Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and methods
3. Results and discussion
References
Tables and Figures
Author biography

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2005; 1(1):13-18. doi:10.7150/ijbs.1.13 This issue Cite
Short Research Paper
Partial mitochondrial genome sequences of Ostrinia nubilalis and Ostrinia furnicalis
Brad S. Coates1 2, Douglas V. Sumerford1, Richard L. Hellmich1 3, Leslie C. Lewis1 3 ![]()
1 USDA-ARS, Corn Insect and Crop Genetics Research Unit, Genetics Laboratory, c/o Insectary, Iowa State University, Ames, Iowa, 50011
2 Interdepartmental Genetics Program, Iowa State University, Ames, IA, 50011
3 Department of Entomology, Iowa State University, Ames, IA, 50011
Received 2004-8-15; Accepted 2004-10-15; Published 2005-1-5
Abstract
Contiguous 14,535 and 14,536 nt near complete mitochondrial genome sequences respectively were obtained for Ostrinia nubilalis and Ostrinia furnicalis. Mitochondrial gene order was identical to that observed from Bombyx. Sequences comparatively showed 186 substitutions (1.3% sequence divergence), 170 CDS substitutions (131 at 3rd codon positions), and an excess of transition mutation likely resulting by purifying selection (dN/dS = ω ≅ 0.15). Overall substitution rates were significantly higher at 4-fold (5.2%) compared to 2-fold degenerate codons (2.6%). These are the 3rd and 4th lepidopteran mitochondrial genome reference sequences in GenBank and useful for comparative mitochondrial studies.
Keywords: Ostrinia nubilalis, Ostrinia furnicalis, mitochondrial variation
1. Introduction
Mitochondrial genomes of 16 insect species are completely sequenced and published with a majority from the order Diptera; D. yakuba [1], Ades gambiae [2], Anopheles quadrimaculatus [3], D. melanogaster [4], Ceratitis capitata [5], Cochliomiyia hominivorax [6], D. simulins [7], and Bactrocera oleae [8]. Complete sequences also have been published from a hymenopteran, Apis mellifera [9], an orthopteran, Locusta migratoria [10], a phthirapteran H. macropus [11], thysanuran, T. inaginis [12], a hemipteran Triatoma dimidiata [13], coleopteran Crioceris duodecipunctata [14], and lepidopterans Bombyx mori and B. mandarina [15].
Larvae from corn borer species Ostrinia nubilalis and Ostrinia furnicalis (Lepidoptera: Crambidae) are pests of agricultural crop plants and cause major crop production losses [16, 17]. Ostrinia nubilalis and O. furnicalis are sister species [18, 19], with difference residing in female O. nubilalis and O. furnicalis emission of E- and Z- stereoisomers of Δ11- [20], and Δ12-tetradecenyl acetates [21], respectively. The pheromone binding protein gene sequences showed little nucleotide variance between O. nubilalis and O. furnicalis [22], and 7 allozyme markers indicated a high similarity between Chinese populations of O. nubilalis and O. furnicalis suggesting recent speciation [18]. Similarly, mitochondrial cytochrome c oxidase subunit II (coxI) gene alignment estimated 1.63% interspecies divergence [19]. The present study compares GenBank annotated mitochondrial genomes from O. nubilalis (accession AF442957) and O. furnicalis (AF467260).
2. Materials and methods
2.1 Samples and amplification
A single bivoltine female Z-pheromone race O. nubilalis adult was collected from the Iowa State University Uthe Farm, Ames, Iowa, USA. One adult multivoltine O. furnicalis female of indeterminate pheromone composition collected from Hengshui, Hebei Province, China was contributed by Dr. Wang Zhen-ying, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China. DNA extractions used Qiagen DNeasy kits (Qiagen, Valencia, CA).
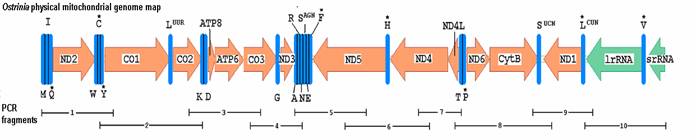
Primers combinations TY-J-1460 with TK-N-3785, J-11545 with N-12854, and N1-J-12585 with SR-N-14588 [23] were used to PCR amplify fragments 2, 9, and 10 (Fig. 1). Bombyx mori (GenBank:AF149768 and AY048187) and D. yakuba (GenBank: MIDYRRN) [1] mitochondrial genomes were aligned using AlignX software (Informax, San Francisco, CA) to identify regions of sequence similarity, from which regions PCR primers were designed to amplify remaining fragments using Primer3 [24]. All PCR reactions were performed in a 50 μl volume with 1.7 U of Tli polymerase (Promega Corp., Madison, WI), 100 ng of DNA, 5 μl 10X thermal polymerase buffer (Promega), 2.5 mM MgCl2, 200 μM dNTPs, and 20 pmol of each primer. Fragments 1, and 3 to 8 were amplified on a PTC-100 thermocycler (MJ Research, Watertown, MA) with denaturation at 95ºC for 2 m, followed by 40 cycles at 94ºC for 30 s, 50 to 54 ºC for 40 s, a 2.5 ºC/s ramp for +15 ºC, and 70 ºC for 1.5 to 3 m depending on fragment length. Fragments 2, 9, and 10 were amplified by denaturing template at 95ºC for 2 m, followed by 40 cycles at 94ºC for 30 s, 44 ºC for 1 m, a 2.0 ºC/s ramp for +23 ºC, and 70 ºC for 3 m.
Ostrinia mitochondrial genome map of sequenced regions. Protein coding genes represented by arrows indicating direction with left-facing arrows on major strand. The tRNA genes are labeled by single letter codes and * indicating coding sequence on minor strand. Underscores indicate positions of ten overlapping PCR amplified genome fragments.
2.2 DNA sequence and analysis
PCR reaction products for fragments 1 to 10 were purified using Qiaquick PCR purification columns (Qiagen), and diluted to 2.5ng/μl/100 bp of product length. Sequencing was performed in duplicate at the DNA sequencing core facility at Iowa State University, Ames, IA. Overlapping fragments were assembled into a single contiguous sequence using Contig Express software (Informax). Ostrinia nubilalis and O. furnicalis mitochondrial genome sequences were aligned with B. mori (GenBank AF149768, and GenBank AY048187) using AlignX software (Informax), and gene features were annotated using Vector NTI 7.0 (Informax). Contiguous mitochondrial DNA sequence of 14535 and 14536 nt were respectively submitted to GenBank for O. nubilalis (AF442957) and O. furnicalis (AF467260).
Substitution rate and transition/transversion ratio for Ostrinia mitochondrial DNA sequences were calculated with MacClade 4.03 [25]. Twenty one tRNA gene structures were predicted with M-fold 3.1 [26], and viewed using RNAviz 2.0 [27]. Codon usage was evaluated by the Countcodon program version 4 (http://www.kazusa.or.jp/codon/ countcodon.html). Average per site rates of synonymous (dS) and nonsynonymous nucleotide substitution (dN) were calculated according to [28] using MEGA [29].
3. Results and discussion
3.1 Ostrinia mitochondrial genomes
Contiguous O. nubilalis (GenBank accession: AF442957) and O. furnicalis (AF467260) mitochondrial genomes were assembled from overlapping PCR product sequence (Fig. 1). Each GenBank record includes 13 open reading frames (ORFs), a large ribosomal RNA (rrnL) gene, 21 tRNAs, and part of trnM and small ribosomal RNA (rrnS) genes (Fig. 1). Gene order and orientation were identical to Drosophila [1, 4], except for translocation of trnM to a position preceding trnI as was observed in Bombyx [15]. Major strand of O. nubilalis (41.3% A, 38.8% T, 8.0% G, and 11.8% C; 80.2% AT) and O. furnicalis (41.5% A, 38.9% T, 7.9% G, and 11.7% C; 80.4% AT) showed a bias toward A and T nucleotides that is typical of insect mitochondrial genomes [30].
The O. nubilalis and O. furnicalis mitochondrial genomes have 3731 codons; 3718 amino acid encoding and 13 termination codons (Table 1). Codons had a prevalence of A and T in 3rd positions and bias may reflect selection for optimal tRNA use [31], speed of genome replication, genome bias, or DNA repair efficacy (Table 2). The O. nubilalis and O. furnicalis mitochondrial peptides comparatively showed 24 predicted amino acid changes (24 of 3718; 0.646%; peptide similarity ≅ 99.22%, and identity ≅ 99.78%) [32]. All ORFs were initiated by ATA or ATT codons, except cox1. Initiation of coxI translation is ambiguous, but may occur by a TATTAG sequence in O. nubilalis and O. furnicalis, that is similar to TTTTAG in the B. mori. Hexanucleotides, initiation signals TATCTA from Penaeus monodon [33], or ATTTAA from A. gambiae [2], A. quadrimaculatus [3] and C. capitata [5] have been proposed. Alternatively, an ATAA tetranucleotide sequence was predicted to initiate cox1 translation in Drosophila, L. migratoria [10], and Daphnia pulex [34]. Termination codons were either TAA or TAG in O. nubilalis and O. furnicalis, except for cox2 and atp6 that have incomplete stop codons T and TA, respectively. Incomplete stop codons may become function after polycistronic transcript cleavage and polyadenylation mechanisms [35, 36].
Codon usage for 3718 amino acid residues and 13 nonsense codons among protein coding regions from each O. nubilalis (On) and O. furnicalis (Of) using the invertebrate mitochondrial genetic code.
| Codon | On | Of | Codon | On | Of | Codon | On | Of | Codon | On | Of |
| UUU-Phe | 347 | 354 | UCU-Ser | 93 | 95 | UAU-Tyr | 175 | 170 | UGU-Cys | 29 | 30 |
| UUC-Phe | 31 | 28 | UCC-Ser | 12 | 10 | UAC-Tyr | 12 | 16 | UGC-Cys | 2 | 1 |
| UUA-Leu | 459 | 454 | UCA-Ser | 94 | 94 | UAA-Ter | *12 | *12 | UGA-Trp | 89 | 89 |
| UUG-Leu | 15 | 17 | UCG-Ser | 5 | 4 | UAG-Ter | 1 | 1 | UGG-Trp | 5 | 5 |
| CUU-Leu | 20 | 22 | CCU-Pro | 60 | 60 | CAU-His | 55 | 58 | CGU-Arg | 14 | 14 |
| CUC-Leu | 0 | 1 | CCC-Pro | 8 | 7 | CAC-His | 7 | 4 | CGC-Arg | 1 | 1 |
| CUA-Leu | 32 | 32 | CCA-Pro | 55 | 52 | CAA-Gln | 61 | 61 | CGA-Arg | 33 | 33 |
| CUG-Leu | 0 | 0 | CCG-Pro | 1 | 2 | CAG-Gln | 3 | 3 | CGG-Arg | 3 | 3 |
| AUU-Ile | 449 | 455 | ACU-Thr | 71 | 72 | AAU-Asn | 232 | 234 | AGU-Ser | 23 | 25 |
| AUC-Ile | 28 | 28 | ACC-Thr | 14 | 10 | AAC-Asn | 24 | 22 | AGC-Ser | 2 | 2 |
| AUA-Met | 262 | 265 | ACA-Thr | 71 | 75 | AAA-Lys | 92 | 94 | AGA-Ser | 90 | 89 |
| AUG-Met | 26 | 20 | ACG-Thr | 1 | 0 | AAG-Lys | 9 | 8 | AGG-Ser | 0 | 1 |
| GUU-Val | 72 | 70 | GCU-Ala | 70 | 71 | GAU-Asp | 59 | 59 | GGU-Gly | 56 | 56 |
| GUC-Val | 1 | 2 | GCC-Ala | 9 | 8 | GAC-Asp | 4 | 4 | GGC-Gly | 1 | 5 |
| GUA-Val | 58 | 62 | GCA-Ala | 46 | 47 | GAA-Glu | 65 | 62 | GGA-Gly | 117 | 115 |
| GUG-Val | 6 | 1 | GCG-Ala | 2 | 1 | GAG-Glu | 8 | 10 | GGG-Gly | 29 | 25 |
* Includes stop codons from cox2 (T) and atp6 (TA), completed by adenylation.
Nucleotide frequencies partitioned among O. nubilalis (On) and O. furnicalis (Of) mitochondrial genome regions. IGS = non-coding intergenic spacer regions.
| Protein Coding Sequence | ||||||||||||||
| 1st position | 2nd position | 3rd position | rrnL | rrnS | tRNAs | IGS | ||||||||
| % nt | On | Of | On | Of | On | Of | On | Of | On | Of | On | Of | On | Of |
| % AT | 74.4 | 74.5 | 70.4 | 70.5 | 92.8 | 93.3 | 84.9 | 85.0 | 82.3 | 82.8 | 82.2 | 82.1 | 91.9 | 93.4 |
| % GC | 25.6 | 25.5 | 29.6 | 29.5 | 7.2 | 6.7 | 15.1 | 15.0 | 17.7 | 17.2 | 17.8 | 17.9 | 8.1 | 7.6 |
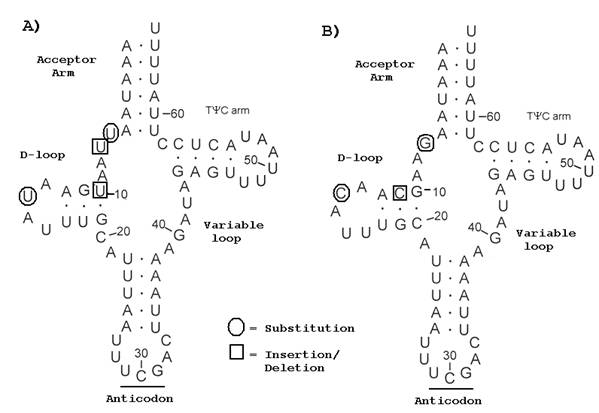
Complete nucleotide sequence was obtained for 21 O. nubilalis and O. furnicalis mitochondrial tRNAs. Seven substitutions were observed, and 0.49% sequence divergence was estimated from 1429-shared sites. Insertion-deletion (indel) mutation occurred in loop structures of trnA, trnD, trnG, and trnT, and, except for trnR, did not affect predicted two-dimensional tRNA structures (Fig. 2). Variable mitochondrial tRNA loops in Bombyx were assumed not to affect biological function [15]. The complete rrnL gene sequence was 1339 nt for O. nubilalis and O. furnicalis, and alignment comparatively showed a single C to T transition. A partial rrnS sequence was obtained from O. nubilalis (434 nt) and O. furnicalis (435 nt), and comparatively showed a single nucleotide deletion.
Predicted secondary structures for A) O. nubilalis and B) O. furnicalis trnR.
3.2 Nucleotide substitution pattern
A 14543 nt consensus mitochondrial genome alignment identified 186 substitutions between O. nubilalis and O. furnicalis: 138 transition (ts) and 48 transversion (tv) mutations (ts:tv = κ ≅ 2.88). This ratio deviated significantly from neutral expectation (1:2; χ2 = 141.447, d.f. = 1, P < 0.001), indicating evolutionary pressures are acting upon O. nubilalis and O. furnicalis mitochondrial genomes. Excess transition mutation also was reported between D. melanogaster subgroup members (κ= 761/180 ≅ 4.23) and attributed to non-neutral evolutionary forces or population effects [7].
Additionally, mitochondrial protein coding sequences (CDS) comparatively showed 170 substitutions between O. nubilalis and O. furnicalis; 131 at 3rd codon positions. The ratio of the rate of nonsynonymous changes at nonsynonymous sites (dN) to synonymous changes at synonymous sites (dS) in Ostrinia ORFs indicated a 7-fold excess of silent mutation (dN/dS = ω ≅ 0.15) [28]. High peptide similarity (≅ 99.22%) may reflect regency O. nubilalis and O. furnicalis speciation, but effects of purifying selection can be inferred since synonymous substitutions are very prevalent. Alternatively, similar environmental selection after speciation could lead to peptide conservation co-occurring with a background of random genetic drift at neutral nucleotide positions. The observed mutation rate at Ostrinia 4-fold degenerate codons (μ4-fold = 5.22%) was significantly higher than at 2-fold degenerate codons (μ2-fold = 2.60%; χ2 = 35.157, d.f. = 1, P < 0.001). Results suggest a greater susceptibility of 4-fold degenerate codons to synonymous substitution.
3.3 Divergence time estimates
The divergence time between O. nubilalis and O. furnicalis mitochondrial was estimated by assuming a linear rate of substitution in short-term evolution (molecular clock) [37] of 2% per million years [38]. Nucleotides in rRNA and tRNA may lack independence due to structural dependence, and purifying selection may act at 1st and 2nd codon positions. The intergenic sequence (IGS) and 3rd codon positions only sites that are nearly neutral. IGS region and 3rd codon positions showed 3.54% nucleotide difference between O. nubilalis and O. furnicalis, indicating that speciation occurred 1.8 mya [38]. Alternatively, 3rd position and IGS region data give a pairwise genetic distance of 0.3284 ± 0.0348 using the Kimura-2-parameter model [39]. Estimates of 0.1 distance unit (D) per 1.0 myr [40] suggest divergence at 3.3 mya. These molecular-based divergence time estimates are supported by highly similar morphology of O. nubilalis and O. furnicalis [41].
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Clary DO, Wolstenholme DR. The mitochondrial DNA molecule of Drosophila yakuba: nucleotide sequence, gene organization, and genetic code. J Mol Evol. 1985 ;22(1):252-271
2. Beard CB, Hamm DM, Collins FH. The mitochondrial genome of the mosquito Anopheles gambiae, DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects. Insect Mol Biol. 1993 ;2(3):103-114
3. Mitchell SE, Cockburn AF, Seawright JA. The mitochondrial genome of Anopheles quadrimaculatus species A: complete nucleotide sequence and gene organization. Genome. 1993 ;36(1):1058-1073
4. Lewis DL, Farr CL, Kaguni LS. Drosophila melanogaster mitochondrial DNA: completion of the nucleotide sequence and evolutionary comparisons. Insect Mol Biol. 1995 ;4(4):263-278
5. Spanos L, Koutroumbras G, Kotsyfakis M, Louis C. The mitochondrial genome of the Mediteranian fruit fly, Ceratitis capitata. Insect Mol Biol. 2000 ;9(2):139-144
6. Lessinger AC, Martins Junqueira AC, Lemos TA, Kemper EL, Da Silva FR, Vettore AL, Arruda P, Azeredo-Espin AML. The mitochondrial genome of the primary screwworm fly Cochliomyia hominivorax (Diptera: Calliphoridae). Insect Mol Biol. 2000 ;9(5):521-529
7. Ballard JWO. Comparative genomics of mitochondrial DNA in Drosophila simulans. J Mol Biol. 2000 ;51(1):64-75
8. Nardi F, Carapelli A, Dallai R, Frati F. The mitochondrial genome of the olive fly Bactrocera oleae: two haplotypes from distant geographical locations. Insect Mol Biol. 2003 ;12(6):605-611
9. Crozier RH, Crozier YC. The mitochondrial genome of the honeybee Apis mellifera: complete sequence and genome organization. Genetics. 1993 ;133(1):97-117
10. Flook PK, Rowell CHF, Gellissen G. The sequence, organization, and evolution of the Locusta migratoria mitochondrial genome. J Mol Evol. 1995 ;41(1):928-941
11. Shao R, Campbell NJH, Baker SC. Numerous gene rearrangements in the mitochondrial genome of the Wallaby louse, Heterodoxus macropus (Pthiraptera). Mol Biol Evol. 2001 ;18(8):58-865
12. Shao R, Barker SC. The highly rearranged mitochondrial genome of the plague thrips, Thrips imagines (Insecta: Thysanoptera): Convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol Biol Evol. 2003 ;20(3):362-370
13. Dotson EM, Beard CB. Sequence and organization of the mitochondrial genome of the Chagas disease vector, Triatoma dimidiata. Insect Mol Biol. 2001 ;10(3):205-215
14. Stewart JB, Beckenbach AT. Phylogenetic and genomic analysis of the complete mitochondrial DNA sequence of the spotted asparagus beetle Crioceris duodecimpunctata. Mol Phylogenet Evol. 2003 ;26(3):513-526
15. Yukuhiro K, Sezutsu H, Itoh M, Shimizu K, Banno Y. Significant levels of sequence divergence and gene rearrangements have occurred between the mitochondrial genomes of the wild mulberry silkmoth, Bombyx mandarina, and its close relative, the domesticated silkmoth, Bombyx mori. Mol Biol Evol. 2002 ;19(8):1385-1389
16. O'Sullivan DF, Bourke RM. Effectiveness of lindane, DDT and monocrotophos for the control of the corn borer Ostrinia furnicalis Guenee (Lepidoptera: Pyralidae) in maize in New Britain. Papua New Guinea Agricult. J. 1975 ;26(1):17-19
17. Mason CE, Rice ME, Calvin DD, Van Duyn JW, Showers WB, Hutchison WD, Witkowski JF, Higgins RA, Onstad DW, Dively GP. European corn borer: Ecology and Management. North Central Regional Publication 327. Ames, IA: Iowa State University. 1996:-
18. Wang R, Yen F, Li S, Li S. Allozyme differentiation among nine populations of the corn borer (Ostrinia) in China. Biochem Genet. 1995 ;33(11-12):413-420
19. Kim C, Hoshizaki S, Huang Y, Tastuki S, Ishikawa Y. Usefulness of mitochondrial COII gene sequences in examining phylogenetic relationships in the Asian corn borer, Ostrinia furnacalis and the allied species (Lepidoptera: Pyralidae). Appl Entomol Zool. 1999 ;34(4):405-412
20. Klun JA, Huettel MD. Genetic regulation of sex pheromone production and response: interaction of sympatric pheromonal race of the European corn borer, Ostrinia nubilalis (Lepidoptera; Pyrallidae). J Chem Ecol. 1988 ;14(11):2047-2061
21. Huang YP, Takanashi T, Hoshizaki S, Tatsuki S, Honda H, Yoshiyasu Y, Ishikawa Y. Geographical variation in the sex pheromone of Asian corn borer, Ostrinia furnacalis in Japan. J Chem Ecol. 1998 ;24(12):2079-2088
22. Willett CS, Harrison RG. Pheromone binding proteins of the European and Asian corn borers: No protein change associated with pheromone differences. Insect Biochem Mol Biol. 1999 ;29(1):277-284
23. Simon C, Frati F, Bechenback A, Crespi B, Liu H, Flook P. Evolution, weighting, and phylogentic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am. 1994 ;87(6):651-701
24. Primer3. 1998 : Rozen S, Skaletsky HJ. http://www-genome.wi.mit.edu/genome_software/other/primer3.html
25. Maddison DR, Maddison WP. MaClade 4.03. Sunderland, MA: Sinaur Associates. 2001
26. Zuker M, Mathews DH, Turner DH. Algorithms and Thermodynamics for RNA Secondary Structure Prediction: A Practical Guide. In: (ed.) Barciszewski B, Clark FC. RNA biochemistry and biotechnology, NATO ASI Series. New York: Kluwer Academic Publishers. 1999:11-43
27. De Rijk P, De Wachter R. RnaViz, a program for the visualization of RNA secondary structure. Nucleic Acids Res. 1997 ;25(22):4679-4684
28. Nei M, Gojobori T. Simple methods for estimating the number of synonymous and nonsynonymous substitutions. Mol Biol Evol. 1986 ;3(5):418-426
29. Kumar S, Tamura K, Nei M. MEGA: Molecular Evolutionary Genetics Analysis. University Park, PA: Pennsylvania State University. 1993
30. Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999 ;27(8):1767-1780
31. Xia X. Maximizing transcription efficiency causes codon usage bias. Genetics. 1996 ;144(3):1309-1320
32. Li WH. Molecular Evolution. Sunderland, MA: Sinauer Associates. 1997
33. Wilson K, Cahill V, Ballment E, Benzie J. The complete sequence of the mitochondrial genome of the crustacean Penaeus monodon: Are Malacostracan crustaceans more closely related to insects than to Branchiopods. Mol Biol Evol. 2000 ;17(6):863-874
34. Crease TJ. The complete sequence of the mitochondrial genome of Daphnia pulex (Cladocera: Crustacea). Gene. 1999 ;233(1-2):89-99
35. Ojala D, Merkel C, Gelfand R, Attaridi G. The tRNA genes punctuate the reading of genetic information in human mitochondrial DNA. Cell. 1980 ;22(2):393-403
36. Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981 ;290(5806):457-465
37. Zuckerland E, Pauling L. Evolutionary divergence and convergence in proteins. In: (ed.) Bryson V, Vogel HJ. Evolving genes and proteins. New York: Academic Press. 1965:97-166
38. Powell JR. Rates of nucleotide substitution in Drosophila mitochondrial DNA and nuclear DNA are similar. Proc Natl Acad Sci USA. 1986 ;83(23):9090-9093
39. Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980 ;16(2):111-120
40. Osawa S, Su ZH, Kim CG, Okamoto M, Tominaga O, Imamura Y. Evolution of the carabid ground beetles. Adv Biophys. 1999 ;36(1):65-106
41. Mutuura A, Munroe E. Taxonomy and distribution of the European corn borer and allied species: genus Ostrinia (Lepidoptera: Pyralidae). Mem Entomol Soc Canada. 1970 ;71:1-112
Tables and Figures
Author biography
Brad S. Coates (M.S. Iowa State University 2001): Graduate student in Genetics program at Iowa State University, Ames, Iowa, USA. Current research includes population genetics (mitochondrial and microsatellites) and molecular mechanisms of European corn borer resistance to transgenic maize.
Douglas V. Sumerford (Ph.D. North Carolina State University, 1999): Currently Research Entomologist at USDA-ARS, Corn Insect and Crop Genetics Research Unit, Ames, Iowa, USA. Research interests include population genetics and population ecology of the European corn borer to better understand strategies to slow the development of resistance to transgenic crops. Previous work history include a Research Entomologist position at the Southern Insect Management Research Unit, Stoneville, MS.
Richard L. Hellmich (Ph.D. The Ohio State University, 1983): Currently Research Entomologist at USDA-ARS, Corn Insect and Crop Genetics Research Unit, Ames, Iowa, USA. Research includes investigation of genetics and ecology of corn pest species, management of European corn borer resistance to transgenic corn, and evaluation of effects of transgenic corn on non-target organisms. Previously worked as a Research Entomologist (Africanized honey bee genetics and ecology) at the USDA-ARS, Honey Bee Breeding, Genetics and Physiology Laboratory, Baton Rouge, LA.
Leslie C. Lewis (Ph.D. Iowa State University, 1970): Currently Research Leader at USDA-ARS, Corn Insect and Crop Genetics Research Unit, Ames, Iowa, USA. Research interests include study of insect pathogens Beauveria bassiana and Nosema pyrausta, and compatibility of insect pathogens with beneficial insects. Also studies endophytic relationship between Beauveria and maize, and incorporates insect pathogens into sustainable agriculture programs.
![]() Corresponding address:
Corresponding address:
Brad S. Coates, USDA-ARS, Corn Insects & Crop Genetics Research, 113 Genetics Lab, Iowa State University, Ames, IA 50010. (515)-294-0668