ-1-/- mice fed on NCD and HFD for 24 weeks. (B)A representative photograph of visceral adipose of WT and DJ-1-/- mice at the end of 24th week of HFD feeding. (C)eWAT weight and eWAT weight to body weight ratio of WT and DJ-1-/- mice after 24 weeks HFD treatment. (D、E) Cumulative (dark, 17:00 to 8:00; light, 8:00 to 17:00) daily food intake per mouse of WT or DJ-1-/- mice treated with HFD for 24 weeks. *P < 0.05, **P < 0.01, *** P < 0.001.
Deletion of DJ-1 improved HFD-induced insulin resistance and glucose metabolic disorder. (A)Random blood glucose and (B) fasting blood glucose (fasted for 6 h) of WT and DJ-1-/- mice that were fed an NCD or HFD for 24 weeks. (C) Fasting blood serum insulin levels of DJ-1-/- and WT mice that after fed an HFD for 24 weeks. (D, E) The level of blood glucose in a glucose tolerance test (GTT) and an insulin tolerance test (ITT) in WT and DJ-1-/- mice after 24 weeks HFD treatment. (F) Glycogen accumulation in liver sections by PAS staining in WT and DJ-1-/- mice that fed HFD for 24 weeks. (G) Liver glycogen contents in WT and Dj-1-/- mice after HFD administration for 24 weeks. (H) Representative western blot for phosphorylation and total IRS1 and AKT, in the liver tissues from DJ-1-/- and WT mice after HFD administration for 24 weeks. β-actin served as the loading control. *P < 0.05, **P < 0.01, *** P < 0.001.
DJ-1 deficiency does not directly affect hepatic lipid synthesis but augmented hepatic fatty acids oxidation in HFD-mediated mice models
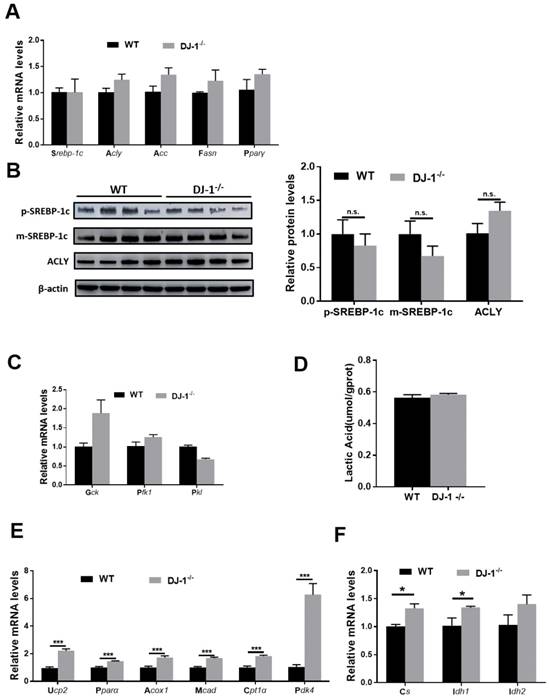
Unbalanced lipid synthesis and break down lead to hepatic steatosis. In order to explore the role of DJ-1 in hepatic lipid synthesis, we first measured fatty acids synthesis-related genes: targets of the key transcriptional factor of lipogenesis (sterol regulatory element binding protein 1C(Srebp1c)) in WT and DJ-1-/- mice liver after fed with HFD for 24 weeks by quantitative real-time PCR. No significant difference was observed in HFD-fed WT mice compare with those in DJ-1-/- mice (figure 4A). We further detected the protein expressions of the precursor (p-SREBP1C) and mature (m-SREBP1C) forms of SREBP1C and ATP citrate lyase (ACLY), and consistent with genes expression, there were also no significant differences between two groups (figure 4B). Further, we also confirmed these results by using in vitro primary hepatocytes treated with palmitic acid (PA) and oleic acid (OA), and there was also no significant difference in intracellular TG contents in two genotypes primary hepatocytes which indicated the lipid synthesis in hepatocytes were comparable in two groups (figure S5). In additions, the mRNA expression of glycolysis-related genes and the levels of lactic acid also showing no significant difference in two groups (figure 4C and 4D). Next, we moved to analyze the catabolism of lipid, the expression of Ucp2 and fatty acid oxidation related genes were higher in DJ-1-/- mice compare to WT mice after HFD feeding for 24 weeks (figure 4E). The mRNA expression of the key enzymes of the tricarboxylic acid (TCA) cycle in HFD-fed DJ-1-/-mice were also higher than that in WT mice (figure 4F). Similarly, we found that deletion of DJ-1 increased body weight loss during starvation (figure S6). Overall, these results suggested that DJ-1 deficiency ameliorated hepatic TG accumulation by promoting fatty acid oxidation.
Discussion
In this work, we have demonstrated that deletion of DJ-1 decreased HFD-induced obesity and visceral adipose accumulation in mice. Furthermore, we found that deletion of DJ-1 does not affect mice food intake and hepatic lipid synthesis. However, genes involved in lipid catabolism showing significantly different, indicate that DJ-1 promotes liver steatosis through decreasing fatty acid oxidation in mice. Thus, our study discovered a novel role of DJ-1 on hepatic lipid metabolism in an HFD-induced fatty liver mice model.
Previous studies have shown that DJ-1 can promote 3T3-L1 cells differentiate into adipocytes and fat accumulation [12]. To check the relationship between DJ-1 and fat accumulation in mice, we fed mice with an NCD or HFD for different time periods and confirmed that DJ-1 deficiency ameliorated diet-dependent obesity and visceral adipose accumulation, and it has no significant effect on age-dependent. In order to explore how DJ-1 promotes fat accumulation in mice, we first measured the food intake between day and night. In the present study, there was no significant change of food intake between DJ-1-/- and WT mice, which suggested that the obesity and fat accumulation were not induced by food intake. Previous studies attributed to no significant difference in respiratory quotients, physical activity, body temperature between DJ-1-/- and WT mice; Yet they also observed that the energy expenditure was increased in DJ-1-/- mice compared with WT mice [11, 13]. In addition, we found that deletion of DJ-1 increased the body weight loss during starvation in our present study, which indicated that deletion of DJ-1 inhibited energy expenditure. All results above and previous articles showed that DJ-1 might promote diet-mediated obesity by decreasing energy expenditure.
It is well known that obesity is one of the most important risk factors for T2DM, and ectopic fat accumulation can induce glucose metabolism disorder and insulin resistance[18]. Previous articles also reported that DJ-1 has a moderating effect on T2DM [10, 12, 19, 20]. In the present study, deletion of DJ-1 ameliorated glucose metabolism and insulin sensitivity in HFD-induced mice model. Our results indicated that deletion of DJ-1 protects T2DM in HFD-induced obesity mice model, which were similar to other studies that DJ-1 has been expected to be a target of therapy for T2DM [11-13].
Obesity and T2DM are closely related to NAFLD and which are the main risk factors of it [21]. NAFLD is defined as liver diseases with hepatic excess lipid accumulation, especially TG, due to non-alcoholic causes. When the pace of synthesis exceeds that of lipolysis in liver, excessive lipid accumulation induces NAFLD [22]. Our results showed that deletion of DJ-1 ameliorated hepatic TG accumulation in HFD-mediated mice model. And the fatty acids synthesis-related genes have no significant difference in mRNA and protein levels between WT and DJ-1-/- mice, which means that the pace of lipid synthesis has no significant changes. Liver plays an important role in the maintenance of glucose metabolism, when liver takes up excess amounts of glucose or the glucose metabolic disorder and it converts glucose to fatty acids[23]. Shi et al.[13] showed that DJ-1 inhibited glycolysis through ROS-mediated MAPK and mitochondrial uncoupling in muscle. Glycolysis have no significant changes in our study. These results indicated that participated pathways in glycolysis in liver might be different from those in muscle. DJ-1 is closely related to uncoupling proteins (Ucps), namely Ucp1, Ucp3, Ucp4 and Ucp5 [11, 13, 24, 25]. Ucp2 is the unique uncoupling protein expressed in liver [26] and it regulates NAFLD by promoting fatty acid oxidation [27, 28]. Next, we investigated whether DJ-1 can regulate hepatic lipid oxidation. Our study found that DJ-1 deficiency does promote the mRNA expression of Ucp2 and lipid oxidation-related genes. Mitochondrial oxidative phosphorylation is not perfectly transfer energy to ATP, and some of the energy is released in the form of proton-leak [29]. Ucps reduce energy utilization by increasing mitochondrial proton-leak[29]. And there was no difference in hepatic ATP contents (data not shown). Given that deletion of DJ-1 increased hepatic lipid oxidation might through Ucp2. Further work was need to elucidate how DJ-1 regulate Ucp2.
In summary, we have demonstrated that deletion of DJ-1 decreased HFD-induced obesity and visceral adipose accumulation in mice and the potential mechanism may through promoting fatty acid oxidation in mice. However, although DJ-1 promoted HFD-induced hepatic TG accumulation by promoting hepatic lipid oxidation, we did not observe similar results in in vitro primary hepatocytes model, which indicated there were complex networks involves in the liver steatosis formation in DJ-1-/- mice, including: indirect modulation of hepatocytes from obesity and insulin resistance condition.
Ablation of DJ-1 prevents diet-induced hepatic TG accumulation. (A)Morphology of liver from WT and DJ-1-/- mice that were fed a NCD or HFD for 24 weeks.(B)Liver weight of WT and DJ-1-/- mice after HFD administration for 24 weeks.(C) A representative image of H&E(top) and ORO(bottom) staining of liver sections after NCD or HFD treatment for 24 weeks.(D) TG (left), TC (right) contents in the liver of WT and DJ-1-/- mice.(E)Serum lipid contents of TG、TC、LDL-C and HDL-C in WT and DJ-1-/- mice after NCD or HFD administration for 24 weeks. *P < 0.05, **P < 0.01, *** P < 0.001.
DJ-1 enhances hepatic fatty acid oxidation in HFD-mediated mice model. (A) The relative mRNA expression of genes related to fatty acid synthesis in WT and DJ-1-/- mice after HFD administration for 24 weeks. (B) Representative Western blots (left) and quantification (right) for the expression of p-SREBP-1C, m-SREBP-1C and ACLY in the liver of WT and DJ-1-/- mice after HFD administration for 24 weeks. β-actin was used as the loading control. (C) The mRNA expression of glycolysis-related genes was analyzed by RT-PCR. (D) Hepatic lactic acid contents in WT and DJ-1-/- mice at the end of 24th week of HFD feeding. (E) The relative mRNA expression of genes related to fatty acid oxidation in WT and DJ-1-/- mice after HFD administration for 24 weeks. (F) The expression of TCA cycle related gene was analyzed by RT-PCR in WT and DJ-1-/- mice after HFD administration for 24 weeks. *P < 0.05, **P < 0.01, *** P < 0.001.
Abbreviations
NAFLD: nonalcoholic fatty liver diseases; NASH: nonalcoholic steatohepatitis; T2DM: type 2 diabetes mellitus; HFD: high fat diet; NCD: Normal chow diet; eWAT: epididymal white adipose tissue; TG: triglyceride; TC: total cholesterol; LDL-C: low-density lipoprotein cholesterol; HDL-C: high-density lipoprotein cholesterol; GTT: glucose tolerance tests; ITT: insulin tolerance tests; TCA: tricarboxylic acid; PAS: periodic acid-Schiff; H&E: hematoxylin-eosin; ORO: oil red O; WT: wild-type; DJ-1-/-: DJ-1 knockout; SREBP-1c: sterol regulatory element binding protein-1c; ACLY: ATP citrate lyase; Ucps: uncoupling proteins; ROS: reactive oxygen species; Park7: Parkinson disease 7; HPC: hepatic progenitor cell; HCC: hepatocellular carcinoma; DEN: diethylnitrosamine; IL6: interleukin 6; STAT3: signal transducer and activator of transcription 3; PA: palmitic acid; OA : oleic acid.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81670562 and 81873582 to X Kong, 81670598 to Q Xia, 31671453 and 31870905 to H. Wu), and Shanghai Municipal Education Commission—Gaofeng Clinical Medicine Grant Support (20171911) to X Kong, grant from the Committee of Science and Technology of Shanghai Municipal Government (16401970600-03) to X Kong, the National Key Research and Development Program of China (2017YFC0908100) to Q Xia.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. Journal of gastroenterology. 2013;48:434-41
2. Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nature reviews Molecular cell biology. 2015;16:678-89
3. Feige JN, Auwerx J. Transcriptional coregulators in the control of energy homeostasis. Trends in cell biology. 2007;17:292-301
4. Bonifati V, Rizzu P, Squitieri F, Krieger E, Vanacore N, van Swieten JC. et al. DJ-1( PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurological sciences: official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2003;24:159-60
5. Liu W, Wu H, Chen L, Wen Y, Kong X, Gao WQ. Park7 interacts with p47(phox) to direct NADPH oxidase-dependent ROS production and protect against sepsis. Cell research. 2015;25:691-706
6. Xu M, Chen L, Li J, Wu H, Xia Q, Kong X. Emerging roles of DJ-1 in liver diseases through regulation of oxidative stress and immune response. Liver Research. 2018
7. Chen L, Luo M, Sun X, Qin J, Yu C, Wen Y. et al. DJ-1 deficiency attenuates expansion of liver progenitor cells through modulating the inflammatory and fibrogenic niches. Cell death & disease. 2016;7:e2257
8. Yu Y, Sun X, Gu J, Yu C, Wen Y, Gao Y. et al. Deficiency of DJ-1 Ameliorates Liver Fibrosis through Inhibition of Hepatic ROS Production and Inflammation. International journal of biological sciences. 2016;12:1225-35
9. Qiu B, Wang J, Yu Y, Zhen C, Gu J, Liu W. et al. DJ-1 promotes development of DEN-induced hepatocellular carcinoma and proliferation of liver cancer cells. Oncotarget. 2017;8:8499-511
10. Eberhard D, Lammert E. The Role of the Antioxidant Protein DJ-1 in Type 2 Diabetes Mellitus. Advances in experimental medicine and biology. 2017;1037:173-86
11. Wu R, Liu XM, Sun JG, Chen H, Ma J, Dong M. et al. DJ-1 maintains energy and glucose homeostasis by regulating the function of brown adipose tissue. Cell discovery. 2017;3:16054
12. Kim JM, Jang HJ, Choi SY, Park SA, Kim IS, Yang YR. et al. DJ-1 contributes to adipogenesis and obesity-induced inflammation. Scientific reports. 2014;4:4805
13. Shi SY, Lu SY, Sivasubramaniyam T, Revelo XS, Cai EP, Luk CT. et al. DJ-1 links muscle ROS production with metabolic reprogramming and systemic energy homeostasis in mice. Nature communications. 2015;6:7415
14. Mehlem A, Hagberg CE, Muhl L, Eriksson U, Falkevall A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nature protocols. 2013;8:1149-54
15. Wobser H, Dorn C, Weiss TS, Amann T, Bollheimer C, Buttner R. et al. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell research. 2009;19:996-1005
16. Leclercq IA, Da Silva Morais A, Schroyen B, Van Hul N, Geerts A. Insulin resistance in hepatocytes and sinusoidal liver cells: mechanisms and consequences. Journal of hepatology. 2007;47:142-56
17. Sanyal AJ, American Gastroenterological A. AGA technical review on nonalcoholic fatty liver disease. Gastroenterology. 2002;123:1705-25
18. Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. Lancet. 2010;375:2267-77
19. Jain D, Jain R, Eberhard D, Eglinger J, Bugliani M, Piemonti L. et al. Age- and diet-dependent requirement of DJ-1 for glucose homeostasis in mice with implications for human type 2 diabetes. Journal of molecular cell biology. 2012;4:221-30
20. Jain D, Weber G, Eberhard D, Mehana AE, Eglinger J, Welters A. et al. DJ-1 Protects Pancreatic Beta Cells from Cytokine- and Streptozotocin-Mediated Cell Death. PloS one. 2015;10:e0138535
21. Yki-Jarvinen H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. The lancet Diabetes & endocrinology. 2014;2:901-10
22. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism: clinical and experimental. 2016;65:1038-48
23. Hijmans BS, Grefhorst A, Oosterveer MH, Groen AK. Zonation of glucose and fatty acid metabolism in the liver: mechanism and metabolic consequences. Biochimie. 2014;96:121-9
24. Xu S, Yang X, Qian Y, Xiao Q. Parkinson's disease-related DJ-1 modulates the expression of uncoupling protein 4 against oxidative stress. Journal of neurochemistry. 2018
25. Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT. et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696-700
26. Ricquier D, Bouillaud F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. The Biochemical journal. 2000;345(Pt 2):161-79
27. Baffy G. Uncoupling protein-2 and non-alcoholic fatty liver disease. Frontiers in bioscience: a journal and virtual library. 2005;10:2082-96
28. Cho KW, Kim YO, Andrade JE, Burgess JR, Kim YC. Dietary naringenin increases hepatic peroxisome proliferators-activated receptor alpha protein expression and decreases plasma triglyceride and adiposity in rats. European journal of nutrition. 2011;50:81-8
29. Busiello RA, Savarese S, Lombardi A. Mitochondrial uncoupling proteins and energy metabolism. Frontiers in physiology. 2015;6:36
Author contact
![]() Corresponding authors: Xiaoni Kong, PhD, Department of Liver Surgery, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, 160 Pujian Road, Shanghai, China. 200127. Telephone: +86 21 68383283. E-mail: xiaoni-kongcom; Or Qiang Xia, MD, PhD, E-mail: xiaqiangedu.cn; or Lei Xia, MD, drxialeicom
Corresponding authors: Xiaoni Kong, PhD, Department of Liver Surgery, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, 160 Pujian Road, Shanghai, China. 200127. Telephone: +86 21 68383283. E-mail: xiaoni-kongcom; Or Qiang Xia, MD, PhD, E-mail: xiaqiangedu.cn; or Lei Xia, MD, drxialeicom
Received 2018-7-18
Accepted 2018-9-7
Published 2018-10-20