ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
1. Introduction
2. Materials and methods
3. Results
4. Discussion
Aknowledgements
References
Figures
Author biography

Int J Biol Sci 2005; 1(1):1-12. doi:10.7150/ijbs.1.1 This issue Cite
Research Paper
RNA interference is ineffective as a routine method for gene silencing in chick embryos as monitored by fgf8 silencing
Victor Hernández Hernández, David Bueno ![]()
Departament de Genètica, Facultat de Biologia, Universitat de Barcelona, Av. Diagonal 645, 08028 Barcelona, Catalonia, Spain
Abstract
The in vivo accessibility of the chick embryo makes it a favoured model system for experimental developmental biology. Although the range of available techniques now extends to miss-expression of genes through in ovo electroporation, it remains difficult to knock out individual gene expression. Recently, the possibility of silencing gene expression by RNAi in chick embryos has been reported. However, published studies show only discrete quantitative differences in the expression of the endogenous targeted genes and unclear morphological alterations. To elucidate whether the tools currently available are adequate to silence gene expression sufficiently to produce a clear and specific null-like mutant phenotype, we have performed several experiments with different molecules that trigger RNAi: dsRNA, siRNA, and shRNA produced from a plasmid coexpressing green fluorescent protein as an internal marker. Focussing on fgf8 expression in the developing isthmus, we show that no morphological defects are observed, and that fgf8 expression is neither silenced in embryos microinjected with dsRNA nor in embryos microinjected and electroporated with a pool of siRNAs. Moreover, fgf8 expression was not significantly silenced in most isthmic cells transformed with a plasmid producing engineered shRNAs to fgf8. We also show that siRNA molecules do not spread significantly from cell to cell as reported for invertebrates, suggesting the existence of molecular differences between different model systems that may explain the different responses to RNAi. Although our results are basically in agreement with previously reported studies, we suggest, in contrast to them, that with currently available tools and techniques the number of cells in which fgf8 gene expression is decreased, if any, is not sufficient to generate a detectable mutant phenotype, thus making RNAi useless as a routine method for functional gene analysis in chick embryos.
Keywords: RNA interference (RNAi), small interfering RNA (siRNA), short hairpin RNA (shRNA), chick embryo, isthmus, fgf8
1. Introduction
The in ovo accessibility of the chick embryo makes it a favoured model system for in vivo studies of developmental processes. Since the development of in ovo gene transfer by electroporation [1], it has been possible to analyse the function of a gene by overexpressing it in specific tissues. However, it is still difficult to knock out the function of a gene of interest because it is not yet possible to make mutant animals by gene targeting. Recently however, the possibility of silencing gene expression by RNA interference (RNAi) has been reported.
RNAi is thought to be an evolutionarily conserved process that responds to the presence of double-stranded RNA (dsRNA) [2, 3, 4, 5]. The mechanism through which RNAi functions is based on the action of a set of enzymes that cleave the dsRNA into small interfering 21-23mer RNAs (siRNA) that act as templates for the specific degradation of the corresponding mRNAs [3, 6, 7, 8]. The key enzyme in this process is Dicer, a Ribonuclease III family molecule containing a PAZ domain and two dsRNA binding domains [9].
RNAi has been successfully used in several invertebrates, e.g. in C. elegans and Drosophila, both through dsRNA and constructs expressing siRNA [10, 11, 12, 13, 14]. It was initially postulated that this technique could not be used in vertebrates because the intracellular presence of dsRNA may trigger an antiviral response mediated by a dsRNA-dependent protein kinase (PKR) and the interferon system, which inhibits transcription [15]. However, the discovery of siRNAs, which are thought to escape the PKR, raised the possibility of using RNAi in vertebrates, although there are some reports suggesting that in mammalian cells siRNAs may actually upregulate some interferon response genes [16, 17].
Several recent papers have suggested that RNAi is effective in zebrafish, mouse and chick embryos. In zebrafish, Wargelius [18] and Li [19] reported the application of RNAi by injecting dsRNAs at 1-2 cell stage. However, Oates [20], using dsRNAs from several genes, were not able to distinguish the phenotypes produced by them, suggesting a non-specific effect of dsRNA on gene silencing. In mice, the use of RNAi has been reported in cultured cell lines [21, 22, 23], in oocytes [24, 25], and in embryos [23, 26, 27]. In these studies, the expression of the gene to be silenced was only partially reduced in a population of cells, and no morphological alterations were reported, with the exception of a mild mutant phenotype observed in the derivates of some oocytes when compared with corresponding null mutants [23].
Finally, although the few published works on RNAi in chick embryos are very optimistic, they show discrete quantitative differences in the expression of endogenous target genes and limited, if any, morphological alterations [28, 29, 30, 31]. It is important to note that in all of these studies the transfection efficiency was assessed by using independent reporter plasmids as markers, thereby precluding the identification of the individual cells containing the molecules that trigger RNAi.
We have performed several experiments to elucidate whether the currently available tools are adequate to silence gene expression in chick embryos to a sufficient extent to produce a clear and specific null-like mutant phenotype using internal markers for siRNA and shRNA localisation. These experiments focused on RNAi of fgf8 in the isthmus. Fgf8 is a member of the fibroblast growth factor gene family, which codes for ligands that act to promote the growth and differentiation of many mesodermal and ectodermal cells by binding to specific receptors. Its expression is spatially regulated during development, e.g. in the isthmic organizer [32, 33]. The isthmic organizer, located at the mid-hindbrain boundary, exhibits a morphogenetic activity that acts on the midbrain neuroectoderm [34, 35]. Both fgf8 hypomorph homozygote mutant mice and zebrafish acerebellar mutants, which carry a mutation affecting fgf8, lack posterior midbrain and cerebellar tissue [36, 37, 38].
We microinjected different potential triggers for RNAi into the neural tube: fgf8 dsRNA; a pool of siRNAs to fgf8 obtained in vitro by Dicer-digestion, some of which were labelled with digoxigenin (DIG) to monitor their uptake at a cellular level; and the psiRNA-hH1GFPzeo G2 plasmid engineered to express short hairpin RNAs (shRNAs) to fgf8. The shRNA-producing vector also expresses green fluorescent protein (GFP) as an internal transfection marker. Although our results are basically in agreement with previously reported studies on the silencing of endogenous genes in chick embryos, i.e. some cells expressing shRNA to fgf8 may show a reduction in fgf8 expression, we suggest that the currently available tools and techniques are inadequate to generate a detectable mutant fgf8 phenotype at the embryo level, thus making RNAi useless as a routine method to study gene function in chick embryos.
2. Materials and methods
Production of dsRNA and in vitro Dicer-digestion
Fgf8 dsRNA was produced from a fragment of cDNA encoding fgf8b cloned in pBluescript KS [39]. After linearization with EcoRI or XhoI, RNA synthesis was carried out in vitro from T7 and T3 promoters for antisense and sense RNAs respectively. For each direction, 2 μg DNA was mixed with a final concentration of 4mM rNTPs or DIG-rNTPs (Roche) to synthesise dsRNA or DIG-labelled dsRNA respectively, and with the appropriate concentration of T3 and T7 polymerases (Roche). After 2 h transcription at 37ºC for the T7 promoter and 25ºC for T3 promoter (to produce equimolar concentrations), 1 μl of DNAse I (Roche) was added and the mixture incubated for 15 min at 37ºC. The reaction was stopped with 380 μl of STOP solution (1M NH4OAc, 10mM EDTA, 0.2% SDS). The RNA was extracted first with phenol:chloroform 1:1 (v/v) and then chloroform. Equal amounts of sense and antisense RNA from the aqueous layer were mixed, heated to 70ºC for 10 min and annealed at 37ºC for 30 min. After precipitation in ethanol, the dsRNA was dissolved in 10 μl of DMEM (Gibco) supplemented with 0.3 μg/ml Vital Red/DMEM to visualize microinjection, or in 10 μl of H20 for in vitro Dicer-digestion. For quality control, 1μl of each sample was analyzed by gel electrophoresis (0.8% agarose).
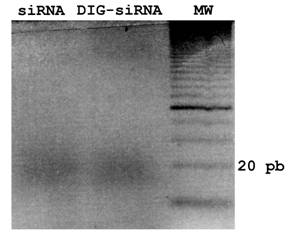
To obtain a pool of siRNAs or DIG-labelled siRNAs from fgf8 dsRNA, dsRNA or DIG-labelled dsRNA dissolved in H20 were digested and purified using the Dicer Kit (Gene Therapy Systems), according to the manufacturers instructions. For quality controls, samples were analyzed by gel electrophoresis (3% agarose). The efficiency of in vitro Dicer-digestion of DIG-dsRNAs was also tested on an agarose gel, showing no differences with respect to control siRNAs (Fig. 1).
Plasmids and shRNA constructs
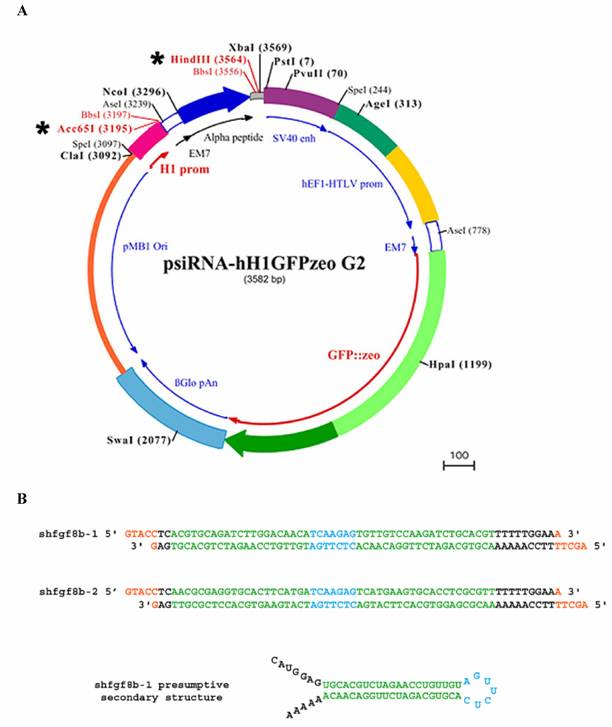
To monitor siRNA electroporation efficiency we used a GFP expression vector (pEGFP-C2, Clontech; see below). The shRNAs used to interfere with fgf8 expression were designed according to the general rules of the siRNA WizardTM program (Invivogen; www.invivogen.com), and the interfering sequence was cloned into the psiRNA-hH1GFPzeo G2 vector (Invivogen). This vector coexpresses the cloned shRNA, under the control of the H1 promoter, with GFP, under the control of the hEF1/HTLV promoter and SV40 enhancer (Fig. 2A).
Two sets of 21-mer oligonucleotides were selected from the fgf8b open reading frame (ORF) sequence. The 21-mer sense and antisense RNA sequences were linked to a seven nucleotide spacer (TCAAGAG) as a loop, and nucleotides corresponding to HindIII and Acc65I restriction sites were added to the 5' and 3' end of the reverse oligonucleotides respectively. The engineered DNAs for the corresponding shRNAs are shown in Fig. 2B. They were annealed and cloned into the psiRNA-hH1GFPzeo G2 vector using the HindIII/Acc65I restriction sites. Following the instructions of the plasmid supplier, vectors were transformed into the E.coli strain GT116, in which sbcC and sbcD genes are deleted, to increase shRNA stability.
In ovo microinjection and electroporation
Fertilized eggs obtained from a local farm (Granja Gibert) were incubated at 38ºC and windowed using Scotch tape. They were staged according to Hamburger and Hamilton [40]. DsRNA, the pool of siRNAs or the shRNA-producing plasmid were microinjected into the neural tube of embryos at HH 9-11 using a Drummond piston microinjector (32 nl at 3-5 μg/μl). Embryos were contrasted with 0.3 mg/ml Fast Green (Sigma) solution, and the pool of siRNAs and the plasmid producing shRNA were mixed with Fast Green at 3 mg/ml (3:1 v/v) prior to microinjection to monitor their localisation in the neural tube. Just after microinjection, the pool of siRNAs and the shRNA-producing plasmid were electroporated using a TSS20 Ovodyne electroporator (Intracel; 25V, 50ms pulse-width, 5 pulses with 30ms intervals between the pulses) and platinum electrodes (Nepagene; 5mm in length, 0.5mm wide) in 0.01x PBS as electroporation medium. After electroporation, the embryo was covered with DMEM (Gibco), the window was sealed with Scotch tape and the egg was incubated at 38ºC. The pool of siRNAs was co-electroporated with pEGFP-C2 (U57606) to assess electroporation efficiency and localisation. As described previously [41, 42], transfection occurs on one side of the neural tube, the other side serving as a control.
In situ hybridisation, immunohistochemistry and TUNEL assay
Embryos were fixed overnight (ON) at 4ºC in 4% paraformaldehyde in 1xPBS, dehydrated in a methanol series and stored at -20ºC prior to use. In situ hybridisation was performed as described by Bueno et al. [43] for single probe development, with minor modifications (hybridisation temperature was 55ºC, and the incubation time of the anti-DIG antibody was 3 hours at room temperature). The Fgf8 construct used to produce the antisense riboprobe was from Crossley et al. [39]. The α-peptide construct for antisense riboprobe production was generated by PCR cloning of 237 nt of the α-peptide sequence into pBluescript KS (EcoRI/XhoI restriction sites). The primers used were the following: forward 5'ccggaattcatggaccctgttgtgctgcaa3' and reverse 5'ccgctcgagcacagtgtcagcctctgggag3'. Antisense riboprobe was transcribed with T7 polymerase after EcoRI digestion.
As in situ hybridization treatments destroy GFP fluorescence, immunohistochemistry with a polyclonal antibody to GFP (Molecular Probes) was performed to detect its presence after fgf8 in situ hybridisation. Briefly, hybridised embryos were post-fixed at 4ºC for 2 hours with 4% paraformaldehyde in 1xPBS. For immunohistochemistry on sections, the embryos were dehydrated in an ethanol series, passed through xylene and embedded in paraffin. 10 μm sections were dewaxed in xylene and rehydrated in an ethanol series. For both immunohistochemistry on sections and on whole mount embryos, samples were blocked and incubated with the anti-GFP antibody, at 1/250 or 1/500 respectively, for 48 h at 4ºC. After extensive washes, samples were incubated with a FITC-conjugated donkey anti-rabbit polyclonal antibody (Molecular Probes).
To detect the presence of DIG-labelled siRNAs, embryos were processed as for in situ hybridisation. After permeabilisation, they were blocked, incubated with an anti-DIG antibody (Roche) and developed with NBT/BCIP.
Apoptotic cells were detected by TUNEL assay on whole mount embryos using standard procedures. Briefly, embryos were fixed overnight at 4ºC in 4% paraformaldehyde and permeabilised with PBT (PBS + 0.1% Triton X-100 + 0.1% Sodium Citrate). The 3'-OH ends of DNA were labelled for two hours at 37ºC by addition of digoxigenin 11-UTPs using the enzyme TdT. Then, the samples were blocked, the presence of DIG was detected with an anti-DIG antibody coupled to alkaline phosphatase and they were developed with NBT/BCIP.
3. Results
dsRNA microinjection into the neural tube
First of all, to test whether dsRNA triggers specific gene silencing in chick embryos, as reported for invertebrates, fgf8 dsRNA was microinjected into the neural tube of embryos at HH9-11. After 24 h or 48 h of in vivo (in ovo) embryonic development, i.e. at HH18-20 or HH23-24 respectively, embryos were killed and examined under the dissecting microscope to search for morphological defects, especially at the midbrain/hindbrain junction. In these experiments no morphological defects were observed, as compared with control embryos (not shown).
To examine whether fgf8 expression was silenced in these embryos, whole mount in situ hybridization with an fgf8 ribroprobe was performed. Fgf8 expression was detected in all neuroepithelial areas where it has previously been reported to be expressed, i.e. the isthmus and the frontal end of the telencephalon, as well as in the other embryonic sites, e.g. the eyes, branchial arches, the narrow pit and the apical ectodermal ridge of the limbs in HH23-24 embryos, as in control embryos (not shown). Finally, careful microscopic examination of serial sections from the hybridized embryos revealed no subtle morphological defects in the neuroectoderm (not shown).
Electroporation of siRNAs into the isthmus and RNAi spreading
To force the penetration of the putative triggering molecules for fgf8 gene silencing, a pool of siRNAs to fgf8 obtained in vitro by Dicer-digestion from the corresponding dsRNA was microinjected into the neural tube of embryos at HH9-11 and immediately electroporated into the isthmic cells. In these experiments the siRNAS were co-injected and co-electroporated with the pEGFP-C2 plasmid as an independent marker of electroporation efficiency. After 24 h of in vivo development the embryos were killed. Those embryos showing GFP fluorescence on the electroporated side of the isthmus were examined under the dissecting microscope to search for morphological defects. Again, no defects were observed when compared with control embryos (Fig. 3, and data not shown).
To examine whether fgf8 was silenced on the electroporated side of the isthmus, whole mount in situ hybridization with an fgf8 ribroprobe was performed. Fgf8 expression showed no significant differences between the two sides of the isthmus. Careful microscopic examination of serial sections from the hybridized embryos did not reveal any subtle morphological defect (not shown).
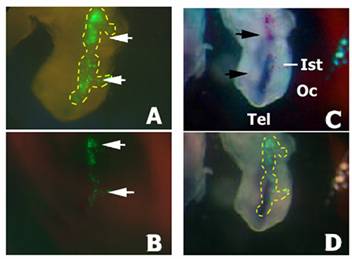
As the pEGFP-C2 plasmid has a different size and overall structure to the siRNAs (circular 4.7kb or linear 21mers respectively) that may affect the electroporation efficiency, we monitored whether the isthmic cells had actually been transfected with the siRNAs by using DIG-labelled siRNAs. The DIG-siRNAs were also co-injected and co-electroporated together with the pEGFP-C2 plasmid. After 24 h of in vivo embryonic development embryos showing GFP fluorescence on the electroporated side of the neural tube were immunostained with an antibody to DIG. Most of the GFP positive cells also showed the presence of siRNAs in their cytoplasm (Fig. 3). Interestingly, no significant spreading of the siRNAs from the transfected cells was observed. From these experiments we conclude that the electroporation profile used is appropriate for the transfection of the siRNAs, and thus that electroporation of a pool of siRNAs to fgf8 is not able to silence fgf8 expression in the isthmus.
Electroporation of a plasmid producing shRNAs
To test the possibility that the results obtained by electroporation of a pool of siRNAs were due to an insufficient number of siRNA molecules per cell to trigger the degradation of all existing and newly synthesised fgf8 mRNA molecules, the psiRNA-hH1GFPzeo G2 shRNA-producing plasmid was microinjected into the neural tube and electroporated into the isthmus.
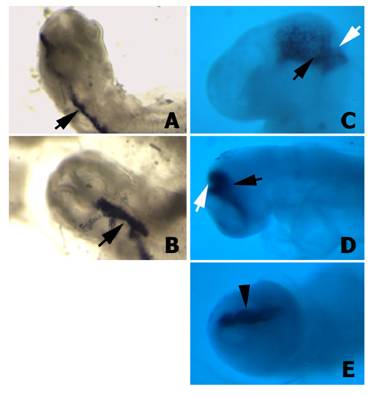
First of all we tested whether the H1 promoter for the shRNA was properly directing the expression of the downstream cloned sequences in chick embryos. Wild type plasmids, which carry the α-peptide sequence of the β-gal gene located under the control of the H1 promoter, were microinjected and electroporated into the isthmus. After 12 h or 24 h of in vivo development, embryos showing GFP fluorescence on the electroporated side of the isthmus were hybridized with a riboprobe to the α-peptide. These embryos exhibited a very high level of α-peptide expression (Fig. 4), suggesting that the H1 promoter also directs the expression of the shRNAs in the cells of the chick neural tube. In this way the number of shRNA molecules present in each particular cell will be much higher than the number of siRNA molecules electroporated after Dicer-digestion of the corresponding dsRNA. Moreover, this experiment also proved that both promoters (i.e. H1 promoter for the shRNA and hEF1/HTLV promoter for GFP) were working simultaneously, with no interferences.
Two different shRNAs to fgf8, the best shRNAs to silence fgf8 expression according the general rules of the siRNA WizardTM program (Invivogen), were engineered and independently cloned into the psiRNA-hH1GFPzeo G2 plasmid. These constructs were either independently or simultaneously microinjected and electroporated into the isthmus of chick embryos at HH9-11, in order to maximize the possible gene silencing effect. After 24 h of in vivo development, embryos showing GFP fluorescence on the electroporated side of the isthmus were examined under the dissecting microscope to search for morphological defects. Once again, no morphological defects were observed, as compared with control embryos electroporated with the wild type plasmid (Figs. 5 and 6). To examine whether fgf8 expression was silenced on the electroporated side of the isthmus, whole mount in situ hybridization with an fgf8 ribroprobe was performed. Fgf8 expression examined at whole embryo level showed a slight decrease on the electroporated side of the isthmus as compared with the control side (Figs. 5 and 6), although careful microscopic examination of serial sections from the hybridized embryos did not reveal any subtle morphological defect (Figs. 5 and 6).
To check whether fgf8 expression was actually silenced in the transformed cells, immunohistochemistry with an antibody to GFP was performed on whole mount embryos and on sections after fgf8 in situ hybridization. This experiment allowed us to visualize individual transfected cells and fgf8 expressing cells simultaneously. Although some GFP positive cells were clearly not expressing fgf8, most of the transfected cells did, suggesting that if RNAi occurs in chick embryos, its efficiency is very low (Fig. 7).
Finally, as it has been reported that dsRNA and siRNA molecules may trigger an apoptotic response in vertebrate cells that may mask and/or simulate RNAi effects, we monitored apoptosis to check whether the slight decrease in fgf8 expression was caused by the death of some of the transfected cells. The pattern of apoptotic cells in adjacent non-transfected structures of the electroporated side as the eye and the otic vesicle were used as controls. Embryos electroporated with the shRNA-producing plasmid showed few apoptotic cells that affect mostly the ectoderm, and no significant differences were observed between these embryos and either control embryos electroporated with the wild type plasmid or non-electroporated embryos (Fig. 8).
4. Discussion
We have analysed the possible use of RNAi as a routine method for specific gene silencing in chick embryos by assessing the capacity of different potential triggers of RNAi (dsRNA, a pool of siRNAs obtained in vitro by Dicer-digestion of the corresponding dsRNA, and a plasmid producing shRNA to the gene to be silenced) to silence fgf8 expression in the isthmus, as well as to provoke morphological malformations in midbrain/hindbrain junction derivates.
Action of RNAi
All published studies focused on the possible use of RNAi to silence gene expression in chick embryos, in which dsRNA molecules or non-viral vectors producing shRNAs have been electroporated into target cells, have taken advantage of the sealed cavity of the neural tube to introduce the putative trigger molecules prior to electroporation [28, 29, 31]. To facilitate the experimental manipulation of the embryos we also introduced the putative trigger molecules for RNAi into the neural tube. We have focused on fgf8 expression in the isthmus to monitor gene silencing for several practical reasons. Firstly, the isthmus is easily accessible from the neural cavity, and clearly recognizable under a dissecting microscope to properly localize the molecules that trigger RNAi. Secondly, when the neural tube is completely sealed, at HH9, fgf8 expression in the isthmus is still faint and is detected in relatively few cells that are restricted to a small area [32, 33], a fact that may facilitate the degradation of all existing mRNA molecules. And thirdly, the phenotype of fgf8 null-mutant and allelic series of mutations affecting the isthmus has been reported in other vertebrate model systems, and is easily morphologically recognizable as a deletion of the isthmus derivatives [36].
We observed no morphological defects in any of the experiments performed. We have also shown that fgf8 expression is neither silenced in embryos microinjected with dsRNA nor in embryos microinjected and electroporated with a pool of siRNAs. Moreover, fgf8 expression is also not significantly silenced in most isthmic cells transformed with the psiRNA-hH1GFPzeo G2 plasmid producing the engineered shRNAs to fgf8, and an increase in cell death is not detected either in the transfected neuroectoderm or in adjacent structures serving as controls for the possible toxic effects of the electric field.
Our results are basically in agreement with previously reported studies on RNAi of endogenous genes in chick embryos. Thus, the electroporation of a pSilencer 1.0-U6 vector (Ambion) producing shRNA to en-1 or en-2 [29] or to Islet1 [31] does not produce any recognizable morphological defect and only a discrete reduction of gene expression. In apparent contrast, electroporation of dsRNA to axonin-1 and NrCAM, which are involved in axonal guidance, produces some morphological defects affecting this process in 20% of the neurons [28], although the possibility that this arises from an increase in cell death due to the presence of dsRNA molecules was not analysed. Nevertheless, the presence of just 20% of affected neurons is unlikely to be sufficient to see any long-term defect, as in many cases embryos are able to balance mild developmental defects.
In a similar way, microinjection of the viral pSuper vector (OligoEngine) engineered to produce shRNA to silence mkp-3 expression in the limb bud mesenchyme [30] caused an increase in cell death compared with controls microinjected with an RCAS virus expressing GFP, although unfortunately no other shRNAs were used to assess the non-specific effect of these molecules on cell death in the limb mesenchyme.
In these studies the electroporation efficiency and the localisation of the molecules that may trigger RNAi were assessed by co-electroporation of an independent reporter plasmid, thereby precluding analysis of which particular cells were effectively transfected with the RNAi triggering molecules. This is especially important when considering that a maximum of 60% of the cells of the electroporated side are actually transfected with the reporter plasmid [28]. This handicap is avoided with the psiRNA-hH1GFPzeo G2 plasmid used in this study, as it coexpresses GFP as a marker. This allowed us to clearly determine that although some transfected cells do not express fgf8, most of them do. This raises the question of whether the reduction of fgf8 expression in some transfected cells occurs irrespective of the presence of the specific shRNA, or instead whether the presence of shRNA is triggering gene silencing only in certain cells due to an unknown intrinsic difference between them. Nevertheless, the small reduction in gene expression, if any, accounts for the lack of morphological defects in RNAi treated embryos.
A similar situation occurs in mice [21, 22, 23, 24, 25, 27]. Here, with the exception of a mild phenotype reported by Paddison et al. [23] in the derivates of some oocytes, no phenotypic alterations have been reported.
Better results are obtained when attempting to silence the expression of exogenous genes in chick embryos, although the gene silencing is incomplete, Pekarik [28] reported that co-electroporation of a vector expressing yellow fluorescent protein (YFP) together with dsRNA to YFP in the neural tube causes a reduction of YFP fluorescence in a substantial number of cells. Moreover, Chesnutt and Niswander [31] also reported a significant but incomplete reduction of GFP expression when co-electroporating a vector expressing GFP and a pSilencer 1.0-U6 vector expressing shRNA to this gene.
Taking into account our results and the published works discussed above, we suggest that the currently available tools and techniques for RNAi are not suitable as a routine method for functional gene analysis in chick embryos, as it is in some invertebrates [11, 12, 13, 14]. This suggestion does not preclude a possible use of RNAi to analyze the effect of gene silencing at a molecular level in individual transfected cells, the ones that may show a reduction in gene expression.
Is RNAi an evolutionarily conserved mechanism?
Although it is commonly said that RNAi is an evolutionarily conserved mechanism, reports of molecular differences between several model systems are appearing. Thus, most invertebrates in which RNAi has been successfully used are able to take up dsRNA molecules by themselves, the exception being Drosophila. The molecule responsible for dsRNA uptake and siRNA spreading is SID-1, a specific transmembrane protein [44, 45, 46]. Drosophila shows RNAi but lacks a sid-1 ortholog, thus not showing the spreading characteristic [45, 47]. In this way, the amount of dsRNA needed for RNAi in this organism is 10,000 times higher than in controls expressing sid-1 ectopically.
The presence of sid homologs has been reported in mice, although these organisms do not seem to be able to take up dsRNA by themselves, a similar situation occurring in chick embryos. In fact, all reports on the use of dsRNA or siRNA for RNAi in vertebrates have forced these molecules to enter the cells, either by calcium precipitation, electroporation or cytoplasmic microinjection. Moreover, our results have also shown that siRNA molecules do not spread from the transfected cells to the rest of the tissue or the embryo, as has been reported to occur in most invertebrates [2], suggesting the existence of molecular differences between different model systems that may explain the different responses to RNAi. To develop a general procedure for RNAi in chick embryos for routine functional gene analysis, if it is technically and molecularly possible, a better knowledge of the mechanisms leading to gene silencing in vertebrates is needed.
Aknowledgements
This work was funded by the Instituto de Salud Carlos III, Ministerio de Sanidad y Consumo (FIS PI020912 to D.B.) and the Ministerio de Educación y Cultura (DGICYT BCM2000-0546 to R. Romero -Dep. de Genètica, U. de Barcelona-, to whom we are grateful).
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Muramatsu T, Mizutani Y, Ohmori Y, Okumura J. Comparison of three nonviral transfection methods for foreign gene expression in early chicken embryos in ovo. Biochem Biophys Res Commun. 1997 ;230:376-380
2. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998 ;391:806-811
3. Bass BL. Double-stranded RNA as a template for gene silencing. Cell. 2000 ;101:235-238
4. Bueno D, Romero R, Hernández Hernández V, Sancho V, Fernández-Rodríguez J, Cardona AT, Vila-Farré M. RNA interference: a new and powerful tool for functional genomic analysis. C Sci. 2001 ;2:23-33
5. Hanon GJ. RNA interference. Nature. 2002 ;418:244-250
6. Tuschl T, Zamore PD, Lehmann R, Bartel DP, Sharp PA. Targeted mRNA degradation by double-stranded RNA in vitro. Gens Dev. 1999 ;13:3191-3197
7. Zamore PD, Tuschl T, Sharp PA, Bertel DP. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000 ;101:25-33
8. Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000 ;404:293-296
9. Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001 ;409:363-366
10. Fire A. RNA-triggered gene silencing. Trends Genet. 1999 ;15:358-363
11. Fraser AG, Kamath RS, Zipperlen P, Martínez-Campos M, Sohrmann M, Ahringer J. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature. 2000 ;408:325-330
12. Gonczy P, Echeverri C, Oegema K, Coulson A, Jones SJM, Copley RR, Duperon J, Oegema J, Brehm M, Cassin E, Hannak E, Kirkham M, Pichler S, Flohrs K, Goessen A, Leidel S, Alleaume AM, Martin C, Ozlu N, Bork P, Hyman AA. Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III. Nature. 2000 ;408:331-336
13. Clemens JC, Worby CA, Simonson LN, Muda M, Maehama T, Hemmings BA. Use of double stranded RNA interference in Drosophila cell lines to dissect signal transduction pathways. Proc Natl Acad Sci USA. 2000 ;97:6499-6503
14. Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003 ;421:231-237
15. Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells responds to interferons. Annu. Rev. Biochem. 1998 ;67:264-277
16. Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R. Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet. 2003 ;34:263-264
17. Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR. Activation of the interferon system by short-interfering RNAs. Nat Cell Biol. 2003 ;5:834-839
18. Wargelius A, Ellingsen S, Fjose A. Double-stranded RNA induces specific developmental defects in zebrafish embryos. Biochem Biophys Res Comm. 1999 ;263:156-161
19. Li YX, Farrell MJ, Liu R, Mohanty N, Kirby ML. Double-stranded RNA injection produces null phenotypes in zebrafish. Dev Biol. 2000 ;217:394-405
20. Oates AC, Bruce AE, Ho RK. Too much interference: injection of double-stranded RNA has nonspecific effects in the zebrafish embryo. Dev Biol. 2000 ;224:20-28
21. Donzé O, Picard D. RNA interference in mammalian cells using siRNAs synthesized with T7 RNA polymerase. Nucleic Acids Res. 2002 ;30(10):e46-
22. Omi K, Tokunaga K, Hohjoh H. Long-lasting RNAi activity in mammalian neurons. FEBS Lett. 2004 ;558:89-95
23. Paddison PJ, Silva JM, Conklin DS, Schlabach M, Li M, Aruleba S, Balija V, O'Shaughnessy A, Gnoj L, Scobie K, Chang K, Westbrook T, Cleary M, Sachidanandam R, McCombie WR, Elledge SJ, Hannon GJ. A resource for large-scale RNA-interference-based screens in mammals. Nature. 2004 ;428:427-431
24. Wianny F, Zernicka-Goetz M. Specific interference with gene function by double-stranded RNA in early mouse development. Nat Cell Biol. 2000 ;2:70-75
25. Svoboda P, Stein P, Hayashi H, Schultz RM. Selective reduction of dormant maternal mRNAs in mouse oocytes by RNA interference. Development. 2000 ;127:4147-56
26. Calegari F, Haubensak W, Yang D, Huttner WB, Buchholz F. Tissue-specific RNA interference in postimplantation mouse embryos with endoribonuclease-prepared short interfering RNA. Proc Natl Acad Sci USA. 2002 ;99(22):14236-14240
27. Gratsch TE, de Boer LS, O'Shea KS. RNA inhibition of BMP-4 gene expression in postimplantation mouse embryos. Genesis. 2003 ;37:12-17
28. Pekarik V, Bourikas D, Miglino N, Joset P, Preiswerk S, Stoeckli ET. Screening for gene function in chicken embryo using RNAi and electroporation. Nat Biotechnol. 2003 ;21:93-96
29. Katahira T, Nakamura H. Gene silencing in chick embryos with a vector-based small interfering RNA system. Dev Growth Differ. 2003 ;45:361-367
30. Kawakami Y, Rodriguez-Leon J, Koth CM, Buscher D, Itoh T, Raya A, Ng JK, Esteban CR, Takahashi S, Henrique D, Schwarz MF, Asahara H, Izpisua Belmonte JC. MKP3 mediates the cellular response to FGF8 signalling in the vertebrate limb. Nat Cell Biol. 2003 ;5:513-519
31. Chesnutt C, Niswander L. Plasmid-based short-hairpin RNA interference in the chicken embryo. Genesis. 2004 ;39:73-78
32. Bueno D, Skinner J, Abud H, Heath JK. Spatial and temporal relationships between Shh, Fgf4 and Fgf8 gene expression at diverse signalling centres during mouse development. Developmental Dynamics. 1996 ;207:291-299
33. Martínez S. The isthmic organizer and brain regionalization. Int J Dev Biol. 2001 ;45:367-371
34. Crossley PH, Martinez S, Martin GR. Midbrain development induced by FGF8 in the chick embryo. Nature. 1996 ;380:66-8
35. Martínez S, Crossley PH, Cobos I, Rubenstein JLR, Martin GR. FGF-8 induces an isthmic organizer and isthmocerebellar development in the caudal forebrain via a repressive effect on Otx2 expression. Development. 1999 ;126:1189-1200
36. Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat Genet. 1998 ;18:136-41
37. Reifers F, Bohli H, Walsh EC, Crossley PH, Stainier DY, Brand M. Fgf8 is mutated in zebrafish acerebellar (ace) mutants and is required for maintenance of midbrain-hindbrain boundary development and somitogenesis. Development. 1998 ;125:2381-2395
38. Jázsai J, Reifers F, Picker A, Langenberg T, Brand M. Isthmus-to-midbrain transformation in the absence of midbrain-hindbrain organizer activity. Development. 2003 ;130:6611-6623
39. Crossley PH, Minowada G, MacArthur CA, Martin GR. Roles for FGF8 in the induction, initiation, and maintenance of chick limb development. Cell. 1996 ;12:127-136
40. Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. J. Morph. 1951 ;88:49-92
41. Funahashi J, Okafuji T, Ohuchi H, Noji S, Tanaka H, Nakamura H. Role of Pax-5 in the regulation of a mid-hindbrain organizer's activity. Dev Growth Differ. 1999 ;41:59-72
42. Nakamura H, Funahashi J. Introduction of DNA into chick embryos by in ovo electroporation. Methods. 2001 ;24:43-48
43. Bueno D, Skinner J, Abud H, Heath JK. Double in situ hybridisation on mouse embryos for detection of overlapping regions of gene expression. TIGs. 1996 ;12:385-387
44. Winson WM, Molodowitch C, Hunter CP. Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science. 2002 ;295:2456-2459
45. Feinberg EH, Hunter CP. Transport of dsRNA into cells by the transmembrane protein SID-1. Science. 2003 ;12:1545-1547
46. Van Roessel P, Brand AH. Spreading silence with Sid. Genome Biol. 2004 ;5:208-
47. Kennerdell JR, Carthew RW. Heritable gene silencing in Drosophila using double-stranded RNA. Nature Biotech. 2000 ;17:896-897
Figures
siRNA and DIG-siRNA obtained by in vitro Dicer-digestion. A 3% agarose gel electrophoresis of siRNA and DIG-labelled siRNA to fgf8 is shown. Note that Dicer is able to generate a pool of siRNAs from both dsRNA and DIG-labelled dsRNA.
Plasmids and shRNA constructs. A) Wild-type psiRNA-hH1GFPzeo G2 vector map showing Acc65I/HindIII restriction sites (*), the α-peptide coding sequence located downstream of the H1 promoter, and the GFP coding sequence located downstream of hEF1-HTLV promoter. B) Engineered DNA oligonucleotides for the shRNAs: 21-mers are shown in green; the spacer is shown in blue; HindIII and Acc65I restriction sites are shown in orange; and the poly-A sequence is shown in black. The presumptive secondary structure of shfgf8b-1 is also shown.
Localisation of DIG-siRNA 24 h after electroporation. Embryos co-electroporated with a pool of DIG-labelled siRNAs and the pEGFP-C2 plasmid. A) GFP fluorescence into the electroporated side of the neural tube before DIG immunodetection (arrows). Yellow dashed line is delimiting the GFP expression area. B) GFP fluorescence into the electroporated side of the neural tube of the embryo shown in A) after DIG immunodetection (arrows). Note a decrease in GFP fluorescence due to the presence of the NBT/BCIP precipitate. C) Immunodetection of DIG-labelled siRNA in the neural tube (in blue; arrows) in the same embryo than A) and B). D) Merging of B) and C). Yellow dashed line has been extrapolated from A) and is delimiting the area of GFP expression before NBT/BCIP precipitation. Note that most cells exhibiting GFP fluorescence also show the presence of the DIG-siRNAs, although they do not colocalize in all electroporated cells. Also note the absence of siRNA spreading from the electroporated area. Abbreviations: Ist, isthmus; Oc, optic cup; Tel, telencephalon.
α-peptide expression in embryos transfected with the psiRNA-hH1GFPzeo G2 vector. A-B) Two different embryos 12 h after electroporation hybridised with the α-peptide antisense riboprobe. Note the expression of the α-peptide sequence in the electroporated half-side of the neural tube (arrows). C-D) Two different embryos 24 h after electroporation hybridised with the α-peptide antisense riboprobe. Note that the expression of the α-peptide is properly localized into the isthmus (arrows). E) Facial view of the embryo shown in D. Note that the α-peptide is expressed only in electroporated side (arrowhead).
Fgf8 and isthmic morphology in flattened isthmus of embryos transfected with shfgf8b-1 and shfgf8b-2. Dorsal view of a flattened isthmus hybridised with the fgf8 riboprobe 24 h after plasmids electroporation. The black line follows the dorsal midline. Note that no significant differences either in fgf8 expression or neuroectoderm morphology are observed when comparing both sides of the isthmus.

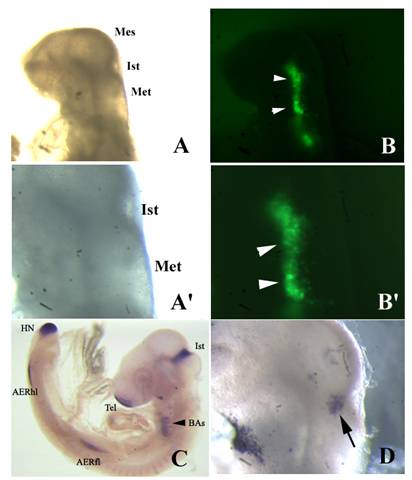
Fgf8 expression and isthmic morphology in whole mount embryos transfected with shfgf8b-1 and shfgf8b-2. Rostral is to the left. A) Embryo 24 h after electroporation, prior to fgf8 in situ hybridisation. Note that the morphology of the isthmic derivatives is completely normal (compare with control embryo shown in C). A') Magnification of A) showing the isthmic area. B) GFP fluorescence (arrowheads) in the electroporated side of the same embryo shown in A). B') Magnification of B) showing the area of GFP expression (arrowheads). C) Fgf8 in situ hybridisation of a control embryo. D) Fgf8 in situ hybridisation in the isthmus of the embryo shown in A) (arrow). Note that fgf8 expression in the isthmus is very similar to that shown in C). Abbreviations: AERfl, forelimb apical ectodermal ridge; AERhl, hindlimb apical ectodermal ridge; BA, branchial arches; Ist, isthmus; Met, metencephalon; Mes, mesencephalon; Tel, telencephalon.
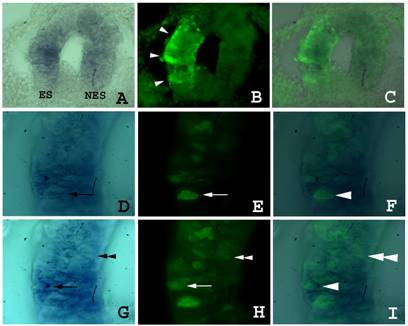
Fgf8 expression and isthmic morphology in sections of embryos transfected with shfgf8b-1 and shfgf8b-2. Transversal sections of the neural tube at the isthmus level. Both sides of neural tube are shown. The electroporated side is to the left. A) Fgf8 expression detected by in situ hybridisation. B) GFP expression in the same section shown in A) detected by GFP immunostaining. Note that GFP is detected only in the electroporated side (arrowheads). C) Merging of A) and B). The expression of fgf8 is very similar in electroporated and control neuroepithelium. D to F) Magnification of sections shown in A to C respectively. Note the presence of cells showing fgf8 and GFP expression simultaneously (arrows and arrowhead). G to I) Magnification of sections shown in A to C respectively in a different focal plane. Note the presence of cells showing GFP expression but not fgf8 expression (double arrowhead).
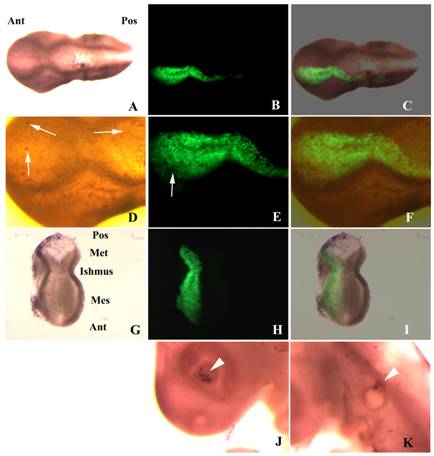
Apoptosis in embryos transfected with shfgf8b-1 and shfgf8b-2. A) Dorsal view of a transfected embryo in which apoptotic cells have been detected by TUNEL assay (brown nuclei). Note the absence of morphological defects (the transfected side is to the bottom). B) GFP fluorescence in the same embryo. The transfected area includes the isthmus. C) Merging of B) and C). D) Magnification of the isthmic area of A). Note that both sides of the embryo exhibit a similar amount of apoptotic cells (arrows), which affect mostly the ectoderm. E) Magnification of the isthmic area of B). The arrow points an apoptotic cell not expressing GFP. F) Magnification of the isthmic area of C). G) Dissected cephalic cavity showing the internal side of the mesencephalon, the metencephalon and the isthmus. Apoptotic cells have been detected by TUNEL assay (brown nuclei). The morphology of the transfected side (left hand side) is completely normal, and that apoptotic cells are equally distributed at both sides of the embryo. H) GFP fluorescence in the same embryo. The transfected area includes the isthmus. I) Merging of G) and H). J) Apoptosis control in the eye of the electroporated side. Note that apoptotic cells are located in the dorsal portion of the optic cup as described in the literature for normal embryos (Trousse et al., 2001). K) Apoptosis control in the ear of the electroporated side. Note that apoptotic cells are located in the dorsal portion of the otic vesicle as described in the literature for normal embryos (Lang et al., 2000). Abbreviations: Ant, anterior; Mes, mesencephalon; Met, metencephalon; Pos, posterior.
Author biography
David Bueno (Barcelona, 1965) received PhD in Genetics in 1994 for work on planarian regeneration at cellular and molecular level. His PhD thesis was awarded cum laude and PhD Extraordinary Award. He did postdoctoral work at the University of Oxford, focused on the analysis of Fibroblast Growth Factor functions during vertebrate development, using transgenic mice as a model system. Since 1996 he has worked at the Department of Genetics of the University of Barcelona and became a Lecturer in 2003. He is a founder member of Spanish Society of Developmental Biology. His current research focuses on central nervous system development and neuronal proliferation and differentiation in vertebrates, analysing molecules involved in these processes that may also be involved in human neurodegenerative diseases. He is author or co-author of over 28 articles in peer-review journals, 35 papers to international congresses, and 2 chapters in books.
Victor Hernández Hernández (Barcelona, 1973) received Master degree in Experimental Biology from the UB in 2001. The same year he started the PhD studies in the Department of Genetics, working on genes involved in the development of the central nervous system in chick embryos. He is author or co-author of 6 articles on planarian regeneration and chick development in international peer-review journals.
![]() Corresponding address:
Corresponding address:
Victor Hernández, Departament de Genètica, Facultat de Biologia, Universitat de Barcelona, Av. Diagonal 645, 08028 Barcelona, Catalonia, SPAIN. Tel: 34-93-402.15.00 / 34-93-403.70.70. e-mail: victorhernandezedu
Received 2004-9-1
Accepted 2004-10-15
Published 2005-1-5