Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. PIKE-S signaling in the...
3. PIKE-L signaling and its role...
4. PIKE-A signaling and its role...
5. PIKE-A homologs
6. Homer/PIKE-L/protein 4.1...
7. Conclusion/perspectives
Acknowledgements
References
Figures
Author biography

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2005; 1(2):44-50. doi:10.7150/ijbs.1.44 This issue Cite
Review
PIKE GTPase Signaling and Function
Jee-Yin Ahn, Keqiang Ye ![]()
Department of Pathology and Laboratory Medicine, Emory University School of Medicine, Atlanta, GA 30322, USA.
Received 2004-10-1; Accepted 2005-2-1; Published 2005-4-1
Abstract
PIKE (PI 3-Kinase Enhancer) is a recently identified brain specific nuclear GTPase, which binds PI 3-kinase and stimulates its lipid kinase activity. Nerve growth factor treatment leads to PIKE activation by triggering the nuclear translocation of phospholipase C-γ1 (PLC-γ1), which acts as a physiologic guanine nucleotide exchange factor (GEF) for PIKE through its SH3 domain. To date, three forms of PIKE have been characterized: PIKE-S, PIKE-L and PIKE-A. PIKE-S is initially identified shorter isoform. PIKE-L, a longer isoform of PIKE gene, differs from PIKE-S by C-terminal extension containing Arf-GAP (ADP ribosylation factor-GTPase Activating Protein) and two ankyrin repeats domains. In contrast to the exclusive nuclear localization of PIKE-S, PIKE-L occurs in both the nucleus and the cytoplasm. PIKE-L physiologically associates with Homer 1, an mGluR I binding adaptor protein. The Homer/PIKE-L complex couples PI 3-kinase to mGluR I and regulates a major action of group I mGluRs, prevention of neuronal apoptosis. More recently, a third PIKE isoform, PIKE-A was identified in human glioblastoma multiforme brain cancers. Unlike the brain specific PIKE-L and -S isoforms, PIKE-A distributes in various tissues. PIKE-A contains the same domains present in PIKE-L but lacks N-terminal proline-rich domain (PRD), which binds PI 3-kinase and PLC-γ1. Instead, PIKE-A specifically binds to active Akt and upregulates its activity in a GTP-dependent manner, mediating human cancer cell invasion and preventing apoptosis. Thus, PIKE extends its roles from the nucleus to the cytoplasm, mediating cellular processes from cell invasion to programmed cell death.
Keywords: PIKE, GTPase, PI 3-kinase, PLC-γ1, GEF, ArfGAP
1. Introduction
The GTPase family members such as Ras are major regulators of cell growth. The GTPase intermediated signal transduction pathways have been well defined in the cytoplasm. However, little is known about signaling aspects of nuclear GTPases. Previously, we identified a brain specific nuclear GTPase called PIKE, which binds PI 3-kinase and enhances its kinase activity. NGF treatment elicits the membrane-associated 4.1N nuclear translocation and binds PIKE, which was initially identified as a 4.1N binding partner in a yeast two-hybrid screening. The binding of 4.1N to PIKE prevents its interactions with nuclear PI 3-kinase, leading to diminish the activation of PI 3-kinase by NGF [1] . PIKE GTPase is activated by nuclear translocated phospholipase C-γ1 (PLC-γ1), which acts as a guanine nucleotide exchange factor (GEF) for PIKE. This action is not dependent on PLC-γ1's lipase activity but appears instead to involve its SH3 domain [2] .
Recently, we have identified and cloned a few isoforms of PIKE: PIKE-L and PIKE-A. Both of them contain an ArfGAP domain and two ankyrin repeats domains. The C-terminal protein structure resembles that of Centaurin family, which are GTPase–activating proteins (GAPs) for Arfs or have homology to ArfGAPs and Arf effectors [3, 4] . In rat brain and hippocampal neurons, PIKE-L physiologically binds to Homer, an adaptor protein known to link metabotropic glutamate receptors (mGluR I) to multiple intracellular targets including the inositol 1,4,5 trisphosphate (IP3) receptor. The activation of mGluR I by its agonists enhances formation of an mGluR I-Homer-PIKE-L complex leading to activation of PI 3-kinase activity and prevention of neuronal apoptosis [5] .
PIKE-A differs from PIKE-L and -S by lacking the N-terminal proline-rich domain (PRD), which binds PI 3-kinase. The comparison of the three protein structures is depicted in Figure 1. Interestingly, PIKE-A directly binds to activated Akt but not PI 3-kinase in a guanine nucleotide-dependent way and stimulates the kinase activity of Akt. PIKE-A is co-amplified with CDK4 in a variety of human cancers. The overexpressed PIKE-A enhances Akt activity, promotes cancer cell invasion and prevents apoptosis [6] . Mounting evidence demonstrates that Akt is a crucial regulator of diverse cellular processes and contributes to cancer progression. Activation of Akt is essentially dependent on PI 3-kinase signaling. Nevertheless, PIKE-A-mediated Akt activation is independent of PI 3-kinase. Therefore, PIKE-A provides a new mechanism whereby growth factors and other cellular stimuli regulate Akt, and functions as an oncogenic effector of cell invasion. Here, we discuss the recent advances in biochemical and molecular studies and future directions that might further our understanding of the roles of nuclear PI 3-kinase and PIKE family.
Diagram of three PIKE isoforms.PIKE-L, an alternatively spliced form of PIKE, which is several hundred amino acids longer than the original form of PIKE, designated as PIKE-S (bankit475414 (AY128689, human)). In addition to the GTPase and PH domains shared by PIKE-S and PIKE-L, PIKE-L contains an ArfGAP domain and two Ankyrin repeats. PIKE-A contains the GTPase, PH, ArfGAP and Ankyrin repeats domains present in PIKE-L but lacks the proline-rich domains (PRD) containing N-terminus, which protein 4.1N, PI 3-kinase and PLC-γ1 bind to.

2. PIKE-S signaling in the nucleus
PIKE-S comprises 753 residues, containing three proline-rich domains (PRD) in the N-terminus, followed by a GTPase domain and a partial pleckstrin homology (PH) domain in the C-terminus. NGF treatment of PC12 cells activates PIKE GTPase with peak at 30 min and decays after 1 h. PIKE-S binds nuclear PI 3-kinase in a GTP-dependent manner. PIKE-S activates PI 3-kinase depending on the presence of its two subunits with PIKE-S being able to bind independently to each of the two subunits p85 and p110. Transfected by itself, p110 lacks catalytic activity but acquires it following co-transfection with p85. PIKE-S transfection quadruples this activity. PIKE-S can activate PI 3-kinase only when both p110 and p85 are expressed, not with p110 alone [1] .
PIKE-S is a nuclear GTPase. Normally, GTPases cycles between the active, GTP-bound and inactive, GDP-bound states [7, 8] . Guanine-nucleotide-exchange factors (GEFs) act as positive regulators that promote release of GDP and consequent formation of the active GTP-bound state, whereas GTPase-activating proteins (GAPs) act as negative regulators, stimulating the intrinsic GTPase activity to generate the inactive GDP-bound form. PRD is a protein-interaction module for SH3 domain. Indeed, PLC-γ1 binding to PIKE involves the SH3 domain of PLC-γ1 interacting with the PRD of PIKE-S [2] . We have demonstrated that PLC-γ1 acts as a physiologic guanine nucleotide exchange factor (GEF) for PIKE-S. This GEF activity is mediated by the SH3 domain with phospholipase catalytic activity not required. Moreover, the SH3 domain but not phospholipase catalytic activity of PLC-γ1 is necessary for its mitogenic actions, probably through activating the nuclear PI 3-kinase [2] .
PLC-γ1 is a tyrosine kinase substrate for many receptor and nonreceptor tyrosine kinases. The activation of PLC-γ1 is essential for cell proliferation and differentiation. The activated PLC-γ1 translocates to the plasma membrane and triggers the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into two second messengers, inositol 1,4,5-triphosphate (IP3) and diacyglycerol (DAG), which regulate the intracellular Ca2+ and activation of protein kinase C respectively. PLC-γ1 does not possess any known nuclear localization signal and predominantly localizes in the cytoplasm. Nevertheless, it has been reported to be present in the nucleus [9-12] . It has been shown that PLC-γ1 localizes to the nucleus of highly transformed and proliferating cell lines (A431, HeLa, mouse hepatoma MH 22A, rat Zajdela ascitic hepatoma) but not to the nucleus of primary embryo skin or lung fibroblasts, where the enzyme is primarily cytoplasmic. Differential subcellular localizations in normal or highly transformed cell lines may reflect the degree of transformation of the cell type or phase of the cell cycle [10] . This finding is consistent with the observation of an increased amount of PLC-γ1 in nuclei of 22 h regenerating rat liver, suggesting a relationship between the S-phase of the cell cycle and intranuclear localization of PLC-γ1 [13] .
PI 3-kinase is a key regulator of many cellular processes including cell proliferation, survival, motility, vesicular trafficking and carbohydrate metabolism. PI 3-kinase is activated by both receptor tyrosine kinases (RTK) and cytoplasma membrane associated GTPase proteins. The activated PI 3-kinase generates the critical second messengers: the D-3 position phosphorylated phosphoinositides, which bind to the Pleckstrin Homology domain (PH) of numerous signaling molecules including Phosphoinositol-Dependent Kinase 1 (PDK1), Akt/PKB and PLC-γ1, etc. While much has been learned about PI 3-kinase signal transduction in the cytoplasm, almost nothing is known about it in the nucleus. Previous studies show that PI 3-kinase also occurs in the nucleus [1, 14-19] . Stimulation of cells with NGF activates nuclear PI 3-kinase with nuclear accumulation of 3-phosphorylated phosphoinositide lipids [18, 20] . However, the physiological role of nuclear PI 3-kinase and its mediated signaling pathways remain obscure. The interaction between PIKE-S and PI 3-kinase is GTP-dependent – like that between Ras and PI 3-kinase [1, 21] . NGF treatment of PC12 cells activates PIKE and nuclear PI 3-kinase with peaks at 30 min, whereas NGF activates the cytoplasmic GTPases of the ras family as well as cytoplasmic PI 3-kinase much more rapidly with peak activity in 5-10 min [21] . Moreover, in dominant-negative PIKE-S (K413AS414N) retrovirus infected PC12 cells, activation by NGF of nuclear PI 3-kinase is abolished, suggesting that PIKE-S is the major mediator of nuclear PI 3-kinase.
Cytoplasmic PI 3-kinase activation requires activated receptor tyrosine kinases (e.g. PDGFR, EGFR, CD28, etc.) or GTPase proteins such as Ras. However, none of these known PI 3-k activators are present in nucleus. Our discovery that the nuclear GTPase, PIKE, enhances nuclear PI 3-kinase activity indicates that PIKE-S may be the nuclear counterpart of Ras. These findings might provide a molecular basis for the regulation of nuclear PI 3-kinase. The extreme N-terminus of PIKE-S associates with the C-terminal domain (CTD) of protein 4.1N, a neuronal isoform of the erythrocyte membrane cytoskeletal protein 4.1R. NGF treatment elicits PIKE-S interactions with nuclear translocated 4.1N. Overexpression of 4.1N abolishes PIKE effects on PI 3-kinase. Therefore, activation of nuclear PI 3-Kinase by PIKE is inhibited by the NGF-stimulated 4.1N translocation to the nucleus [1] . The nuclear PLC-γ1/PIKE-S/ nuclear PI 3-kinase cascade is depicted in Figure 2.
PLC-γ1/PIKE-S/nuclear PI 3-kinase signalling: NGF treatment of PC12 cells provokes PLC-γ1 nuclear translocation, and stimulates PIKE-S GTPase to bind GTP. The activated PIKE-S binds and elevates nuclear translocated PI 3-kinase activity. NGF also causes 4.1N to translocate to the nucleus over a period of hours, lagging behind the translocation of PI 3-kinase and the peak activation of PIKE elicited by NGF. The decline of activated nuclear PI 3-kinase, which coincides with the appearance of nuclear 4.1N, might involve 4.1N sequestering PIKE away from nuclear PI 3-kinase. The decline of PIKE's NGF-induced GTPase activation takes place at about the same time and so also may participate in the decline of nuclear PI 3-kinase.
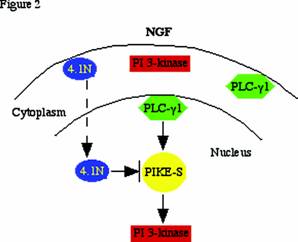
PLC-γ1 and PI 3-kinase share the same substrate PI (4,5) P2, and both enzymes are concomitantly recruited to the plasma membrane and activated, where they mediate each other's enzymatic activity. Several studies have suggested cross-talk between PLC-γ1 and PI 3-kinase in the cytoplasm. For example, PI (3,4,5) P3 generated by PI 3-kinase influences PLC-γ1 membrane translocation and activation by binding to its PH domain and a C-terminal SH2 domain [22, 23] , and activation of PLC downregulates PI 3-kinase by at least two mechanisms: (1) inhibition of IRS-1-associated PI 3-kinase; and (2) acute activation of a PtdIns (3,4,5) P3 5-phosphatase. NGF elicits PLC-γ1 nuclear translocation and acts as a GEF for PIKE through it SH3 domain. The activated PIKE GTPase subsequently provokes nuclear PI 3-kinase activation. Thus, the nuclear PLC-γ1/PIKE/PI 3-kinase signaling pathway seems to be the extension of the cross-talk between the cytoplasmic PLC-γ1 and PI 3-kinase.
3. PIKE-L signaling and its role in neuronal survival
PIKE-L was identified in searching databases for sequences that might resemble PIKE-S. PIKE-L differs from PIKE-S in containing a 40 kDa C-terminal extension which includes an Arf-GAP and two ankyrin repeats domains [5] . PIKE-L and PIKE-S are alternatively spliced isoforms and brain-specific. However, whereas PIKE-S occurs in all brain regions examined, PIKE-L is uniquely absent from the cerebellum. The subcellular localization of the two proteins differs. PIKE-S is exclusively nuclear, whereas PIKE-L occurs in multiple subcellular fractions and, by immunohistochemistry, is observed throughout the cell body and all neuronal processes [5] .
Sequence analysis led to the discovery that PIKE-L binds to Homer 1C, an adaptor protein localized to postsynaptic densities coupling cytoplasmic regions of Group I coupling metabotropic glutamate receptors (mGluRs) to inositol-1,4,5-trisphosphate receptors (IP3Rs) as well as SHANK proteins [24] . The mGluRs comprise three groups: group I (mGluR 1 and 5), group II (mGluR 2 and 3) and group III (mGluR 4, 6, 7 and 8). Via G proteins, Group I receptors stimulate phospholipase β (PLCβ) leading to the formation of IP3 and calcium mobilization. By contrast, Group II and Group III receptors are negatively coupled to adenylyl cyclase [25] . One particularly prominent action of Group I mGluRs is to protect neurons from apoptotic death [26] . Binding of mGluRI with Homer and thereby with PIKE-L occurs only with the Group I class of mGluRs, which are the only forms that contain the Homer ligand PxxF motif. Amino acids 187-190 of PIKE-L are PKPF which corresponds to a domain recently appreciated as a consensus motif (PxxF) present in proteins that bind to the EVH1 domain of Homer [24] . PIKE-L and Homer 1C coprecipitate robustly, and the interaction is dependent upon the portion of PIKE-L containing the PKPF sequence. Mutation of proline-187 of PIKE-L abolishes binding of PIKE-L to Homer 1C and provides a useful tool to analyze the importance of this binding in various signal cascades.
PIKE-L/Homer 1C complex couples PI 3-kinase to metabotropic glutamate receptor 1 (mGluR1), suggesting that mGluR1 might activate PI 3-kinase separately from its well-known activation of phospholipase C. This pathway has been verified by the demonstration that mGluR5 transfection of HEK293 cells stimulates PI 3-kinase activity while mutants of mGluR5 that do not bind Homer fail to activate PI 3-kinase [5] . Moreover, Homer mutants that do not bind mGluR5 block PI 3-kinase activation. In hippocampal cultures mGluR I activation increases PI 3-kinase activity, while infection with an adenovirus containing a dominant-negative form of PIKE-L blocks such activation. Delivery into hippocampal cultures of the Homer-binding motif of PIKE as a dominant-negative, blocks mGluR I stimulation of PI 3-kinase, while mutant peptides that do not bind Homer have no effect. Finally, mGluR I activation fails to stimulate PI 3-kinase in the cerebellum, a brain region that is devoid of PIKE-L.
Prevention of neuronal apoptosis is a major action of group I mGluRs, and this process appears to be mediated by PIKE-L and PI 3-kinase [5] . In hippocampal cultures mGluR I activation and PIKE-L transfection block the apoptotic effects of staurosporine and other agents. By contrast, infection with a PIKE-L dominant-negative construct augments apoptosis, and a PIKE construct which cannot bind Homer prevents the anti-apoptotic actions of mGluR activation. Additionally, mGluR I activation does not block apoptosis in the cerebellum, which lacks PIKE-L.
PIKE-L may have other activities besides mediating actions of mGluR I on PI 3-kinase and apoptosis. Some possibilities are suggested by the structure of PIKE-L, whose C-terminal region contains an Arf-GAP domain and two ankyrin repeats. GTPases are typically activated by GAP proteins, but whether the Arf-GAP domain functions as an internal GAP for the GTPase activity of PIKE-L remains to be determined, though there are instances of Arf-GAP domains exerting such activity [27] . Mediation of anti-apoptotic actions is the only mGluR I function thus far examined in connection with PIKE-L. Group I mGluRs exert a variety of neuronal functions, influencing motor learning and coordination via cerebellar Purkinje cells [28] and influencing long term potentiation in the hippocampus [29] . Conceivably, PIKE-L also participates in these activities.
4. PIKE-A signaling and its role in human cancers
In our evaluation of PIKE amplification in human glioblastoma multiforme, we have identified a third PIKE form (PIKE-A), which was originally identified in the human genome sequencing effort as KIAA0167 [30] , and was independently identified by Liu and collaborators [27] . The chromosome 12q13-15 region is frequently amplified in human sarcomas and brain tumors [31, 32] . 12q13-15 amplicon contains two separate core regions, one containing MDM2 and the other containing Cyclin-Dependent-Kinase 4 (CDK4) [32, 33] . Blast search reveals the PIKE gene is localized at 12q13.3 adjacent to CDK4 [32] . PIKE-A, -L and -S arise from the same PIKE gene. Gene structure analysis reveals that PIKE-A results from different transcription initiation site from that of PIKE-L and -S. PIKE-A contains the GTPase, PH, ArfGAP and two ankyrin repeats domains present in PIKE-L but lacks the PRD containing N-terminus, which binds protein 4.1N, PI 3-kinase and PLC-γ1. Fluorescent in-situ hybridization (FISH) and Northern blotting analysis reveal that PIKE gene is amplified in a variety of human sarcoma and brain tumor cell lines [6] .
Unlike PIKE-L or -S, PIKE-A does not associate with PI 3-kinase, instead, it binds to active but not inactive Akt. Truncation analysis demonstrates that GTPase and ankyrin repeats domains interact with partial catalytic and regulatory domains of Akt. Binding by Akt to purified recombinant GST-PIKE-A is evident in cells treated with the mitogenic stimulus IGF-1 compared to untreated cells. GTP-γ-S and GDP-β-S strongly stimulate binding of activated Akt and PIKE-A compared to faint binding in the absence of guanine nucleotide treatment [6] . Interestingly, dominant-negative PIKE-A-DN binds Akt promiscuously even in cells not treated by growth factor IGF-1 and, in treated cells, binding occurs even in the absence of guanine nucleotides. PIKE-A strongly binds to active Akt, indicating that Akt phosphorylation might play a role in mediating its association with PIKE-A. In vitro Akt kinase assay demonstrates that PIKE-A substantially enhances Akt activity, for which GTPase domain but not ankyrin repeats domain is responsible. To date, numerous PIKE binding proteins have been identified and are now depicted in Figure 3.
PIKE binding proteins: Protein 4.1N and PI 3-kinase share the same binding motif on the N-terminus of PIKE. The third proline-rich domain of PIKE binds SH3 domain of PLC-γ1. mGluR I adaptor protein Homer 1 interacts with PIKE through “Homer binding motif PXPF (a.a. 187-190)”. Akt associates with PIKE-A through its GTPase and C-terminal ankyrin repeats domains.

Functional analysis of the interaction between PIKE-A and Akt demonstrates that PIKE-A mediates invasion of cancer cells through Akt. Two human glioblastoma cell lines, LN-Z308, in which PIKE-A is heavily overexpressed, and U87MG that does not overexpress PIKE-A, were utilized to dissect the physiological consequence of PIKE amplification. Infection of U87MG cells with an adenovirus containing wild-type PIKE-A elicits almost a doubling of invasive cell number. Only a modest increase occurs with the LN-Z308 cells, presumably because endogenous PIKE-A is already highly expressed. In both LN-Z308 and U87MG cells, infection with PIKE-A-DN reduces cell invasion by about 50% compared with control cells. The same results are observed in other human cancer cell lines. Thus, PIKE-A is a physiologic regulator of Akt and an oncogenic effector of cell invasion [6] .
More recently, we show that overexpression of PIKE-A wild-type but not dominant-negative mutant prevents apoptosis through mediating Akt activity. Knockdown of PIKE-A diminishes Akt activity and increases apoptosis. Collectively, these observations suggest that PIKE-A amplification contributes to cancer cell survival and progression by inhibiting apoptosis through upregulating Akt [34] .
5. PIKE-A homologs
AGAP1/GGAP1/KIAA1099 (accession number AB029022), M-RIP (accession number AF359283), and AGAP2/GGAP2/KIAA0167, we have now designated it as PIKE-A (accession number D79989), consist of the identical gene structure. GGAP1 and GGAP2 share approximately 50% sequence homology, while GGAP1 and MRIP1 share 70% sequence homology at the amino acid level. The complete protein sequences of this family encode an N-terminal Ras-related GTPase domain, a PH domain, followed by a C-terminal GAP domain and an ANK repeat domain. The GTPase domain in the GGAP proteins reveals 60% homology to the Ras family of GTPases. The GTPase domain of AGAP1 is 51% similar and 27% identical to K-Ras, AGAP2 is 42% similar and 28% identical to Rab9, and M-RIP is 47% similar and 27% identical to H-Ras [35] .
Northern blotting analysis demonstrates that GGAP1 mRNA was found in most of the tissues although the level of expression is varied, with enriched expression in skeletal muscle, brain, placenta, and kidney. Most tissues contain two messages at 5 kb and 8.5 kb, while the peripheral blood leukocytes (PBL) have only one message with the size of 5.5 kb. The expression of PIKE-A is highly enriched in the brain, heart, skeletal muscle, and immune tissues [27, 32] . Northern blotting analysis of human brain regions reveals that a prominent band was found in cerebellum, cerebral cortex, occipital pole, frontal lobe, temporal lobe, and putamen for GGAP1 and GGAP2 (PIKE-A), while low level of expression was detected in medulla and spinal cord for PIKE-A [27] .
Subcellular localization reveals AGAP/GGAPs are expressed in different cellular compartments. In transfected COS-7 cells, Flag-tagged AGAP1/GGAP1 staining occurs in the cytosol, possibly in the internal membrane systems, such as the endoplasmic reticulum (ER) and the Golgi apparatus. On the other hand, the Flag-tagged GGAP2/PIKE-A was stained in both the cytoplasm and the nucleus. This observation is consistent with the subcellular distribution of amplified PIKE-A in human glioblastoma cells [6] . Furthermore, cells overexpressing AGAP1/GGAP1 are more flat and have much more lamellipodia than control cells. In contrast, cells overexpressing AGAP1GGAP2/PIKE-A are smaller and more rounded than control cells.
Although the similarity of the GTPase domain in AGAP1/GGAP1 to K-Ras, the purified recombinant protein does not bind to GDP, GTP, ADP, or ATP. No GTPase activity was detected in full-length AGAP1 or the isolated GTPase domain. Similarly, no any effect on the Ras phosphorylation cascade was detected either. However, AGAP1/GGAP1 displays ArfGAP activity toward Arf1>Arf5>Arf6 in vitro. Phosphatidylinositol 4,5-bisphosphate and phosphatidic acid synergistically stimulated GAP activity. Deletion of the GTPase domain affects lipid dependence of ArfGAP activity. Overexpressed AGAP1 induced the formation of and was associated with punctate structures containing the endocytic markers transferrin and Rab4 [35] . Thus, AGAP1 is a phosphoinositide-dependent ArfGAP that impacts both the endocytic compartment and actin, consistent with the observations described [27] .
Different from the findings on AGAP1/GGAP1 by Nie et al, Xia and his colleagues demonstrated GTPase domain of AGAP1/GGAP1 and GGAP2/PIKE-A binds to [32P]GTP, but the C-terminal segment of the proteins fail to display GTP-binding. Moreover, they showed the prominent GTPase activity from the full-length AGAP1/GGAP1 and AGAp2/GGAP2/PIKE-A immunopurified from the cells, transiently transfected with the cDNAs encoding the Flag-tagged proteins into COS-7 cells. The turnover number of AGAP1/GGAP1 and AGAP2/GGAP2/PIKE-A for GTPase activity is similar to GTPase Ras (0.01 min-1) [27] . Furthermore, the authors showed that the C-terminal ArfGAP domain could regulate the activity of the N-terminal GTPase domain via direct intramolecular interaction. Bacterially purified GTPase domains from AGAP1/GGAP1 and AGAP2/GGAP2/PIKE-A have very low intrinsic GTPase activity. Addition of the ArfGAP domain from the same protein (GGAP1 or GGAP2) significantly increased the GTPase activity [27] .
Phosphoinositide lipids bind to PH domain and activate dynamin GTPase activity, which is important in dynamin function during vesicle budding [36] . To test this possibility that phosphatidylinositol lipids also affect PIKE GTPase activity, we treated PIKE-S with different phospholipids including phosphatidylinositol 4,5-bisphosphate (PI-4,5-P2) and phosphatidylinositol 3,4,5-trisphosphate (PI-3,4,5-P3), but failed to detect its activation, presumably because it just contains a partial PH domain. However, PIKE-L contains a full module of PH domain and displays high homology with phosphatidylinositol bi- or tri- phosphates. In fact, our recent data reveal that it selectively binds to PI (3,4)P2, PI (3,4,5)P3 and PI (3,5)P2 and this interaction mediates PIKE-L's GTPase activity (unpublished data). This observation raises an intriguing and provocative possibility that PIKE-L might regulates its own GTPase activity through a negative feed-back mechanism using the PI 3-kinase lipid product.
6. Homer/PIKE-L/protein 4.1 complex in postsynaptic assembly and signaling
Neuronal activity-regulated synaptic α-amino-3-hydroxy-5-methylisoxazole-4-propionate receptor (AMPAR) trafficking plays a critical role in synaptic plasticity, a molecular model underlying the learning and memory. Synaptic AMPAR insertion and removal from postsynaptic membrane mediates N-methyl-D-aspartate receptors (NMDAR) and mGluR I-dependent long-term potentiation (LTP) and long-term depression (LTD) [37, 38] . While numerous proteins coupling NMDAR to AMPAR have been demonstrated, little is known the molecular partners that mediate the crosstalk between mGluR I and AMPAR. We have previously shown that protein 4.1N, which binds to the cytoplasmic tail of AMPAR [39] , interacts with the extreme N-terminus of PIKE [1] . Moreover, mGluR I activation stimulates the formation of Homer/PIKE-L complex, which couples PI 3-kinase to mGluR I [5] . These observations support an intriguing notion that PIKE-L-Homer interaction might couple AMPA receptor to mGluRs through PIKE-L association with the postsynaptic proteins 4.1N.
Protein 4.1 are a family of multifunctional cytoskeletal components (4.1R, 4.1G, 4.1N and 4.1B) derived from four related genes, each of which is expressed in the nervous system [40] . Mouse 4.1R knockout supports the notion that protein 4.1 plays an important role in the nervous system, since multiple disturbances in behavior indicate the cerebellar and hippocampal defects, correlating well with the absence of 4.1R in cerebellar granule cells, and hippocampal dentate gyrus cells [41, 42] . Previous studies indicate that proteins 4.1 exist in both presynaptic and postsynaptic density [39, 43, 44, 45, 46] . Immunohistochemical studies using a polyclonal 4.1N antibody revealed several patterns of neuronal staining, with localizations in the neuronal cell body, dendrites, and axons. In primary hippocampal cultures, mouse 4.1N is enriched at the discrete sites of synaptic contact, colocalizing with the postsynaptic density protein of 95 kDa (a postsynaptic marker) and glutamate receptor type 1 (an excitatory postsynaptic marker) [43] . Previously, 4.1N has been shown to associate with the membrane proximal region of GluR1 in vivo and colocalize with AMPA receptors at excitatory synapses, where it might implicate in AMPA receptors clustering and connecting to the actin cytoskeleton [39] . Conceivably, protein 4.1N may function to confer stability and plasticity to the neuronal membrane via interactions with multiple binding partners, including the spectrin-actin-based cytoskeleton, integral membrane channels and receptors, and postsynaptic protein like PIKE-L. This finding conforms to our current model in postsynaptic glutamate receptors clustering and synaptogenesis mediated by Homer-Shank interaction by docking mGluRs to the NMDARs.
7. Conclusion/perspectives
PIKE-S GTPase is activated by PLC-γ1, which acts as a physiologic guanine nucleotide exchange factor (GEF) for PIKE. The activated PIKE-S in turn stimulates PI 3-kinase activity in the nucleus. The identification of PIKE-L and -A isoforms, which contain an ArfGAP domain, might shed light on the molecular mechanism how PIKE GTPase is negatively regulated. The ArfGAP domain possesses potent GAP activity on Arf GTPase and its internal GTPase domain. Moreover, this GAP activity is mediated by phosphoinositol lipids through PH domain of PIKE, suggesting a negative feed-back mechanism between PIKE/PI 3-kinase singaling. Besides its anti-apoptotic role in mGluR I cascade, PIKE-L might also implicate in synaptic plasticity by coupling mGluR I to AMAP receptor. PIKE-A may have even broader relevance resulting from its subcellular localization (in both the nucleus and the cytoplasm) and tissue distribution. PIKE-A might contribute to cancer progression by promoting cancer cell invasion and survival through up-regulating Akt.
Acknowledgements
This work is supported by grants from American Cancer Society (#RSG-04-077-01-TBE) and NIH (RO1 NS045627).
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Ye K. et al. Pike. A nuclear gtpase that enhances PI3kinase activity and is regulated by protein 4.1N. Cell. 2000 ;103:919-30
2. Ye K. et al. Phospholipase Cγ1 is a physiological guanine nucleotide exchange factor for the nuclear GTPase PIKE. Nature. 2002 ;415:541-544
3. Donaldson JG. Filling in the GAPs in the ADP-ribosylation factor story. Proc Natl Acad Sci U S A. 2000 ;97:3792-4
4. Cukierman E. et al. The ARF1 GTPase-activating protein: zinc finger motif and Golgi complex localization. Science. 1995 ;270:1999-2002
5. Rong R. et al. PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci. 2003 ;6:1153-61
6. Ahn JY. et al. PIKE (PI 3-kinase-enhancer)-A GTPase stimulates Akt activity and regulates cancer cell invasion. J Biol Chem. 2004 ;279:16441-16451
7. Boguski MS, McCormick F. Proteins regulating Ras and its relatives. Nature. 1993 ;366:643-54
8. Quilliam LA. et al. Guanine nucleotide exchange factors: activators of the Ras superfamily of proteins. Bioessays. 1995 ;17:395-404
9. Martelli AM. et al. Phosphoinositide signaling in nuclei of Friend cells: phospholipase C beta down-regulation is related to cell differentiation. Cancer Res. 1994 ;54:2536-40
10. Diakonova M. et al. Intracellular distribution of phospholipase C gamma 1 in cell lines with different levels of transformation. Eur J Cell Biol. 1997 ;73:360-7
11. Neri LM. et al. Nuclear diacylglycerol produced by phosphoinositide-specific phospholipase C is responsible for nuclear translocation of protein kinase C-alpha. J Biol Chem. 1998 ;273:29738-44
12. Zini N. et al. The intranuclear amount of phospholipase C beta 1 decreases following cell differentiation in Friend cells, whereas gamma 1 isoform is not affected. Eur J Cell Biol. 1995 ;68:25-34
13. Neri LM. et al. Changes of nuclear PI-PLC gamma1 during rat liver regeneration. Cell Signal. 1997 ;9:353-62
14. Neri LM. et al. Nuclear translocation of phosphatidylinositol 3-kinase in rat pheochromocytoma PC 12 cells after treatment with nerve growth factor. Cell Mol Biol (Noisy-le-grand). 1994 ;40:619-26
15. Lu PJ. et al. Phosphoinositide 3-kinase in rat liver nuclei. Biochemistry. 1998 ;37:5738-45
16. Kim SJ. Insulin rapidly induces nuclear translocation of PI3-kinase in HepG2 cells. Biochem Mol Biol Int. 1998 ;46:187-96
17. Bavelloni A. et al. Phosphatidylinositol 3-kinase translocation to the nucleus is induced by interleukin 1 and prevented by mutation of interleukin 1 receptor in human osteosarcoma Saos-2 cells. J Cell Sci. 1999 ;112:631-40
18. Tanaka K. et al. Evidence that a phosphatidylinositol 3,4,5-trisphosphate-binding protein can function in nucleus. J Biol Chem. 1999 ;274:3919-22
19. Bacqueville D. et al. Characterization of a G protein-activated phosphoinositide 3-kinase in vascular smooth muscle cell nuclei. J Biol Chem. 2001 ;276:22170-6
20. Neri LM. et al. Increase in nuclear phosphatidylinositol 3-kinase activity and phosphatidylinositol (3,4,5) trisphosphate synthesis precede PKC-zeta translocation to the nucleus of NGF-treated PC12 cells. Faseb J. 1999 ;13:2299-310
21. Rodriguez-Viciana P. et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994 ;370:527-32
22. Bae YS. Activation of phospholipase C-gamma by phosphatidylinositol 3,4,5- trisphosphate. J Biol Chem. 1998 ;273:4465-9
23. Ji QS. et al. Physiological requirement for both SH2 domains for phospholipase C-gamma1 function and interaction with platelet-derived growth factor receptors. Mol Cell Biol. 1999 ;19:4961-70
24. Xiao B. et al. Homer: a link between neural activity and glutamate receptor function. Curr Opin Neurobiol. 2000 ;10:370-4
25. De Blasi A. et al. Molecular determinants of metabotropic glutamate receptor signaling. Trends Pharmacol Sci. 2001 ;22:114-20
26. Copani A. et al. Activation of metabotropic glutamate receptors prevents neuronal apoptosis in culture. J Neurochem. 1995 ;64:101-8
27. Xia C. et al. GGAPs, a new family of bifunctional GTP-binding and GTPase-activating proteins. Mol Cell Biol. 2003 ;23:2476-88
28. Aiba A. et al. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994 ;79:377-88
29. Lu YM. et al. Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J Neurosci. 1997 ;17:5196-205
30. Nagase T. et al. Prediction of the coding sequences of unidentified human genes. V. The coding sequences of 40 new genes (KIAA0161-KIAA0200) deduced by analysis of cDNA clones from human cell line KG-1. DNA Res. 1996 ;3:17-24
31. Elkahloun AG. et al. Molecular cytogenetic characterization and physical mapping of 12q13-15 amplification in human cancers. Genes Chromosomes Cancer. 1996 ;17:205-14
32. Elkahloun AG. et al. Transcript mapping in a 46-kb sequenced region at the core of 12q13.3 amplification in human cancers. Genomics. 1997 ;42:295-301
33. Reifenberger G. et al. Refined mapping of 12q13-q15 amplicons in human malignant gliomas suggests CDK4/SAS and MDM2 as independent amplification targets. Cancer Res. 1996 ;56:5141-5
34. Ahn JY. et al. PIKE-A is amplified in human cancers and prevents apoptosis by up-regulating Akt. Proc Natl Acad Sci U S A. 2004 ;101:6993-8
35. Nie Z. et al. AGAP1, an endosome-associated, phosphoinositide-dependent ADP-ribosylation factor GTPase-activating protein that affects actin cytoskeleton. J Biol Chem. 2002 ;277:48965-75
36. Scaife RM, Margolis RL. The role of the PH domain and SH3 binding domains in dynamin function. Cell Signal. 1997 ;9:395-401
37. Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994 ;4:389-99
38. Oliet SH. et al. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997 ;18:969-82
39. Shen L. et al. Regulation of AMPA receptor GluR1 subunit surface expression by a 4. 1N-linked actin cytoskeletal association. J Neurosci. 2000 ;20:7932-40
40. Parra M. et al. Cloning and characterization of 4.1G (EPB41L2), a new member of the skeletal protein 4.1 (EPB41) gene family. Genomics. 1998 ;49:298-306
41. Walensky LD. et al. Neurobehavioral deficits in mice lacking the erythrocyte membrane cytoskeletal protein 4.1. Curr Biol. 1998 ;8:1269-72
42. Hoover KB, Bryant PJ. The genetics of the protein 4.1 family: organizers of the membrane and cytoskeleton. Curr Opin Cell Biol. 2000 ;12:229-34
43. Walensky LD. et al. A novel neuron-enriched homolog of the erythrocyte membrane cytoskeletal protein 4.1. J Neurosci. 1999 ;19:6457-67
44. Ohara R. et al. Cellular and subcellular localization of a newly identified member of the protein 4.1 family, brain 4.1, in the cerebellum of adult and postnatally developing rats. Brain Res Dev Brain Res. 1999 ;117:127-38
45. Biederer T, Sudhof TC. CASK and protein 4.1 support F-actin nucleation on neurexins. J Biol Chem. 2001 ;276:47869-76
46. Scott C. et al. Protein 4.1 in forebrain postsynaptic density preparations: enrichment of 4.1 gene products and detection of 4.1R binding proteins. Eur J Biochem. 2001 ;268:1084-94
Figures
Author biography
Keqiang Ye obtained his Ph.D. from Emory University in 1998. He performed 3.5-year postdoc training with Dr. Solomon Snyder at The Johns Hopkins University. Currently, he is an assistant professor in Emory University. His research interest is focused on nuclear signaling cascade mediating the anti-apoptotic actions of NGF. Major achievements: 1) identification of nuclear GTPase PIKE, which upregulates nuclear PI 3-kinase (2000). 2) discovery of anti-cancer drug: noscapine (1998).
![]() Corresponding address:
Corresponding address:
Keqiang Ye, Department of Pathology and Laboratory Medicine, Room 145, Whitehead Building, Emory University School of Medicine, 615 Michael Street, Atlanta, GA 30322, USA. Phone: 404-712-2814; Fax: 404-712-2979. kyeedu