Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2006; 2(2):48-53. doi:10.7150/ijbs.2.48 This issue Cite
Research Paper
Getting closer to a pre-vertebrate genome: the non-LTR retrotransposons of Branchiostoma floridae
Jon Permanyer, Ricard Albalat, Roser Gonzàlez-Duarte ![]()
Departament de Genètica. Facultat de Biologia. Universitat de Barcelona. 08028 Barcelona, Spain.
Received 2006-2-6; Accepted 2006-3-10; Published 2006-4-10
Abstract
Non-LTR retrotransposons are common in vertebrate genomes and although present in invertebrates they appear at a much lower frequency. The cephalochordate amphioxus is the closest living relative to vertebrates and has been considered a good model for comparative analyses of genome expansions during vertebrate evolution. With the aim to assess the involvement of transposable elements in these events, we have analysed the non-LTR retrotransposons of Branchiostoma floridae. In silico searches have allowed to reconstruct non-LTR elements of six different clades (CR1, I, L1, L2, NeSL and RTE) and assess their structural features. According to the estimated copy number of these elements they account for less than 1% of the haploid genome, which reminds of the low abundance also encountered in the urochordate Ciona intestinalis. Amphioxus (B. floridae) and Ciona share a pre-vertebrate-like organization for the non-LTR retrotransposons (<150 copies, < 1% of the genome) versus the complexity associated to higher vertebrates (Homo sapiens >1.3·106 copies, > 20% of the genome).
Keywords: transposable elements, non-LTR retrotransposons, cephalochordates, genome evolution.
1. Introduction
Transposable elements (TEs) are almost invariably found in all species that have been studied. TEs are classified according to their degree of self-sufficiency and to their mechanism of transposition [1]. Regarding the first, TEs are divided in autonomous and nonautonomous elements. Based on the mode of transposition, two classes of TEs are defined: class I elements or retroelements (which utilize reverse transcription to amplify) and class II or DNA transposons (which transpose by the cut-and-paste or the rolling circle mode). This work has focussed on the autonomous class I elements non-LTR retrotransposons (also called LINE-like elements, polyA retrotransposons or retroposons) of the cephalochordate Branchiostoma floridae.
Non-LTR retrotransposons are one of the most abundant classes of transposable elements that make up a substantial fraction of the vertebrate genome. They comprise a variety of dispersed sequences that cluster in at least 14 clades and are divided in two groups, old-LINEs or site-specific endonuclease retrotransposons encoded in a single open reading frame (ORF), and young-LINEs or non-site-specific endonuclease retrotransposons that encode two ORFs (ORF1 and ORF2) [1, 2]. Both groups codify a preserved reverse transcriptase (RT), the only common domain, strictly required to achieve transposition and frequently used to analyse phylogenetic relationships. Additional structural motives are, a restriction enzyme-like endonuclease (REL-endo) or an apurinic/apyrimidinic endonuclease (APE), of those, at least one is strictly required and, optionally, several nucleic acid binding domains (NABD) and an RNAse H signature. Irrespective of the type of non-LTR retrotransposons, overall copy number is high enough not to leave them aside when dealing with genome evolution. Regarding TEs in general, their contribution to genome rearrangements has been deeply reported (reviewed in [1]).
Amphioxus (B. floridae) is a key organism to understand the invertebrate to vertebrate transition because it possesses a prototypical chordate body plan and is considered the closest living relative to vertebrates. The genome of this animal is small and relatively unduplicated, as shown by the single cluster of 14 Hox genes vs the four, or even more, clusters described in vertebrates [reviewed in 3]. Moreover, the recent availability of the genome draft of the amphioxus B. floridae has facilitated the analysis and comparison of non-LTR retrotransposons with those of the urochordate Ciona intestinalis and other vertebrate species.
2. Materials and methods
In silico search of non-LTR retrotransposons
The Branchiostoma floridae non-LTR elements were identified through a local TBLASTN [4] search of the first 4,772,554 B. floridae whole genome shotgun sequences (8xcoverage) generated at the JGI (
Copy number
The copy number for each non-LTR retrotransposon per haploid genome was determined as described [6], by multiplying the number of matching shotgun clones with an expected value of <10-200 by 5.8·108bp the size of the B. floridae haploid genome and divided by the length of the composite and the number of shotgun sequences in the local database (4,772,554).
Phylogenetic analysis
The RT deduced sequences of B. floridae were added to a previous alignment [7] and a new one was generated with Clustal X [8], maintaining the same pairwise gap penalties and multiple alignment parameters (Fig 1). Phylogenetic analyses were performed using the neighbor-joining method, rooted with the Neurospora organellar group II intron (accession number S07649) and drawn with the TreeViewPPC program [9]. Confidence in each node was assessed by 1,000 bootstrap replicates.
Alignment of the deduced protein sequence of the consensus contig of each clade. APE domains I, II, III, V, VI, VIII and IX, and the RT structural blocks 0-9 are indicated. Amino acid identities and similarities are shown in black and gray shading, respectively

3. Results
We have screened the non-LTR retrotransposons in the shotgun genome project of B. floridae in order to characterise the type and number of elements and draw a comparison with other known genomes.
Searches identified members of six out of fourteen previously reported clades. According to the phylogeny established and the Genbank comparisons, they will be termed BfCR1, BfI, BfL1, BfL2, BfNeSL and BfRTE. Comparisons of the composites of each element allowed to define two conserved domains: RT and APE. The RT domain, described in all amphioxus clades, contained all the distinctive structural hallmarks defined as block 0, 1, 2, 2a, 3, 4, 5, 6, 7, 8 and 9 [10]. Moreover, the apurinic/apyrimidinic endonuclease (APE) region was identified with reasonable confidence in BfCR1, BfL1, BfL2 and BfRTE (Fig. 1) on the basis of the reported domains I to IX [11] and only the last domain in BfI, which supports the bona fide structure of the defined composite and argues against a non-TE-based assembly. Notwithstanding our exhaustive search, the N-terminal APE domain and the RNaseH (RNH) sequence were not detected in BfI elements; neither REL-endo signatures could be clearly characterised in BfNeSL. For none of the elements identified, either NABD or ORF1 sequences could be detected. Copy number of each element per haploid genome was determined from the whole-length available sequence. In silico estimates showed low copy numbers: 25 for BfCR1, 3 for BfI, 32 for BfL1, 35 for BfL2, 6 for BfNeSL and 42 for BfRTE (Table 1). A rough estimation of the genome fraction harbouring non-LTR retrotransposons could be obtained considering that all the estimated copies (143) correspond to 5 kb full- length elements, and the value obtained would represent less than 1% of the haploid genome.
Non-LTR retrotransposons in protostomes and deuterostomes. The copy number for each clade, clade complexity (clades), total copy number (Copy num.), genomic burden (% Genome), and genome size (Gen. size) is shown.
| C. elegans [12] | D. melanogaster [13] | C. intestinalis [7] and [14] | B. floridae | T. rubripes [15] | R. norvegicus [16] | H. sapiens [17] | |
|---|---|---|---|---|---|---|---|
| CRE | 2,000 | ||||||
| I | 67 | 9 | 3 | ||||
| Jockey | 392 | ||||||
| L1 | 22 | 32 | 500 | 597,000 | 904,000 | ||
| L2 | 24 | 35 | 6,500 | 48,000 | 408,000 | ||
| L3/CR1 | 1,000 | 25 | 11,000 | 55,000 | |||
| LOA | 18 | 69 | |||||
| NeSL | 110 | 6 | 6 | 30 | |||
| R1 | 130 | ||||||
| R2 | 3-60 | 13 | |||||
| R4 | 1,000 | ||||||
| Rex1 | 2,000 | ||||||
| RTE | 15 | 42 | 2,300 | ||||
| Tad1 | |||||||
| Clades | 3 | 5 | 6 | 6 | 7 | 3 | 3 |
| Copy num. | 1,115 | 667 | 143 | 143 | 14,300 | 657,000 | 1,368,000 |
| % Genome | <5 | <3 | <1 | <1 | 1.3 | 23.1 | 21.05 |
| Gen. size | 9.7·107 | 1.6·108 | 1.8·108 | 5.8·108 | 4·108 | 2.9·109 | 3.2·109 |
Intra-sequence variability for each of the 6 clades was assessed from amino acid sequence comparison of the RT domains and expressed by the degree of similarity in percentage (Fig 2C).
Schematic representation of the amphiouxus non-LTR elements and the phylogenetic relationships. (A) Phylogenetic tree based on the reverse transcriptase sequence with only the branch points (and neighbor-joining bootstrap support) leading to the major 14 clades of non-LTR elements. (B) Schematic representation of the characterised domains. The APE domains and RT blocks are numbered below each element. (C) The main features (length, copy number, assembled composites, range of similarity) defining each non-LTR retrotransposon clade are indicated
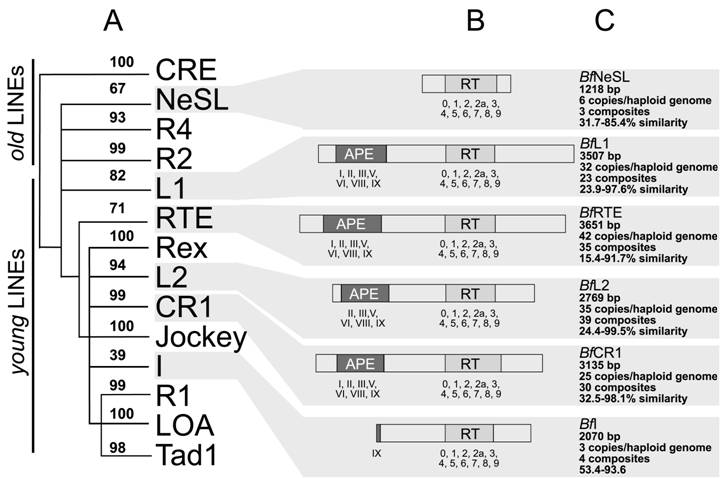
The matrices gave a range of 32.5-98.1% for BfCR1, 53.4-93.6% for BfI, 23.9-97.6% for BfL1, 22.4-99.5% for BfL2, 31.7-85.4% for BfNeSL and 15.4-91.7% for BfRTE.
Not only the reported missing domains of the elements, but the elusive target repeats at the recipient site that would help to identify the borders at 5' and 3' of the elements, together with the fact that, in all genomes, most non-LTR copies are truncated at 5', strongly suggests that only a very reduced proportion of the identified retrotransposons are full-length copies with preserved autonomy. And those few, if any, could have remained undetectable in the raw genome database as, indeed, we have not found any full-length element.
The RT domain of non-LTR elements was used to establish the phylogenetic relationships of the amphioxus elements and the 14 reported non-LTR clades. In the neighbor-joining tree (Figure 2A). Twelve out of 14 clades were supported with significant bootstrap values (≥70%). Clade I, showed the lowest bootstrap value (39%), in agreement with previous analyses [2, 10] whereas clade NeSL, gave bootstrap value close to the cut-off value (67%). Consequently, amphioxus composites were clustered in six different clades: CR1, I, L1, L2, NeSL and RTE (bootstraps: 99%, 39%, 82%, 94%, 67% and 71%, respectively) and were recorded as new members of each group.
4. Discussion
The approach used in this work has allowed the in silico identification and reconstruction of amphioxus non-LTR retrotransposons. The fact that the deduced features of BfCR1, one of the derived elements, are in agreement with previous experimental findings [18] validates the in silico strategy and supports the data generated for other elements. We therefore propose that the B. floridae genome accommodates the old LINE NeSL, and young LINEs such as CR1, I, L1, L2 and RTE, with an overall structure consistent with that reported for each clade. Concerning BfCR1, BfL1, BfL2 and BfRTE retrotransposons, although no structural hallmarks for ORF1 and NABD could be confidently detected, the RT and APE domains were clearly ascertained. On the other hand, although the phylogenetic affiliation of BfI and BfNeSL was poorly supported, their ascription to the I and NeSL clades was established following the BLAST hits with reported retrotransposons (2e-46 for the I element of Biomphalaria glabrata, and 2e-33 for NeSL of C. elegans). The difficulties in BfI and BfNeSL characterization are probably due to their low copy number, 3 and 6 respectively, significantly lower than that of the other elements and because these clades are still weakly defined [6].
The estimation of the copy number of each element suffered from small inaccuracies caused by the cut-off e-value assigned to discriminate the sequences belonging to the same clade and, the fact that a whole genome shotgun sequencing does only yield a fraction of the genome. Nevertheless, the in silico estimates for BfCR1 (25 copies) were in agreement with those obtained following an experimental approach (15) [17]. Our data showed low copy number per haploid genome for all the amphioxus elements, ranging from 3 to 42, a figure clearly similar to the number of the different composites assembled with the TIG clustering tools, thus showing the efficiency of the assembling procedure. Despite this overall scarcity, differences among clades were observed: BfCR1, BfL1, BfL2 and BfRTE were more frequent than BfI and BfNeSL elements. The permissiveness of the APE mediated insertion could account for the relative abundance of the former, whereas self regulatory mechanisms [19] or a high target site specificity [20] could explain the reduced number of the latter.
The mechanisms controlling copy number are still an open question but the values obtained in this work agree with those found in another lower chordate, Ciona intestinalis, and other organisms with small genomes such as Drosophila melanogaster and Anopheles gambiae [13, 21]. The overall copies of non-LTR retrotransposons in lower chordates represent, indeed, a very modest fraction of the genome, if compared to vertebrates (i.e. <1% in ascidians and amphiouxus versus the >20% in human). Then, low copy number in small genomes could easily be under self-control without having to invoke to host-promoted repression through methylation, as it has been shown in vertebrates and already discarded for BfCR1 and C. intestinalis non-LTR retrotransposons [7]. Other mechanisms, such as co-supression for the I elements of Drosophila [19] or RNA silencing in fungi, plants and animals [22-24] could play a major role in the regulation of the expansion of this type of elements.
In summary, the present work shows that the amphioxus genome harbors at least 6 different clades of non-LTR retrotransposons, all present at low copy numbers. Although from our data we cannot assume that the overall structure of the amphioxus genome resembles that of the chordate Ciona intestinalis, it seems clear that both share a comparable burden of non-LTR retrotransposons. The analysis of the non-LTR content of the B. floridae genome here reported provides valuable data to understand the evolution of chordate genomes, enlarges the view of the distribution of the non-LTR clades in eukaryotes and highlights the structural differences between pre-vertebrate and vertebrate genomes.
Acknoledgments
We thank JL Gelpí and M Pignatelli for help in the linux platform installation. This study was supported by the Ministerio de Educación y Ciencia (grant BMC 2003-05211). J.P. was the recipient of a fellowship from the Universitat de Barcelona.
Conflict of interests
The authors have declared that no conflict of interests exists.
References
1. Finnegan DJ. Eukaryotic transposable elements and genome evolution. Trends Genet. 1989 ;5:103-107
2. Lovsin N, Gubensek F & Kordi D. Evolutionary dynamics in a novel L2 clade of non-LTR retrotransposons in Deuterostomia. Mol Biol Evol. 2001 ;18:2213-2224
3. Holland LZ, Laudet V & Schubert M. The chordate amphioxus: an emerging model organism for developmental biology. Cell Mol Life Sci. 2004 ;61:2290-2308
4. Altschul SF, Madden TL. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acid Res. 1997 ;25(17):3389-3402
5. Swindell SR & Plasterer TN. SEQMAN. Contig assembly. Methods Mol Biol. 1997 ;70:75-89
6. Van Dellen K, Field J. et al. LINEs and SINE-like elements of the protist Entamoeba histolytica. Gene. 2002 ;297:229-239
7. Permanyer J, Gonzalez-Duarte R & Albalat R. The non-LTR retrotransposons in Ciona intestinalis: new insights into the evolution of chordate genomes. Genome Biol. 2003 ;4:R73
8. Thompson JD, Gibson TJ. et al. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997 ;25:4876-4882
9. Page RD. TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996 ;12:357-358
10. Malik HS, Burke WD & Eickbush TH. The age and evolution of non-LTR retrotransposable elements. Mol Biol Evol. 1999 ;16:793-805
11. Martin F, Olivares M. et al. Do non-long terminal repeat retrotransposons have nuclease activity? Trends Biochem Sci. 1996 ;21:283-285
12. Zagrobelny M, Jeffares DC & Arctander P. Differences in non-LTR retrotransposons within C. elegans and C. briggsae genomes. Gene. 2004 ;330:61-66
13. Berezikov E, Bucheton A & Busseau I. A search for reverse transcriptase-coding sequences reveals new non-LTR retrotransposons in the genome of Drosophila melanogaster. Genome Biol. 2000 ;1:RESEARCH0012
14. Kojima KK, Fujiwara H. Cross-genome screening of novel sequence-specific non-LTR retrotransposons: various multicopy RNA genes and microsatellites are selected as targets. Mol Biol Evol. 2004 ;21(2):207-217
15. Aparicio S, Chapman J. et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002 ;297:1301-1310
16. Gibbs RA, Weinstock G. et al. Genome sequence of the brown norway rat yields insights into mammalian evolution. Nature. 2004 ;428:493-521
17. Lander ES, Linton LM. et al. Initial sequencing and analysis of the human genome. Nature. 2001 ;409:860-921
18. Albalat R, Permanyer J. et al. The first non-LTR retrotransposon characterised in the cephalochordate amphioxus, BfCR1, shows similarities to CR1-like elements. Cell Mol Life Sci. 2003 ;60:803-809
19. Jensen S, Gassama MP & Heidmann T. Taming of transposable elements by homology-dependent gene silencing. Nat Genet. 1999 ;21:209-212
20. Zingler N, Weichenrieder O, & Schumann GG. APE-type non-LTR retrotransposons: determinants involved in target site recognition. Cytogenet Genome Res. 2005 ;110:250-268
21. Holt RA, Subramanian GM. et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science. 2002 ;298:129-149
22. Rossignol JL & Faugeron G. Gene inactivation triggered by recognition between DNA repeats. Experientia. 1994 ;50:307-317
23. Waterhouse PM, Wang MB & Lough T. Gene silencing as an adaptive defence against viruses. Nature. 2001 ;411:834-842
24. Bingham PM. Cosuppression comes to the animals. Cell. 1997 ;90:385-7
Author contact
![]() Corresponding address:
Corresponding address: