ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2019; 15(3):701-713. doi:10.7150/ijbs.29323 This issue Cite
Research Paper
Sirt6 Suppresses High Glucose-Induced Mitochondrial Dysfunction and Apoptosis in Podocytes through AMPK Activation
Yanqin Fan, Qian Yang, Yingjie Yang, Zhao Gao, Yiqiong Ma, Lu Zhang, Wei Liang, Guohua Ding ![]()
Division of Nephrology, Renmin Hospital of Wuhan University, Wuhan, Hubei, China
Abstract

Previous studies have shown that mitochondrial dysfunction plays an important role in high- glucose(HG)-induced podocyte injury and thus contributes to the progression of diabetic nephropathy(DN). The histone deacetylase Sirtuin6 (Sirt6) has been revealed to have an essential role in the regulation of mitochondrial function in skeletal muscle and cardiomyocytes. However, its specific role in mitochondrial homeostasis in podocytes is undetermined. Here, we aimeds to explore the physiological function of Sirt6 in podocyte mitochondria and apoptosis under HG conditions and explore the possible mechanism. Herein, we observed that Sirt6-WT-1 colocalization was suppressed in the glomeruli of patients with DN. In addition, diabetic mice exhibited reduced Sirt6 expression and AMP kinase (AMPK) dephosphorylation accompanied by mitochondrial morphological abnormalities. In vitro, podocytes exposed to HG presented with mitochondrial morphological alterations and podocyte apoptosis accompanied by Sirt6 and p-AMPK downregulation. In addition, HG promoted a decrease in mitochondrial number and an increase in mitochondrial superoxide production as well as a decreased mitochondrial membrane potential. ROS production was also increased in HG-treated podocytes. Conversely, all these mitochondrial defects induced by HG were significantly alleviated by Sirt6 plasmid transfection. Sirt6 overexpression simultaneously alleviated HG-induced podocyte apoptosis and oxidative stress, as well as increased AMPK phosphorylation. Increased levels of H3K9ac and H3K56ac induced by HG were attenuated in podocytes transfected with Sirt6 plasmids. Therefore, these results elucidated that Sirt6 protects mitochondria of podocytes and exerts anti-apoptotic effects via activating AMPK pathway. The present findings provide key insights into the pivotal role of mitochondria regulation by SIRT6 in its protective effects on podocytes.
Keywords: diabetic nephropathy, Sirt6, podocytes, mitochondrial dysfunction, apoptosis
Introduction
Diabetic nephropathy (DN) is experienced by the majority of individuals with diabetes worldwide and considered as the most common cause of end-stage renal disease [1, 2]. DN imposes a substantial burden on the economy and becomes a severe public health problem [1, 2]. Podocytes are regarded as a crucial component in maintaining the integrity of the glomerular filtration barrier [3]. A reduction in the number of podocytes resulting from apoptosis contributes to the onset of proteinuria and the progression of end-stage renal disease, including DN [4-6]. The extent of podocyte injury is recognized as a major prognostic determinant in chronic glomerular disease [7]. Mitochondrial dysfunction caused by hyperglycemia leads to oxidative damage and cell apoptosis and is increasingly deemed as a contributor to the pathogenesis of DN [1, 8]. Ameliorating mitochondrial dysfunction would be an effective approach to alleviating podocyte apoptosis and thus delaying the progression of DN. Hence, identifying the key molecules involved in the mitochondrial malfunction may provide clues to develop new therapeutic strategies for patients with DN.
Sirt6 is a member of the highly conserved sirtuin family of NAD+-dependent enzymes and has diverse enzymatic activities including deacetylation and ribosylation [9-11]. Sirt6-dependent deacetylation is essential to the regulation of DNA repair, cellular glucose/lipid metabolism, telomere maintenance, cellular senescence and life span [12-16].Until now, only a few studies have revealed that Sirt6 participates in the regulation of podocyte homeostasis and glomerular functional maintenance [17, 18]. These previous studies suggested that Sirt6 was involved in the reduction of pro-inflammatory mediators, amelioration of actin cytoskeleton derangement and attenuation of podocyte apoptosis with high glucose (HG) treatment. However, the effects of Sirt6 on the mitochondrial function in podocytes under HG conditions remain to be elucidated. Recent research has identified that Sirt6 serves as a pivotal mediator of mitochondrial function in skeletal muscle and cardiomyocytes [19, 20]. Therefore, we speculate that the renal protective effects of Sirt6 may be due to its role in maintaining mitochondrial homeostasis.
Cui et al. reported that Sirt6 could activate AMP kinase (AMPK) to maintain metabolic homeostasis in skeletal muscle [19]. As a key enzymatic protein, AMPK links energy sensing to metabolic manipulation. In addition, AMPK exerts mitochondrial protection by regulating the biogenesis and degradation of mitochondria [21-26]. Administration of an AMPK agonist to streptozotocin (STZ)-induced diabetic mice protected the animals from renal damage via improving mitochondrial function [27]. AMPK plays a decisive role in maintaining mitochondrial homeostasis. Thus, we hypothesize that Sirt6 might ameliorate HG-induced mitochondrial dysfunction and podocyte injury through the AMPK pathway.
In this study, we investigated the role of Sirt6 in HG-induced podocyte mitochondrial dysfunction and injury, and further elucidated its potential molecular mechanism.
Materials and methods
Human renal biopsy samples
Renal biopsy samples from patients who had been diagnosed with DN were obtained from the Division of Nephrology, Renmin Hospital of Wuhan University, Wuhan, China. The control samples were para-carcinoma tissues from individuals without diabetes who underwent tumor nephrectomies, and were obtained from the Division of Pathology, Renmin Hospital of Wuhan University, Wuhan, China; histologic examination and biochemical analysis (urine albumin-to-creatinine ratio <30 mg/g) revealed no features of DN or other renal disease except for solitary renal cell carcinoma. Investigation was performed in accordance with the principles of the Declaration of Helsinki. Experiment was performed in accordance with the approved guidelines of Wuhan University and was approved by the Research Ethics Committee of Renmin Hospital of Wuhan University after written informed consent was received from the patients.
Animals
A total of twelve male C57BL/6 mice were obtained from the Hubei Research Center of Experimental Animals. STZ was used to induce a type 1 diabetes mouse model, and the method was employed as described previously [28]. The mice were randomly allocated to the group with diabetes or the control group and were fed a normal chow diet. The animals were raised in an environment with free access to tap water and food pellets. The housing conditions were maintained under an artificial light cycle with controlled temperature and humidity. For the first injection, a dose of STZ (75 mg/kg) was administered to the eight-week-old experimental mice via tail vein injection. Five days later, a second dose of STZ (150 mg/kg) was injected via tail vein. An equal volume of normal saline was administered to age-matched control mice via tail vein injection. Blood glucose from tail vein was measured regularly. Mice with glycemia levels exceeding 16.7 mmol/L were considered diabetic. Twelve weeks after STZ injection, 24 -hour urine samples were collected from metabolic cages for subsequent biochemistry test. ACR (albumin-to-creatinine ratios) was determined by an ADVIA® 2400 Clinical Chemistry System (Siemens, Germany). The mice were sacrificed at 12 weeks post STZ injection. The kidneys were perfused with normal saline before isolation, and part of the kidneys was stored at -80°C for biochemical analysis, part of kidneys was fixed in 4% phosphate-buffered paraformaldehyde for pathological analysis, and the remaining part of the kidneys was fixed in glutaraldehyde prior to further electron microscope examination. The experimental procedures and protocols were approved by the Ethical Committee for the Experimental Use of Animals of Renmin Hospital in Wuhan, China.
Cell culture
Conditionally immortalized human podocytes were kindly provided by Dr. Moin A. Saleem (Academic Renal Unit, Southmead Hospital, Bristol, UK). Podocytes were cultured at 33 °C in medium that consisted of RPMI 1640 medium (HyClone, USA) with 10% fetal bovine serum (BI, Israel), 100 μg /mL streptomycin, 100 U/mL penicillin G, and 1×insulin- transferrin- selenium (ITS) (Invitrogen, USA) for proliferation. To differentiate, the cells were cultured at 37°C for 10-14 days with ITS-free medium. The differrentiated cells were stimulated for 24 h with normal glucose (NG) (5 mM), HG (30 mM) or hypertonic solution (5 mM glucose combined with 25 mM mannitol). All of the experimental results were confirmed in at least three independent cultures of podocytes.
Transfection
The transfection of the Sirt6 plasmid (Addgene) was conducted using the X-tremeGENE HP DNA Transfection Reagent (Roche) in terms of the manufacturer's instructions. A density of 2×105 cells was first seeded in each well of a six-well plate and then transfected with complexes containing 2 μg of Sirt6 plasmid or a negative control with pcDNA3.1 and 2 μl of the X-tremeGENE transfection reagent. Then, the cells were incubated under normal condition for 48 h at 37°C.
Western immunoblotting
Total protein from the glomeruli and podocytes was extracted with RIPA buffer (Beyotime, China) mixed with a protease inhibitor cocktail (Sigma- Aldrich, USA). The extractions were then centrifuged at 13,000 rpm for 10 min at 4°C. The supernatants were mixed with loading buffer prior to being boiled at 100°C for 5 min. Equal amounts of protein samples were separated through SDS-PAGE and then transferred to nitrocellulose membranes (GE Healthcare). Membranes were blocked with milk for 1-2 h before incubation with a primary antibody (Sirt6 rabbit monoclonal antibody, 1:1000, Abcam; p-AMPKα1/2 (Thr172) rabbit polyclonal antibody, 1:100, Santa Cruz Biotechnology; AMPKα1/2 (H-300) rabbit polyclonal antibody, 1:100, Santa Cruz Biotechnology; caspase-3 mouse monoclonal antibody, 1:1000, Novus Biologicals; GAPDH rabbit monoclonal antibody, 1: 1,000, Antgene and GAPDH mouse monoclonal antibody, 1: 1,000, Antgene) overnight at 4°C. An Alexa Fluor 680/ 790-labeled goat anti-rabbit/goat anti-mouse IgG antibody (1:10,000, LI-COR Biosciences, USA) was used as the secondary antibody, and the blots were visualized using a LI-COR Odyssey Infrared Imaging System.
Real-time PCR
Total RNA was extracted from mouse kidney using TRIzol reagent (MRC, USA), and spectrophotometry was used to measure the concentration of the collected RNA. Next, cDNA was synthesized by a reverse transcription kit (TaKaRa, Japan). A real-time fluorescence-based quantitative PCR machine (Illumina Eco, USA) was used to determine the mRNA expression levels. GAPDH was used as control for normalization. The sequences of the primers used are listed in Table 1.
Primers used in real-time PCR.
| Gene | Forward | Reverse |
|---|---|---|
| TFAM | CTCCTAATCTTTACTGTTGC | CTCTAAGCCTCCTCAATACAA |
| NRF1 | CCTTTGGAGAATGTGGTGCG | CCCCGACCTGTGGAATACTTG |
| EER-α | CAGTGAACGCTCTGGTGTCG | GTGAGGAGAAGCCTGGGAT |
| SIRT6 | GAGCGGAAGGTGTGGGAACT | TGAAGTCGGGGATGCCAGAGG |
| GAPDH | TCAAGAAGGTGGTGAAGCAGG | TCAAAGGTGGAGGAGTGGGT |
Immunofluorescence assay
The paraffin-embedded kidney sections were first dewaxed and subjected to antigen retrieval. Then, they were blocked with 5% bovine serum albumin for 30 min at room temperature. The sections were next incubated with a mixture of rabbit anti-Sirt6 polyclonal antibody (1:100, Thermo Fisher Scientific, USA) and mouse anti-WT1 monoclonal antibody (1:100, Novus, USA) overnight at 4°C.The sections were stained with a mixture of Alexa Fluor 488, Donkey anti-mouse IgG (HL) (1:200, Jackson Immuno Research Laboratories, USA) and Alexa Fluor 594, Donkey anti-Rabbit IgG (HL) (1:200, Jackson Immuno Research Laboratories, USA) as the secondary antibodies at 37°C for 60 min in the dark. The nuclei were counterstained with DAPI (Antgene, China) for 5 min.
The cell-climbing films (cells growing on glass slides) were fixed with 4% paraformaldehyde for 30 min at 4°C and stained with rabbit anti-Sirt6 polyclonal antibody (1:100, Thermo Fisher Scientific, USA) overnight at 4°C. The films were then incubated with Alexa Fluor 594, Donkey anti Rabbit IgG (HL) (1:200, Jackson Immuno Research Laboratories, USA) at 37°C for 60 min. The nuclei were counterstained with DAPI (Antgene, China) for 5 min. A confocal microscope (Olympus, Japan) was used to record all of the microscopic images.
Immunohistochemistry
The paraffin-embedded sections were first dewaxed and subjected to antigen retrieval. After blocking with 5% bovine serum albumin for 30 min at room temperature, they were next incubated with Sirt6 primary antibody (1:100, Thermo Fisher Scientific, USA) overnight at 4°C. Subsequently, the sections were incubated with biotin-conjugated secondary antibody and avidin-biotin peroxidase complex (Dako, USA) for 30 min. After DAB (Dako) staining, sections were stained with hematoxylin.
Apoptosis assay
The degree of podocyte apoptosis in vitro was assessed by flow cytometry with AnnexinV-FITC/PI double staining according to the manufacturer's instructions (Annexin V-FITC Apoptosis Detection Kit I, BD Pharmingen, USA).
Podocyte staining for mitochondrial number, mitochondrial superoxide and mitochondrial membrane potential
Human podocytes were cultured on cell- climbing films at 37°C. After culturing with different stimuli, (1) mitochondrial number was determined by MitoTracker Green staining according to the manufacturer's instructions (MitoTracker Green FM, Yeasen, China). In order to count mitochondrial number by Mitotracker Green, ImageJ was employed to measure the total Mito Green fluorescence intensity and the density per area unit. (2) Mitochondrial superoxide was measured by MitoSox Red staining according to the manufacturer's instructions (MitoSox Red Mitochondrial Superoxide Indicator, Yeasen, China). In order to measure mitochondrial superoxide production by MitoSox Red staining, ImageJ was performed to measure the total MitoSox Red fluorescence intensity and the density per area unit. (3) Mitochondrial membrane potential was evaluated by JC-1staining according to the manufacturer's instructions (Mitochondrial membrane potential assay kit with JC-1, Beyotime, China). The images were recorded with a confocal microscope (Olympus, Japan).
Statistical analyses
All values were presented as the mean ± SD and analyzed with SPSS, version 17.0. Differences in mean values were tested using Student's t test or one-way analysis of variance. Differences with P values less than 0.05 were considered statistically significant.
Results
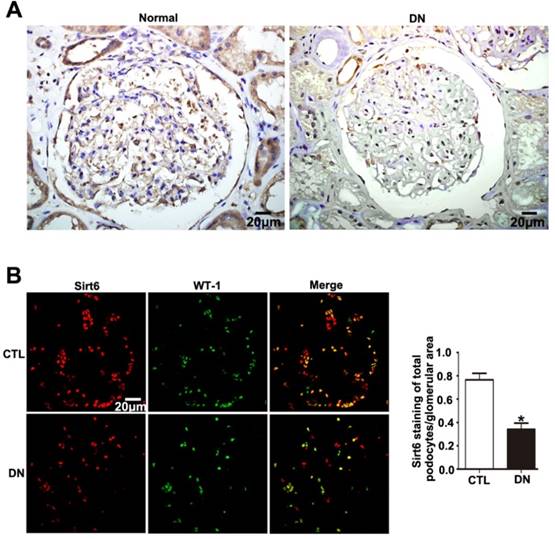
Sirt6 expression in podocytes from patients with DN
Sirt6 was previously suggested to be widely expressed in renal glomeruli of human and was reduced in renal biopsies from DN. In this study, we either observed an obvious reduction in Sirt6 in glomeruli from patients diagnosed with DN compared with glomeruli from healthy subjects by immunohistochemistry staining (Fig. 1A). In addition, we further performed double immunofluorescent staining of Sirt6 and the podocyte marker WT-1 to evaluate Sirt6 expression in glomerular podocytes. The results indicated that Sirt6 was colocalized with WT-1 and mainly localized in the nuclei of podocytes. Decreased Sirt6 expression was detected in podocytes from individuals with DN relative to those from healthy subjects (Fig. 1B). Thus, we speculated that HG may cause a decrease in Sirt6 in glomerular podocytes from patients with DN compared with that in podocytes from healthy subjects.
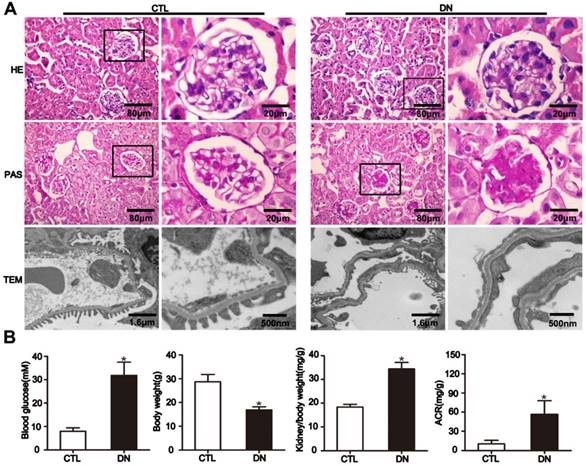
Sirt6 expression in podocytes from mice with DN
To elucidate the effect of HG on Sirt6 expression in podocytes in vivo, we established STZ-induced diabetic mice model. The diabetic mice developed hyperglycemia and a significant increase in albumin- to-creatinine excretion and exhibited a higher kidney/body weight ratio and a lower body weight than their nondiabetic counterparts (Fig. 2B). Mice with diabetes exhibited pathologic changes characteristic of DN including extracellular matrix deposition from 12 weeks after modeling (Fig. 2A, rows 1-2). Furthermore, transmission electron microscopy (TEM) examination revealed evident diffuse foot process fusion and glomerular basement membrane thickening in diabetic mice (Fig. 2A, row 3).
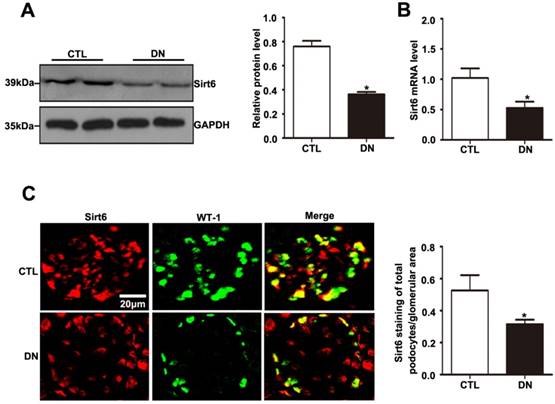
As with the tendency of Sirt6 expression in the kidney in patients with DN, Sirt6 was also reduced in the kidney of STZ-induced diabetic mice (Fig. 3A). The mRNA expression level of Sirt6 was lower in the kidneys of mice in the diabetic group than in those of controls (Fig. 3B). To better understand the changes in Sirt6 expression in the podocytes of mice under hyperglycemic conditions, double immunofluorescent staining of Sirt6 and WT1 was conducted in kidney tissues. A dramatic reduction in Sirt6 was detected in the podocytes in diabetic mice relative to those in control mice (Fig. 3C). Therefore, this result further verified that hyperglycemia-induced Sirt6 levels were decreased in vivo.
Podocyte mitochondrial dysfunction in mice with DN
Mitochondrial dysfunction has been increasingly recognized as a key mediator in DN. However, the in vivo studies related to mitochondria in podocytes under HG conditions have been insufficient. Thus, we focused on the mitochondrial morphological alterations in STZ-induced diabetic mice. Podocyte mitochondria exhibited severe swelling, vacuole formation, and mitochondrial cristae fracture or loss in the diabetic mice (Fig. 4A). Mitochondrial damage might be induced by excessive oxidative stress or impaired mitochondrial biogenesis.
Sirt6 expression in glomeruli from renal biopsies of individuals with DN. (A) Representative images of immunohistochemical staining of Sirt6 in renal cortical tissue of individuals from each group (original magnification, ×400). (B) Representative images of double immunofluorescent staining of WT1 and Sirt6 in kidney sections of individuals from each group (original magnification, ×400) and quantitation of these results (n=4). *P< 0.05 relative to control. Normal=healthy subjects, DN= patients with diabetic nephropathy.
General characteristics of mice with DN. (A) Representative micrographs of HE-stained kidney sections (row 1) (original magnification, ×400), PAS-stained kidney sections (row 2) (original magnification, ×400), and transmission electron micrographs (row 3) (original magnification, ×3 000, ×8 000) from different groups. (B) Metabolic parameters including blood glucose, body weight, kidney/body weight and ACR (albumin-to-creatinine ratios) of mice in each group (n=6). *P< 0.05 relative to control. CTL=control, DN=diabetic nephropathy.
Sirt6 expression in glomeruli from mice with DN. (A) Representative Western blots of glomerular Sirt6 expression and quantitation in the different groups (n=6). *P< 0.05 relative to control. (B) The mRNA level of glomerular Sirt6 expression in each group (n=6). *P< 0.05 relative to control. (C) Representative images of double immunofluorescent staining of glomerular WT1 and Sirt6 in the different groups (original magnification, ×400) and quantitation of these results (n=6). *P< 0.05 relative to control. CTL=control, DN=diabetic nephropathy.
Effects of HG on podocyte mitochondrial morphology and function of mice with DN. (A) Representative images of the ultrastructure of podocyte mitochondria by transmission electron microscopy in each group (original magnification, ×15 000). (B) Representative Western blots of glomerular p-AMPK and AMPK expression in the different groups and quantitation of these results (n=6). *P< 0.05 relative to control. CTL=control, DN=diabetic nephropathy.
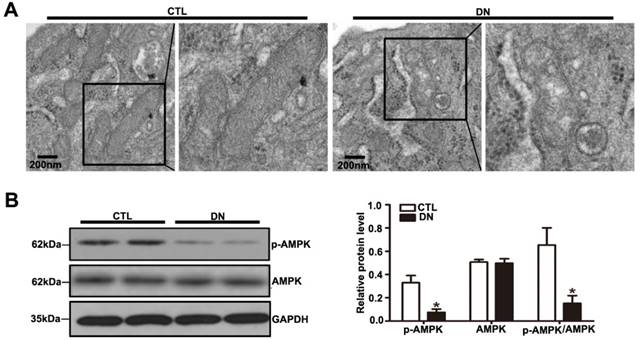
AMPK plays a major role in cellular energy metabolism and redox homeostasis. Previous evidence suggested that AMPK activation can decrease reactive oxygen species (ROS) production and protect mitochondria biogenesis as well [21]. A significant reduction in the p-AMPK / AMPK protein ratio was detected in diabetic mice compared with that in controls by immunoblotting (Fig. 4B). We also assessed the expression of mitochondrial biogenesis molecules in kidney from DN mice. Our results showed that the protein level of PGC-1α, the mRNA levels of NRF-1 and TFAM were decreased in DN mice (Supplementary Fig. 3A and Supplementary Fig. 3B), suggesting that hyperglycemia promoted podocyte mitochondrial dysfunction in mice with DN. Taken together, these suggested that the reduction in Sirt6 expression might be involved in mitochondrial damage in diabetes.
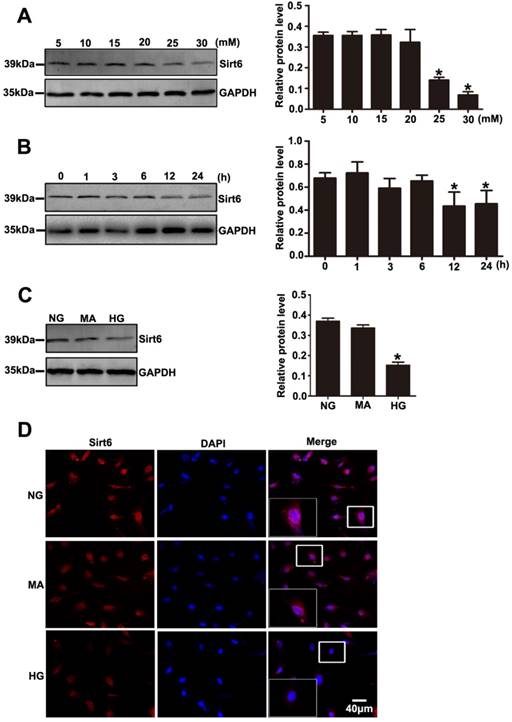
Effects of HG on Sirt6 expression in cultured podocytes
Decreased expression of Sirt6 was also observed in podocytes exposed to HG in vitro. As shown in Fig. 5A, Sirt6 expression was markedly decreased after 24 h in a concentration- dependent manner in the cultured podocytes (5, 10, 15, 20, 25 and 30 mM). We also evaluated the levels of Sirt6 in podocytes incubated in medium without glucose for 24h. The results suggested that Sirt6 was upregulated in podocytes deprived of glucose (Supplementary Fig. 5A). Similarly, a time-dependent reduction in the Sirt6 protein level was exhibited in podocytes under HG (30mM) conditions (0, 1, 3, 6, 12 and 24 h) (Fig. 5B). In addition, HG -treated cells presented a dramatically decreased amount of Sirt6 fluorescence in the nucleus via immunofluorescent assays (Fig. 5D). Furthermore, mannitol had no influence on Sirt6 expression and distribution in podocytes compared with normal culture conditions (Fig. 5C and Fig. 5D). Sirt1 has been confirmed to positively regulate Sirt6 expression [29] .Recently Min Liu et al. reported that Sirt1 also decreased in DN model [18]. Whether Sirt1 is an upstream regulator of SIRT6 in DN model remains unknown. In this study, we used a specific inhibitor of Sirt1 (EX-527) to evaluate the effect of Sirt1 on Sirt6 in HG-treated podocytes. Treatment with EX-527 resulted in a decreased Sirt6 expression in podocytes under HG conditions (Supplementary Fig. 1), which indicated that Sirt6 may be a downstream molecule of Sirt1 in DN model. Taken together, the results suggested that the HG-stimulated alteration in Sirt6 expression might be connected with biological changes in podocytes in diabetes.
Effects of HG on Sirt6 expression in cultured podocytes. (A) Representative Western blots of Sirt6 expression in cultured podocytes stimulated with different concentrations of glucose for 24 h and quantitation of these results (n=4). *P< 0.05 compared with cells treated with NG. (B) Representative Western blots of Sirt6 expression in 30 mM HG-treated podocytes at various times points and quantitation of these results (n=4). *P< 0.05 compared with cells treated with HG for 0 h. (C) Representative Western blots of Sirt6 expression and quantitation of these results in podocytes cultured with different medium (n=4). *P< 0.05 compared with cells in the NG or MA group. (D) Immunofluorescence results of Sirt6 in cultured podocytes in each group(original magnification, ×400). NG=5 mM glucose for 24 h, MA=5 mM glucose+25 mM mannitol for 24 h, HG=30 mM glucose for 24 h.
Effects of HG on mitochondrial dysfunction in cultured podocytes
TEM photomicrographs showed severe mitochondrial swelling, vacuole formation, mitochondrial cristae loss and decreased matrix density in podocytes treated with HG (Fig. 6A). The mitochondrial abnormalities produced by HG in vitro were similar to the mitochondrial morphological alterations observed in diabetic mice. HG induced a decrease in mitochondrial number evaluated by Mito Green immunofluorescent staining (Fig. 6B), while it induced an increase in mitochondrial superoxide production measured by MitoSox Red staining (Fig. 6C).
Effects of HG on mitochondrial morphology and function in cultured podocytes. (A) Representative transmission electron micrographs of mitochondria in cultured podocytes in each group (original magnification, ×8 000, ×15 000). (B) Podocyte mitochondrial numbers were assessed by Mito Green fluorescence staining in different groups (original magnification, ×1 000) and quantitation of these results (n=4). *P< 0.05 compared with cells in the NG or MA group. (C) Mitochondrial oxidative stress and ROS production in podocytes were determined by MitoSox Red fluorescence staining in each group (original magnification, ×1 000) and quantitation of these results (n=4). *P< 0.05 compared with cells in the NG or MA group. (D) Representative images of mitochondrial membrane potential in podocytes by JC-1 staining in different groups (original magnification, ×100) and quantitation of these results.*P< 0.05 compared with cells in the NG or MA group. (E) Western blot results of AMPK and p-AMPK expression in podocytes of different groups and quantitation of these results (n=4).*P< 0.05 compared with cells in the NG or MA group. (F) Western blots detection of cleaved caspase-3 expression in cultured podocytes in each group and quantitation of these results (n=4). *P< 0.05 compared with cells in the NG or MA group. (G) Flow cytometry analysis of apoptosis in cultured podocytes in different groups and quantitation of these results (n=4). *P< 0.05 compared with cells in the NG or MA group. NG=5 mM glucose for 24 h, MA=5 mM glucose+25 mM mannitol for 24 h, HG=30 mM glucose for 24 h.
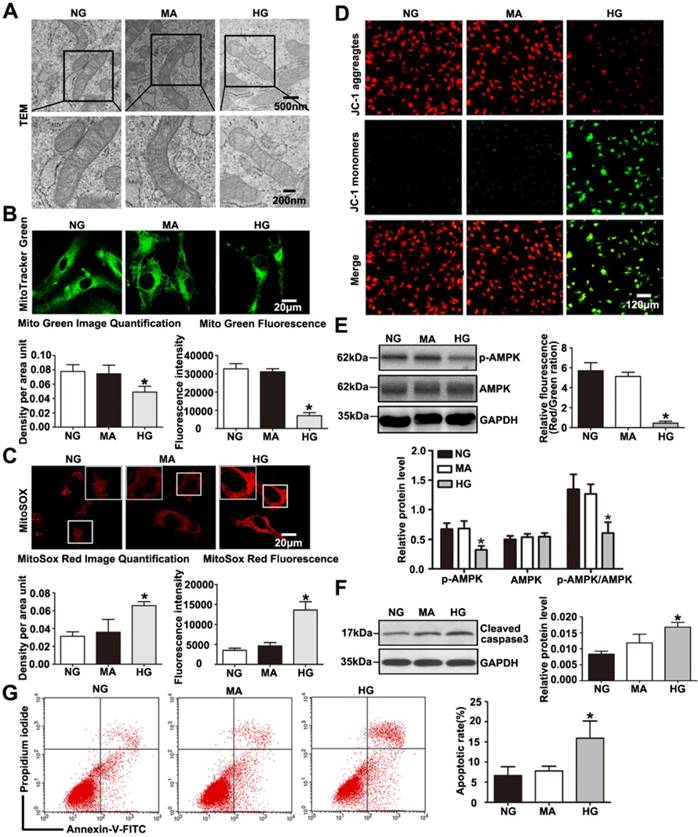
Meanwhile, we found that podocytes with glucose withdrawal also presented an increased mitochondrial superoxide production (Supplementary Fig. 5B). Previous reports have revealed that HG exposure causes injury to mitochondria combined with mitochondrial membrane potential decline [30]. Therefore, we further evaluated the effect of HG on mitochondrial membrane potential by JC-1 staining. As shown in (Fig. 6D), HG markedly provoked a decrease in mitochondrial membrane potential, which signified dramatic mitochondrial damage induced by HG. DCFH-DA fluorescent probe was conducted to detect cellular oxidative stress, we found that intracellular ROS production was markedly increased in podocytes treated by HG (Supplementary Fig. 6). Further, we discovered that HG caused obvious decreased levels of PGC-1α and NRF-1 and TFAM, which were biogenesis molecules related to mitochondria (Supplementary Fig. 3C and Supplementary Fig. 3D). Damaged Podocytes treated with HG presented a reduction in p-AMPK and the p-AMPK /AMPK protein ratio (Fig. 6E). A reduction in p-AMPK expression might be involved in mitochondrial morphological and functional disorders. As shown in (Fig. 6F-6G), HG ultimately markedly caused podocyte apoptosis, which was consistent with our previous observation [31]. Thus, the results demonstrated that HG stimulated- podocyte injury may be attributed to mitochondrial dysfunction, and AMPK alteration might be involved in the regulatory mechanism.
Sirt6 overexpression attenuated HG-induced mitochondrial dysfunction and apoptosis in podocytes through AMPK activation
Sirt6 has been investigated for its ability to suppress mitochondrial defects in cardiomyocytes under hypoxia/reoxygenation treatment and to maintain mitochondria function in skeletal muscle[19, 20]. However, whether Sirt6 modulates mitochondrial function in podocytes with HG stimulation remains unclear. To further validate the mitochondrial functional protection of Sirt6 in HG-treated podocytes, pc-DNA3.1SIRT6 was transfected into cultured podocytes followed by subsequent culture under HG conditions. As shown in Fig. 7A and 7F, pc-DNA3.1SIRT6 transfection significantly reversed the Sirt6 downregulation and dephosphorylation of AMPK observed in HG-treated podocytes compared with the effects observed in untransfected or pcDNA3.1-transfected podocytes. Sirt6 overexpression largely attenuated the mitochondria defects induced by HG (Fig. 7B, 7C, 7D and 7E). Moreover, Sirt6 overexpression alleviated HG produced cellular ROS production in podocytes (Supplementary Fig. 6). It has been demonstrated that Sirt6 protected against podocyte injury and proteinuria via Notch signaling in DN model [18]. Furthermore, we employed AICAR, which was a specific activator of AMPK, to elucidate the relationship between AMPK signaling and Notch signaling in vitro. After the transfection with scrambled or Sirt6 siRNA for 24h, podocytes were treated with AICAR (1mM, 24h). The results showed that gene silencing of Sirt6 resulted in AMPK dephosphorylation and increased Notch1 and Notch4 expression. However, activating AMPK could attenuate the Notch upregulation induced by Sirt6 siRNA (Supplementary Fig. 2). In addition, cell apoptosis was significantly ameliorated after the transfection of the Sirt6 plasmid in HG-stimulated podocytes (Fig. 7G and 7H). However, Sirt6 overexpression had no effects on PGC-1α, NRF-1 and TFAM expression in HG-stimulated podocytes (Supplementary Fig. 3C and Supplementary Fig. 3D). As Sirt6 is a histone deacetylase which has the ability to target specific histone substrates to modifying gene expression [32]. We have measured the acetylation levels of H3K9 and H3K56 in podocytes transfected with WT Sirt6 plasmids or Sirt6 H133Y mutant plasmids under HG conditions (Figure S4). The results indicated that HG-treated podocytes presented increased acetylation of the two specific histone substrates. Overexpression of WT Sirt6 but not the H133Y mutant could attenuate the increase in H3K9 and H3K56 acetylation (Supplementary Fig. 4). To summarize, these findings indicated that Sirt6 protects podocytes against HG-induced mitochondrial dysfunction and apoptosis. AMPK is likely to be a downstream intermediate of Sirt6 to rescue HG-induced mitochondrial abnormalities and podocyte injury. Moreover, such protective effects of Sirt6 may depend on its deacetylation of H3K9 and H3K56.
Discussion
Mitochondrial malfunction has been proposed to play a crucial role in hyperglycemia-induced podocyte injury and promote the development of DN[1, 8, 33, 34]. Current understanding of mitochondrial homestasis in podocytes in the HG envirnoment is still limited , and finding the key molecules involved in mitochondrial regulation in podocytes are meaningful. Here, we demonstrated that Sirt6-mediated AMPK signaling contributes to HG-induced mitochondrial dysfunction and apoptosis in podocyte, which indicates the possibilities of therapies targeting Sirt6 for the treatment of patients with DN.
Sirt6 overexpression attenuated HG induced mitochondrial dysfunction and podocyte injury. Podocytes were transfected with no plasmid, pcDNA3.1, or pcDNA3.1-SIRT6 and then stimulated with HG (30 mM) for 24 h. Untreated and untransfected podocytes were regarded as normal cells. (A) Representative Western blots and quantitative analysis showing the relative protein level of Sirt6 after pcDNA3.1-SIRT6 transfection of podocytes in different groups (n=4). *P< 0.05 compared with normal cells. #P< 0.05 compared with cells treated with HG only. (B) Ultrastructural changes in podocytes by transmission electron microscopy in each group (original magnification, ×8 000, ×15 000). (C) Mitochondrial numbers of podocytes were determined by Mito Green fluorescence staining in different groups (original magnification, ×1 000) and quantitation of these results (n=4). *P< 0.05 compared with normal cells. #P< 0.05 compared with cells treated with HG only. (D) Mitochondrial oxidative stress and ROS production were evaluated by MitoSox Red fluorescence staining in different groups (original magnification, ×1 000) and quantitation of these results (n=4). *P< 0.05 compared with normal cells. #P< 0.05 compared with cells treated with HG only. (E) Representative images of mitochondrial membrane potential of podocytes by JC-1 staining in each group (original magnification, ×100) and quantitation of these results. *P< 0.05 compared with normal cells. #P< 0.05 compared with cells treated with HG only. (F) Western blots detection of AMPK and p-AMPK expression in podocytes for each group and quantitation of these results (n=4). *P< 0.05 compared with normal cells. #P< 0.05 compared with cells treated with HG only. (G) Western blots results of cleaved caspase-3 expression of podocytes in each group and quantitation of these results (n=4). *P< 0.05 compared with normal cells. #P< 0.05 compared with cells treated with HG only. (H) Flow cytometry analysis of apoptosis in the different groups and quantitation of these results (n=4). *P< 0.05 compared with normal cells. #P< 0.05 compared with cells treated with HG only.
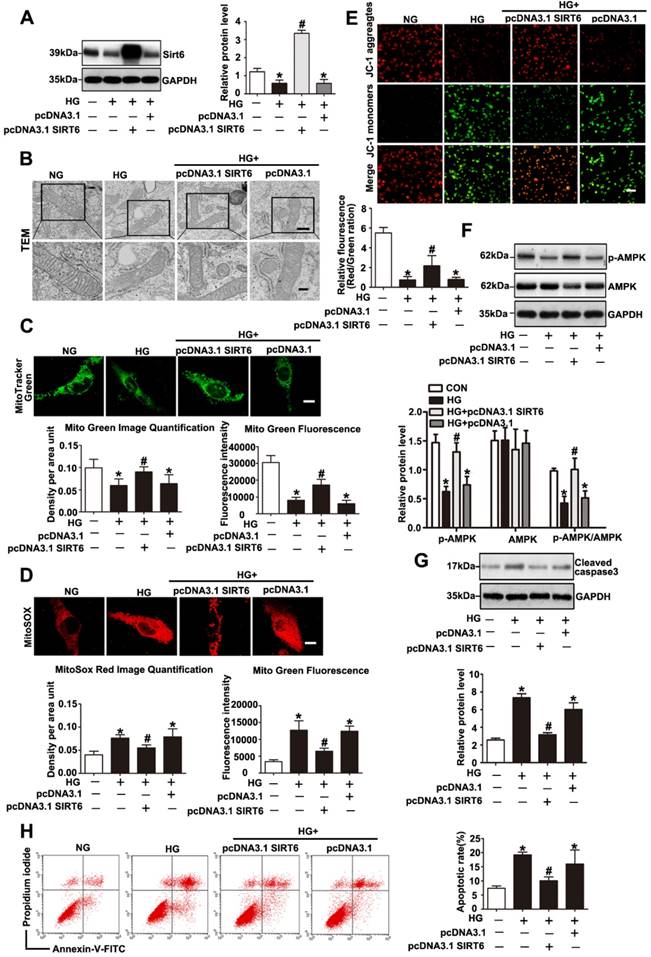
SIRT6 belongs to the sirtuin family of deacetylases, and it has been proved to mediates pleiotropic physiological activities in kidney. Min Liu et al. discovered that HG-induced a reduction in Sirt6 expression both in vivo and in vitro studies, and they detected a reduction in Sirt6 expression in the kidney that was associated with estimated GFR and proteinuria levels from patients with different types of podocytopathies. In this study, we found a downregulation of Sirt6 expression in podocytes of diabetic mice and in podocytes cultured with HG, which was in consistent with that obtained by Min Liu et al.[17]. Further, we put forwarded that Sirt6 was decreased in podocytes in glomerulus from patients with DN by double immunofluorescent staining, which provided the basis for clinical research of Sirt6. We also found that Sirt1 was decreased in HG-treated podocytes and Sirt1 overexpression ameliorated HG-induced Srit6 reduction in vitro. Sirt1 has been demonstrated with a protective role in limiting podocyte injury in DN. We considered that Sirt1 was associated with the reduction of Sirt6 in DN model. Podocyte-specific Sirt6 knockout was previously confirmed to exacerbate the urinary albumin excretion and the glomerular injury in diabetic mice. The renoprotective effects of Sirt6 were related to its inhibition effects on apoptosis, the inflammatory response and renal fibrosis [17, 18]. In addition, Sirt6 was demonstrated to suppress the kidney aging through inhibiting NF-ΚB pathway[17, 18]. These results indicated that the reduction in Sirt6 may be a common feature of podocytes under HG conditions, and we speculated that the expression and distribution of Sirt6 under HG environment might be involved in the biological changes in podocytes in DN.
Mitochondria play pivotal roles in contributing to cell apoptosis and have been demonstrated to have great importance in the pathogenesis of DN[1, 8, 33, 34]. We presently confirmed that diabetic mice exhibited dramatic mitochondrial damage. We observed significant mitochondrial swelling combined with mitochondrial cristae fracture or a loss of cristae membranes. The levels of PGC-1α, TFAM and NRF-1 were decreased in the diabetic mice and HG-treated podocytes, which are important in the control of mitochondrial biogenesis and antioxidant generation [35, 36]. As a metabolic perturbation common in DN, the phenotype of podocytes cultured in HG environments resemble the characteristics of DN. Podocytes treated with HG presented similar mitochondrial morphological abnormities and further exhibited dramatically decreased mitochondrial membrane potentials, which is a marker of apoptosis. Previous studies suggested that mitochondrial dysfunction could be ascribed to the alterations in matrix volume [37]. Mitochondrial swelling is a pivotal step for the occurrence of apoptosis as the inner membrane is disrupted and mitochondrial proapoptotic factors are released [38]. The protein complexes of the electron transport chain are located on the cristae membranes, and shrinking of the cristae membranes would result in reduced ATP production and ultimately lead to apoptosis [39]. Additionally, we found decreased mitochondrial numbers and increased superoxide production in podocytes under HG conditions. HG-induced mitochondrial overproduction of ROS is the primary initial mechanism of DN. We showed the direct evidence that HG stimulated ROS production in podocytes. Mitochondrial damage triggered by hyperglycemia lead to excess production and accumulation of ROS which stimulate oxidative stress and apoptosis. Superoxide production is predominantly generated from electron leaks in the mitochondrial electron transport system [40], and therefore, damaged mitochondrial cristae cause mass superoxide production. Our findings confirmed that mitochondrial dysfunction and apoptosis existed in podocytes under HG envirment and the HG-induced podocyte injury may be partly due to morphological and structural disorders of mitochondria.
Emerging studies have indicated that Sirt6 has an inhibitory effect on mitochondrial damage both in cardiomyocytes and skeletal muscle[19, 20]. Considering that the decrease of Sirt6 expression was accompanied by mitochondrial malfunction in the HG environment, we speculated that Sirt6 is involved in HG-induced mitochondrial dysfunction. In order to explore the role of Sirt6 in this regard, cells were transfected with the Sirt6 plasmid before stimulated with HG. In the present study, it revealed that Sirt6 overexpression could effectively reduce podocyte apoptosis accompanied by AMPK phosphorylation. Upregulation of Sirt6 was demonstrated to significantly reduce mitochondrial superoxide content and cellular ROS production, which revealed that Sirt6 played a pivotal role in reducing cellular oxidative stress. Meanwhile, Sirt6 overexpression could stabilize the integrity of the mitochondrial membrane in HG-treated podocytes. Reductions of the mitochondrial membrane potential in podocytes cultured in HG was alleviated after Sirt6 plasmid transfection. AMPK is a highly conserved cellular bioenergetic sensor that contributes to the regulation of multiple cellular functions ranging from energy homeostasis to cell death regulation[21]. Growing evidence has suggested that AMPK signaling malfunction is proved to have pathological implications in mitochondrial diseases. AMPK has been found to have vital importance to the control of the quantity of mitochondria via controlling both their biogenesis and degradation and thus maintains mitochondrial homeostasis[21-26]. In our study, we have discovered that AMPK was an upstream regulator of Notch1 in podocytes. Notch1 pathway activation controls the mitochondrial proteome and induce proteins alteration involved in common metabolic pathway [41]. It has been proved to promote reprogramming of mitochondrial metabolism for M1 macrophage activation [42]. Notch1 might also contribute to the protection of mitochondrial function regulated by Sirt6. And Notch1 may be a key intermediate molecule involved in the AMPK signaling pathway regulated by Sirt6.
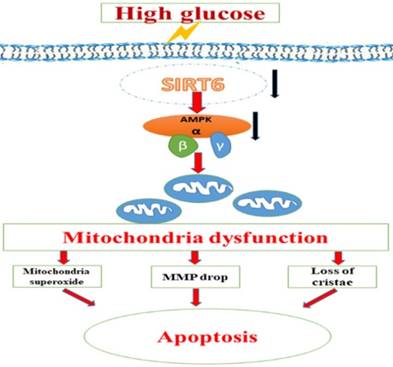
A schematic summary of the effects of Sirt6 on HG-induced mitochondrial dysfunction and apoptosis. HG can inhibit Sirt6 expression and induce AMPK dephosphorylation, which contributes to mitochondrial dysfunction in podocytes including increased mitochondrial superoxide production, lower mitochondrial membrane potential and loss of mitochondrial cristae, which finally lead to apoptosis in podocytes. However, Sirt6 overexpression attenuates HG-induced mitochondrial dysfunction and apoptosis via promoting AMPK phosphorylation.
Sirt6 is a complex enzyme with multiple deacetylase substrates, which can target specific sites on histone H3 to regulate gene expression [32]. Thus, Sirt6 exerts its suppressive effects on several transcription factors implicated in aging, metaboliam and cancer. H3K9 has been proved to play an important role in the Notch signaling pathway regulated by Sirt6 [18]. It has been suggested that Sirt6 suppresses Notch signaling via deacetylating H3K9. Our studies detected that overexpression of Sirt6 weakened the increased levels of H3K9ac and H3K56ac caused by HG in podocytes. We speculated that both H3K9 and H3K56 may be involved in regulation of the AMPK signaling pathway controlled by Sirt6 in podocytes.
Taken together, we speculated that the protective effect of Sirt6 on podocyte injury caused by HG could be attributed to the suppression of mitochondrial dysfunction through AMPK activation, including reducing mitochondrial superoxide production, increasing mitochondrial membrane potential and maintaining intact mitochondrial morphology (Fig 8). Nonetheless, we could not exclude other downstream molecules of Sirt6 that may also contribute to the regulation of mitochondrial function and apoptosis in podocyte. For instance, as lacking of the specific activator of Sirt6, it is unable to further investigate the specific effects of Sirt6 on mitochondrial homestasis in kidney.
Collectively, this study identified that Sirt6 protects against HG induced podocyte injury and mitochondrial malfunction, at least in part via activating AMPK. Our results suggested that Sirt6 might be regarded as a potential therapeutic target for the treatment of DN. These findings could contribute to further studies on the Sirt6 pathways in clinical settings.
Abbreviations
HG: high-glucose; DN: diabetic nephropathy; Sirt6: sirtuin6; AMPK: AMP kinase; STZ: streptozotocin; TEM: transmission electron microscopy; ROS: reactive oxygen species.
Supplementary Material
Supplementary figures.
Acknowledgements
These studies were supported by grants from the National Natural Science Foundation of China (81570617 and 81770687 to G.D., 81700600 to Q.Y., and 81800615 to Y.M.). All authors approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Higgins GC, Coughlan MT. Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy?. Br J Pharmacol. 2014;171:1917-42
2. Ahmad J. Management of diabetic nephropathy: Recent progress and future perspective. Diabetes Metab Syndr. 2015;9:343-58
3. Mathieson PW. The podocyte as a target for therapies-new and old. Nat Rev Nephrol. 2011;8:52-6
4. Zhou L, Liu Y. Wnt/beta-catenin signalling and podocyte dysfunction in proteinuric kidney disease. Nat Rev Nephrol. 2015;11:535-45
5. Burlaka I, Nilsson LM, Scott L, Holtback U, Eklof AC, Fogo AB. et al. Prevention of apoptosis averts glomerular tubular disconnection and podocyte loss in proteinuric kidney disease. Kidney Int. 2016;90:135-48
6. Shankland SJ. The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69:2131-47
7. Anil Kumar P, Welsh GI, Saleem MA, Menon RK. Molecular and cellular events mediating glomerular podocyte dysfunction and depletion in diabetes mellitus. Front Endocrinol (Lausanne). 2014;5:151
8. Yuan Y, Huang S, Wang W, Wang Y, Zhang P, Zhu C. et al. Activation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha ameliorates mitochondrial dysfunction and protects podocytes from aldosterone-induced injury. Kidney Int. 2012;82:771-89
9. Gil R, Barth S, Kanfi Y, Cohen HY. SIRT6 exhibits nucleosome-dependent deacetylase activity. Nucleic Acids Res. 2013;41:8537-45
10. Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C. et al. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013;496:110-3
11. Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A. et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011;332:1443-6
12. Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L. et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218-21
13. Kanfi Y, Peshti V, Gil R, Naiman S, Nahum L, Levin E. et al. SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell. 2010;9:162-73
14. Masri S, Rigor P, Cervantes M, Ceglia N, Sebastian C, Xiao C. et al. Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell. 2014;158:659-72
15. Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V. et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat Med. 2012;18:1643-50
16. Zhong L, D'Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD. et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140:280-93
17. Huang W, Liu H, Zhu S, Woodson M, Liu R, Tilton RG. et al. Sirt6 deficiency results in progression of glomerular injury in the kidney. Aging. 2017;9:1069
18. Liu M, Liang K, Zhen J, Zhou M, Wang X, Wang Z. et al. Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling. Nature Communications. 2017;8:413
19. Cui X, Yao L, Yang X, Gao Y, Fang F, Zhang J. et al. SIRT6 regulates metabolic homeostasis in skeletal muscle through activation of AMPK. American Journal of Physiology Endocrinology & Metabolism. 2017;313:ajpendo.00122.2017
20. Cheng MY, Cheng YW, Yan J, Hu XQ, Zhang H, Wang ZR. et al. SIRT6 suppresses mitochondrial defects and cell death via the NF-kappaB pathway in myocardial hypoxia/reoxygenation induced injury. Am J Transl Res. 2016;8:5005-15
21. Madhavi YV, Gaikwad N, Yerra VG, Kalvala AK, Nanduri S, Kumar A. Targeting AMPK in Diabetes and Diabetic Complications: Energy homeostasis, Autophagy and Mitochondrial health. Curr Med Chem. 2018
22. Hardie D. AMP-activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol. 2007;47:185-210
23. Jäger S, Handschin C, Pierre JS, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12017
24. Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M. et al. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010;11:213-9
25. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016-23
26. Palikaras K, Tavernarakis N. Mitochondrial homeostasis: the interplay between mitophagy and mitochondrial biogenesis. Exp Gerontol. 2014;56:182-8
27. Dugan LL, You YH, Ali SS, Diamond-Stanic M, Miyamoto S, DeCleves AE. et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest. 2013;123:4888-99
28. Kai Z, Kakehi T, Matsumoto M, Iwata K, Ibi M, Ohshima Y. et al. NADPH oxidase NOX1 is involved in activation of protein kinase C and premature senescence in early stage diabetic kidney. Free Radical Biology & Medicine. 2015;83:21-30
29. Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A. et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010;12:224-36
30. Wang RM, Wang ZB, Wang Y, Liu WY, Li Y, Tong LC. et al. Swiprosin-1 Promotes Mitochondria-Dependent Apoptosis of Glomerular Podocytes via P38 MAPK Pathway in Early-Stage Diabetic Nephropathy. Cell Physiol Biochem. 2018;45:899-916
31. Ma Y, Yang Q, Chen X, Liang W, Ren Z, Ding G. c-Abl contributes to glucose-promoted apoptosis via p53 signaling pathway in podocytes. Diabetes research and clinical practice. 2016;113:171-8
32. Tasselli L, Zheng W, Chua KF. SIRT6: Novel Mechanisms and Links to Aging and Disease. Trends Endocrinol Metab. 2017;28:168-85
33. Abe Y, Sakairi T, Kajiyama H, Shrivastav S, Beeson C, Kopp JB. Bioenergetic characterization of mouse podocytes. Am J Physiol Cell Physiol. 2010;299:C464-76
34. Wang L, Li SJ, Sidhu A, Zhu L, Liang Y, Freedman RB. et al. Reconstitution of human Ero1-Lalpha/protein-disulfide isomerase oxidative folding pathway in vitro. Position-dependent differences in role between the a and a' domains of protein-disulfide isomerase. The Journal of biological chemistry. 2009;284:199-206
35. Wang XX, Edelstein MH, Gafter U, Qiu L, Luo Y, Dobrinskikh E. et al. G Protein-Coupled Bile Acid Receptor TGR5 Activation Inhibits Kidney Disease in Obesity and Diabetes. Journal of the American Society of Nephrology Jasn. 2016;27:1362
36. Lee EJ, Kang YC, Park WH, Jeong JH, Pak YK. Negative transcriptional regulation of mitochondrial transcription factor A (TFAM) by nuclear TFAM. Biochemical & Biophysical Research Communications. 2014;450:166-71
37. Cai X, Bao L, Ren J, Li Y, Zhang Z. Grape seed procyanidin B2 protects podocytes from high glucose-induced mitochondrial dysfunction and apoptosis via the AMPK-SIRT1-PGC-1α axis in vitro. Food & Function. 2016;7:805-15
38. Michea L, Combs C, Andrews P, Dmitrieva N, Burg MB. Mitochondrial dysfunction is an early event in high-NaCl-induced apoptosis of mIMCD3 cells. American Journal of Physiology Renal Physiology. 2002;282:F981
39. Szeto HH, Liu S, Yi S, Alam N, Prusky GT, Seshan SV. Protection of mitochondria prevents high fat diet-induced glomerulopathy and proximal tubularinjury. Kidney International. 2016;90:997-1011
40. Murphy MP. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417:1
41. Basak NP, Roy A, Banerjee S. Alteration of mitochondrial proteome due to activation of Notch1 signaling pathway. The Journal of biological chemistry. 2014;289:7320-34
42. Xu J, Chi F, Guo T, Punj V, Lee WN, French SW. et al. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J Clin Invest. 2015;125:1579-90
Author contact
![]() Corresponding author: Guohua Ding, Division of Nephrology, Renmin Hospital of Wuhan University, 238 Jiefang Rd, Wuhan, Hubei 430060, China. Tel: +86 27 88041919; fax: +86 27 88042292;Email:ghxdingcom
Corresponding author: Guohua Ding, Division of Nephrology, Renmin Hospital of Wuhan University, 238 Jiefang Rd, Wuhan, Hubei 430060, China. Tel: +86 27 88041919; fax: +86 27 88042292;Email:ghxdingcom
Received 2018-8-18
Accepted 2018-12-21
Published 2019-1-24