Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2006; 2(2):66-72. doi:10.7150/ijbs.2.66 This issue Cite
Research Paper
Evolutionary genomics of the recently duplicated amphioxus Hairy genes
Senda Jiménez-Delgado1, Miguel Crespo2, Jon Permanyer1, Jordi Garcia-Fernàndez1 #, Miguel Manzanares2 # ![]()
1 Departament de Genètica, Facultat de Biologia, Universitat de Barcelona, Avda. Diagonal 645, 08028 Barcelona, Spain
2 Instituto de Investigaciones Biomédicas CSIC-UAM, Arturo Duperier 4, 28029 Madrid, Spain
# Both authors contributed equally to the work
Received 2006-1-18; Accepted 2006-2-24; Published 2006-4-10
Abstract
Amphioxus Hairy genes have gone through a number of lineage-specific duplications, resulting in eight members, some of which are differentially expressed in the embryo. In order to gain insights into the evolution and function of this gene family we have compared their genomic structure and searched for conserved non-coding sequence elements. We have found that introns have been lost independently from these genes at least twice and after the duplication events. By carrying out phylogenetic footprinting between paralogues expressed in the embryo, we have found a differential distribution of conserved elements that could explain the limited overlap in expression patterns of Hairy genes in the amphioxus embryo. Furthermore, clustering of RBP-Jk binding sites in these conserved elements suggests that amphioxus Hairy genes are downstream targets of the Notch signaling pathway, as occurs in vertebrates. All of this evidence suggests that amphioxus Hairy genes have gone through a process of subfunctionalization shortly after their duplication, representing an extreme and rapid case of the duplication-degeneration-complementation model.
Keywords: amphioxus, DDC model, Hairy, phylogenetic footprinting, evolution
1. INTRODUCTION
Amphioxus is believed to be the closest living invertebrate relative to vertebrates [1]. However, this privileged position does not imply the freezing of its genome. It evolved directly from the last common ancestor of cephalochordates and vertebrates, but it is not the ancestor, extinct for several hundreds of million years. Hence, its genome has been independently evolving since then, as has been that of vertebrates.
In the vertebrate lineage the evolution from the last ancestor most probably involved two rounds of genome duplication [2]. This increase in the number of genes may have been instrumental, by means of neo-functionalization (adoption of new roles) of gene duplicates, for the acquisition of vertebrate novel features, such as migratory neural crest and derivates, skeletal system, cartilaginous tissue, and a complex brain [3].
Studying the prototypical amphioxus genome may help understand the ancestral function of a vertebrate gene family, with typically several members in vertebrates and a single representative in amphioxus [4]. Hence, the latest may be closer to the single gene present in the chordate ancestor. Such analyses in vertebrates are usually hampered by redundancy and neofunctionalization. However, as noticed earlier, the amphioxus genome has been also evolving since the separation from its last common ancestor with vertebrates. One therefore expects to find cephalochordate-specific duplicates for some gene families. Actually, this has been reported [5], and the extreme case is to be found in the Hairy family of helix-loop-helix transcription factors, with eight members in amphioxus (HairyA to H) and only one in mouse (Hes1) and two in chicken, Xenopus and zebrafish [6].
The eight amphioxus Hairy genes conserve their gene structure without excessive accumulation of mutations, expansions or losses, and none of the copies include stop codons in the coding sequence. This indicates that they have not degenerated to pseudogenes, and should be expressed at some point during amphioxus life cycle. For four of them (HairyA to HairyD) their expression during development is known [6]. HairyA to D expression are mostly not overlapping, or overlapping in a subtle manner during particular developmental stages. From the expression data, it has been proposed that amphioxus Hairy genes, after gene duplication from a single ancestor, underwent a process of divergence in the cis-regulatory regions that matches the duplication-degeneration-complementation process (the DDC model). The DDC model tries to explain why a high percentage of gene duplicates (much more than expected by statistics) are maintained (and expressed) in present genomes, by means of subfunctionalization [7]. Under this model, duplicated copies of a single gene suffer differential loss of cis-regulatory regions. Therefore, a complex or pleiotropic function that before duplication was performed by a single gene is subdivided in discrete components. These copies become all necessary and essential, as they keep individual and unique cis-regulatory regions. Although amphioxus HairyA to HairyD have specific expression and minor overlapping, the overall regions or tissues where they are expressed are the same. They are expressed in the neural tube, presomitic mesoderm, somites, endoderm or notochord. For this reason, one expects to find conserved elements in the regulatory sequences of these four genes. These elements could represent enhancers with spatial information for these big regions and then, each gene's expression should be refined via enhancers that we could not recognise because they contain specific spatial information (unique to a particular Amphi-Hairy gene).
One of the developmental processes where Hairy genes play a major role is the molecular clock responsible for the formation of somites. Various factors involved in this process are under the control of the Notch signalling pathway in zebrafish [8, 9], chicken [10] and mice [11-13]. The RBP-Jk binding sites are the primary transcriptional mediators of the Notch signaling. The intracellular domain of Notch (NICD), resulting after the cleavage of Notch upon ligand binding, is able to transactivate the transcription of target genes through binding to RBP- Jk binding sites [14]. One of these targets in mouse is Hes1, a Hairy gene which contains RBP- Jk sites in its 5' regulatory region.In order to gain insights into the process of amplification of the Hairy family in amphioxus, to identify cis-regulatory modules responsible for the maintenance of the duplicates and to unravel whether amphioxus Hairy genes may be under the control of the Notch pathway, we have cloned the full coding region of HairyA to F, plus 3 kb of 5' regulatory regions and performed in silico analysis of potential cis-regulatory control elements.
2. MATERIALS AND METHODS
Isolation and sequencing of genomic clones
Genomic clones containing amphioxus genes were isolated by high stringency screening at 65ºC in Church's buffer [15] of a lambda phage genomic library [16]. Positive clones were isolated, subcloned in plasmids, and relevant regions sequenced.
The probes used to screen the library were derived from previously described cDNA clones [6] and designed to contain the 3' coding region but neither the conserved bHLH domain nor the orange domain from each Hairy gene. We used 6 different probes for amphioxus HairyA to HairyF. Genomic clones for HairyG and HairyH were previously described [6]. The last exon/intron boundary of HairyH was determined by searching the Trace Archives of B. floridae (clone gnl/ti/538762490)
Sequence analysis and comparative genomics
Genomic sequences from amphioxus HairyA, B, C and D were analyzed using both local and multiple alignment algorithms through the Vista (genome.lbl.gov/vista [17]), Pipmaker (bio.cse.psu.edu/pipmaker [18]) and Mulan (mulan.dcode.org [19]) servers. Conserved elements identified this way were further analyzed and corrected alignments generated using Clustal X [20]. Potential binding site for transcription factors, particularly for RBP-Jk, were analyzed using Matinspector (
3. RESULTS AND DISCUSSION
Genomic organization and intron loss in amphioxus Hairy genes
In order to understand the genomic structure and analyze the existence of putative regulatory elements in amphioxus Hairy genes, we screened a genomic library [16] under high stringency conditions using probes specific for six genes from this family from which no genomic clones were previously available [6]. We were able to obtain genomic fragments ranging from 15 to 20 kb containing the full coding region of AmphiHairy A to G. At least 8 kb of genomic sequence was obtained for AmphiHairyA, B, C and D, covering 3 kb of 5' upstream sequence, the full coding region including (accession numbers DQ402480 to DQ402483)
Comparison of the genomic sequence to the predicted peptides from all eight amphioxus Hairy genes allowed us to determine their exon/intron structure and compare it to what is known for members of the family in other chordates. We found that the coding regions of HairyA and HairyE are interrupted by three introns, the two first (introns I and II, Fig. 1) localize within the bHLH domain, and the third (intron III, Fig. 1) in the intervening region between the bHLH and the Orange domain (see Fig. 1A in [6]). This exon-intron organization is remarkably conserved during evolution, and for example the mouse Hes1 [22] and the chick Hairy1 (not shown) genes share the exact location of all three introns. However, and to our surprise, we found that the HairyC, D, F, G and H genes were lacking intron III, and that HairyB was lacking introns I and III (Fig. 1). We also examined the intron-exon organization of the three Hairy genes described in the urochordate Ciona intestinalis [23] and found that while hairya and hairyb share all three intron positions with other vertebrates, hairyc also lacks intron III.
Comparison of the genomic organization of amphioxus (Bf) Hairy and mouse (Mm) Hes1 genes. Dashed lines indicated identical positions in the coding sequence. The diagram is not drawn to scale.
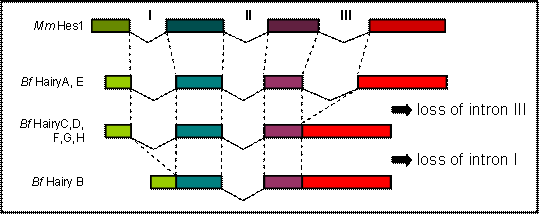
The fact that two amphioxus and Ciona Hairy genes share the exon-intron organization with vertebrates makes this the most likely gene structure in the common ancestor of chordates, as well as of the ancestral gene that gave rise to all amphioxus Hairy genes via tandem gene duplication [6]. The most parsimonious explanation for the observed distribution of intron loss in the remaining genes is that loss of intron III occurred only once, and that this copy was further duplicated in tandem. It is possible that loss of intron III occurred before the divergence of cephalochordates and urochordates, but this is not supported by phylogenetic analysis, as Ciona hairyc does not group with any of the amphioxus HairyC, D, F, G nor H genes, but with the other two Ciona Hairy genes (not shown). Finally, the loss of intron I in HairyB would surely have occurred at a later stage, once all duplicated copies had been fixed in the genome.
Detailed comparative studies have shown that the loss of introns during evolution is much more frequent [24, 25], than generally expected [26]. Two main possible mechanisms have been envisioned that could explain the precise excision of intron sequences, such as we find in amphioxus Hairy genes. On one hand, an RNA mediated mechanisms, by which a spliced mRNA recombines with the endogenous locus, has been described [27, 28]. An alternative mechanism argues that precise excision is mediated by non-homologous recombination between short repeats located at the 5' and 3' end of introns [29, 30]. In any case, our observation of at least two independent events of intron loss in a duplicated gene family in the amphioxus lineage shows that introns are being lost at a high rate even during recent duplication events.
Evolutionary conservation of genomic sequences upstream of amphioxus Hairy genes
Previous analysis of the expression pattern of amphioxus Hairy genes showed that the four genes that are expressed during embryogenesis (HairyA, B, C and D) had undergone a process of subfunctionalization, whereby the sum of all their expression domains in the embryo resembles that of Hairy genes from vertebrates [6]. It has been argued that these processes occur via differential loss of cis-regulatory elements between duplicated genes [7, 31]. In order to examine if this held true for the amphioxus Hairy family, we analyzed the genomic region surrounding these four genes to search for conserved non-coding sequences. This approach, named phylogenetic footprinting [32], has been mainly used to compare ortholog genes and genomic regions between different species [33] as an approach for the identification of cis-regulatory elements [34, 35]. However, it has been rarely used to compare paralogous genes from the same species as we carry out in this study.
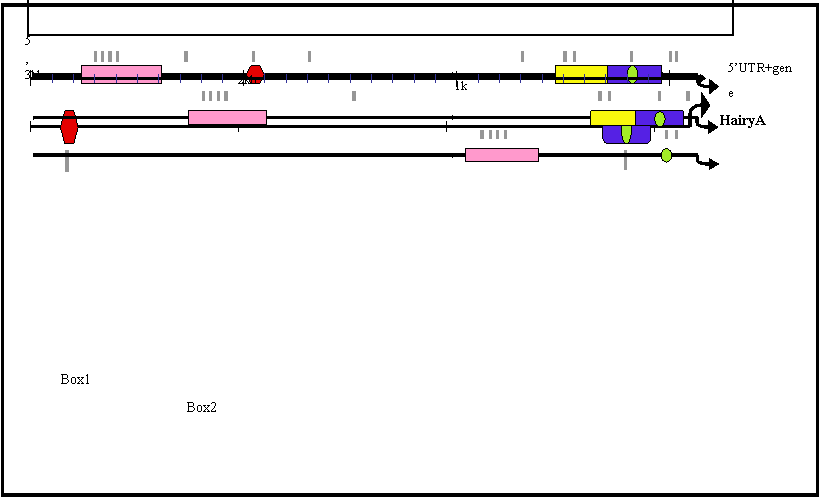
Genomic sequence ranging from 3 kb upstream to 3 kb downstream was obtained for amphioxus HairyA, B, C and D. Other Hairy genes were excluded from the analysis, as they are not expressed in the embryo and therefore not relevant for our analysis. Multiple local and global alignments between these sequences were performed using different comparative genomic tools (see Materials and methods). Using a detection threshold of a minimum of 50 base pairs (bp) and 65% similarity, we did not detect any conserved blocks in introns nor in 3' flanking genomic sequences. However, under these conditions we were able to identify four conserved blocks in the 5' flanking genomic regions that were differentially distributed among the genes under study (boxes 1 to 4; Fig. 2, 3). Extensive Blast [35] searches using these conserved blocks failed to find significant matches in any genomic or cDNA database. Furthermore, we were unable to find any similarity in the available genomic sequences from amphioxus, nor in the urochordate Ciona intestinalis [37], the closest relative to amphioxus whose genome has been sequenced. These negative results allowed us to rule out that these conserved sequences could be repetitive DNA or part of some poorly defined transcription unit. We also performed a more detailed and thorough comparison of these sequences with genomic regions surrounding mouse, chick and Ciona Hairy genes, not finding any significant similarity even at very low sensitivity (data not shown). Therefore, we concluded that these sequence elements are specific for amphioxus Hairy genes, and must have diverged after the duplications events that took place in this lineage.
Schematic representation of the 5' region of amphioxus HairyA, B, C and D genes showing conserved non-coding regions. These are named Box1 to 4 and shown in different colours. The small green oval represents a core region conserved in all four genes and located inside the conserved Box3 (yellow) present in all but the HairyB gene. Putative RBP-Jκ binding sites are shown as grey lines below each gene and are clustered within the conserved boxes.
Sequence alignments of the conserved boxes 1 to 4. Identities are indicated by dots and gaps by dashes. The boxed region inside Box 4 shows the core region conserved in all four genes. Consensus binding sites for RBP-Jk [39] are highlighted in blue.
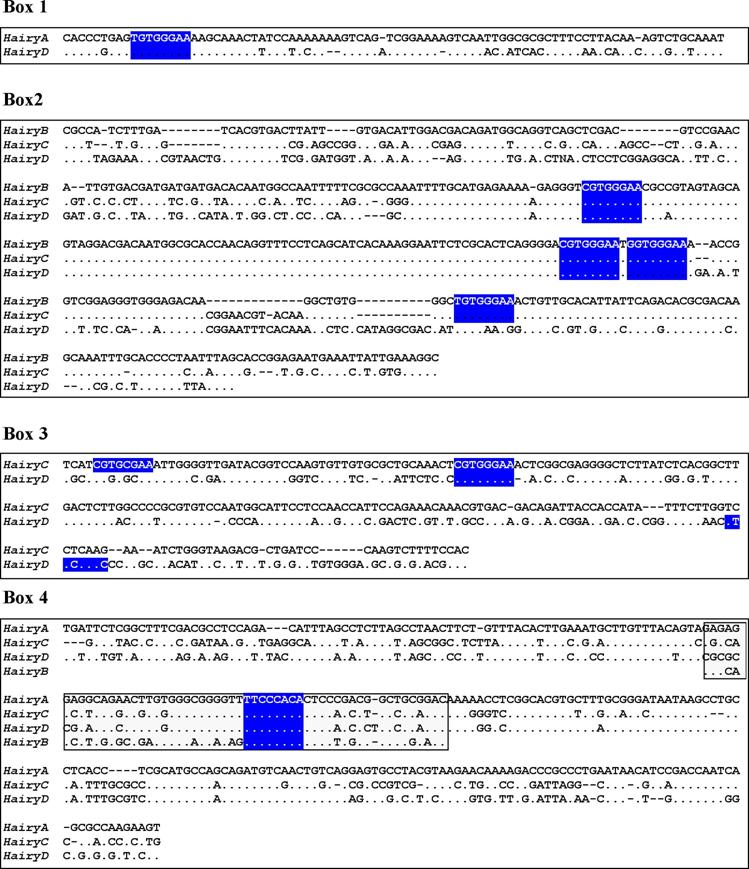
Box1 (red in Fig. 2) is present in HairyA and HairyD, with a length of 87 and 86 bp, respectively, and 76% similarity. Box2 (pink in Fig. 2) is shared by HairyB (361 bp), HairyC (382 bp) and HairyD (375 bp) with pairwise similarities ranging from 76% to 84%. Box3 (yellow in Fig. 2) is found in HairyC (220 bp) and HairyD (234 bp) with 61% similarity. Finally, Box4 (blue in Fig. 2) is present in HairyA (273 bp), HairyC (271 bp) and HairyD (279 bp), with pairwise similarities between 56% and 76%. Moreover, the central part or core of this box, encompassing 55 bp, can also be found in the 5' region of HairyB (green oval in Fig. 2, and boxed region in Fig. 3) making it the only conserved non-coding sequence element shared by all four genes under study. It should also be noted that Box4 and the core conserved region is located immediately 5' upstream of the transcription start site, and that in HairyC and HairyD, boxes 3 and 4 are adjacent (Fig. 2), making them share a stretch of nearly 500 bp of contiguous sequence.
We next searched in these elements for known transcription factors binding sites (see Materials and Methods), with the aim of identifying possible upstream regulators of amphioxus Hairy genes. While being aware of the very high rate of false positives inherent to this type of analysis owing to the promiscuous binding of transcription factors [38], we were particularly interested in consensus sites that were evolutionarily conserved between different Hairy genes. Interestingly, we identified a number of putative binding sites for RBP-Jk in all four conserved boxes, which is known to be the major transcriptional effector of Notch, and it has been shown that Hairy genes from different organisms are downstream targets of this signaling pathway [9, 12]. To examine whether the distribution of RBP-Jk binding sites was random or specific for the conserved elements, we repeated our search for these sites but over the full 3 kb of upstream genomic sequence for all four genes (grey lines in Fig. 2; blue boxes in Fig. 3). We found that the majority (22 out of 29) of sites were located within the conserved boxes, with a maximum of four sites present in Box 2 (Fig. 2). This strongly suggests that the presence of evolutionarily conserved RBP-Jk binding sites could have a functional role in the regulation of amphioxus Hairy genes.
Rapid evolution of amphioxus Hairy genes: an extreme case of DDC?
Hairy genes are highly pleiotropic and have been implicated in numerous functions, such as somitogenesis, neurogenesis, endocrine and T lymphocyte development [40]. Based on the embryonic expression patterns, we can hypothesize that amphioxus Hairy genes will carry out similar roles, as they are predominantly expressed in the neural tube, notochord, endocrine system, and mesoderm [6]. While these functions are performed by one (mouse) or two (chick, Xenopus, zebrafish) genes in vertebrates, in amphioxus there are eight Hairy genes product of a specific duplication in this lineage, of which at least four are expressed in specific domains in the early embryo [6]. These domains are only partially overlapping and not all four genes are expressed in all domains. Therefore, it is tempting to speculate that following duplication there has been a process of subfunctionalization [7] by which regulatory elements responsible for specific expression domains have been differentially retained in the duplicated genes.
Our analysis of conserved non-coding elements fits well with this scenario, as we have been able to identify a number of these that are differentially distributed among HairyA to D genes. Matching expression domains with conserved elements allows us to suggest that these boxes are cis-regulatory elements driving gene expression in specific domains (see Fig. 4 and legend). Furthermore, we have found that these boxes have a statistically over-representation of putative binding sites for the RBP-Jk transcription factor, that is known to directly regulate Hairy genes in vertebrates [9, 12]. Although the ortholog of RBP-Jk has not been identified yet in amphioxus, it is extremely likely that it is present and will carry out similar roles to those described in vertebrates. We will have to wait until robust transgenic methods are developed for amphioxus embryos [41] before we have a definitive answer about the regulatory capacities of these elements in vivo.
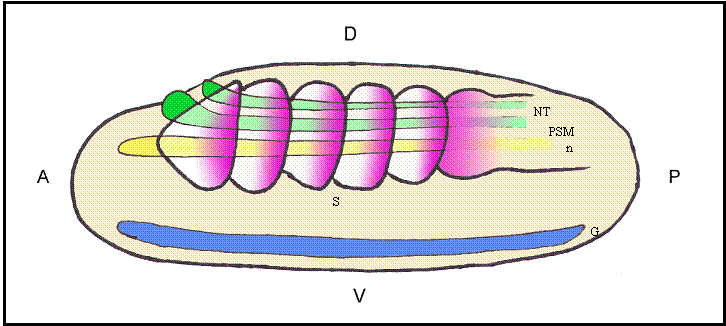
Schematic representation of a lateral view of an amphioxus neurula showing a possible correlation between evolutionarily conserved sequence elements and shared sites of expression for different Hairy genes. The color code matches that of different boxes in Fig.2. Box 2 would correlate with the expression of HairyB, HairyC and HairyD in somites (S) and presomitic mesoderm (PSM; pink); Box 3 with the expression of HairyC and HairyD in the notocord (n; yellow); Box 4 with the expression of HairyA, HairyC and HairyD in the gut endoderm (G; blue); and finally, the core of Box 4 could be related with the expression of all four genes in the neural tube (NT; green). At present, we have not detected any obvious shared expression domain exclusive to HairyA and HairyD that could correlate with conserved Box 1. Positions of the anterior (A)-posterior (P) and dorsal (D)-ventral (V) axes are indicated
This and previous studies [6] highlight the rapid divergence that has occurred after the lineage-specific duplication of amphioxus Hairy genes. We can observe differential gene expression during development, intron loss and distribution of putative cis-regulatory elements in a single gene family. These observations highlight the risk of considering the amphioxus genome as archetypal and representative of the last common ancestor of chordates. The amphioxus genome has continued changing since the divergence from the common ancestor with vertebrates, and specific gene expansions have occurred at multiple times. A thorough understanding of the genome organization and regulation in amphioxus will certainly help understand vertebrate biology, but taking into account that we will be comparing two of the endpoints of the evolutionary bush, and not the branching point and the tip of the branch from a tree.
Acknowledgements
We wish to thank Bárbara Pernaute and Eva Alonso for help with phylogenetic footprinting, Carlos Quijano for help with phylogenetic analysis, Juan Pascual-Anaya for his assistance in images design, and members of the Garcia-Fernàndez and Manzanares labs for helpful discussions. This work was supported by the EMBO Young Investigator Programme and grant BMC2002-03558 (Spanish Ministry of Science and Technology) to M. M., and grants BMC2002-03316 and BFU2005-00252 (Spanish Ministry of Science and Technology) and Distinció de la Generalitat de Catalunya to J. G-F. S. J-D. and M. C. hold FPI predoctoral fellowships.
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exist
References
1. Wada H, Satoh N. Details of the evolutionary history from invertebrates to vertebrates, as deduced from the sequences of 18S rDNA. Proc Natl Acad Sci U S A. 1994 ;91:1801-1804
2. Holland PW, Garcia-Fernandez J, Williams NA, Sidow A. Gene duplications and the origins of vertebrate development. Dev Suppl. 1994 :125-133
3. Shimeld SM, Holland PW. Vertebrate innovations. Proc Natl Acad Sci U S A. 2000 ;97:4449-4452
4. Holland PW. More genes in vertebrates? J Struct Funct Genomics. 2003 ;3:75-84
5. Minguillon C, Ferrier DE, Cebrian C, Garcia-Fernandez J. Gene duplications in the prototypical cephalochordate amphioxus. Gene. 2002 ;287:121-128
6. Minguillon C, Jimenez-Delgado S, Panopoulou G, Garcia-Fernandez J. The amphioxus Hairy family: differential fate after duplication. Development. 2003 ;130:5903-5914
7. Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999 ;151:1531-1545
8. Jiang YJ, Aerne BL, Smithers L, Haddon C, Ish-Horowicz D, Lewis J. Notch signalling and the synchronization of the somite segmentation clock. Nature. 2000 ;408:475-479
9. Oates AC, Ho RK. Hairy/E(spl)-related (Her) genes are central components of the segmentation oscillator and display redundancy with the Delta/Notch signaling pathway in the formation of anterior segmental boundaries in the zebrafish. Development. 2002 ;129:2929-2946
10. Dale JK, Maroto M, Dequeant ML, Malapert P, McGrew M, Pourquie O. Periodic notch inhibition by lunatic fringe underlies the chick segmentation clock. Nature. 2003 ;421:275-278
11. Morales AV, Yasuda Y, Ish-Horowicz D. Periodic Lunatic fringe expression is controlled during segmentation by a cyclic transcriptional enhancer responsive to notch signaling. Dev Cell. 2002 ;3:63-74
12. Jouve C, Palmeirim I, Henrique D, Beckers J, Gossler A, Ish-Horowicz D, Pourquie O. Notch signalling is required for cyclic expression of the hairy-like gene HES1 in the presomitic mesoderm. Development. 2000 ;127:1421-1429
13. Cole SE, Levorse JM, Tilghman SM, Vogt TF. Clock regulatory elements control cyclic expression of Lunatic fringe during somitogenesis. Dev Cell. 2002 ;3:75-84
14. Tamura K, Taniguchi Y, Minoguchi S, Sakai T, Tun T, Furukawa T, Honjo T. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H). Curr Biol. 1995 ;5:1416-1423
15. Shifman MI, Stein DG. A reliable and sensitive method for non-radioactive northern blot analysis of nerve growth factor mRNA from brain tissues. J Neurosci Methods. 1995 ;59:205-208
16. Ferrier DE, Minguillon C, Holland PW, Garcia-Fernandez J. The amphioxus Hox cluster: deuterostome posterior flexibility and Hox14. Evol Dev. 2000 ;2:284-293
17. Shah N, Couronne O, Pennacchio LA, Brudno M, Batzoglou S, Bethel EW, Rubin EM, Hamann B, Dubchak I. Phylo-VISTA: interactive visualization of multiple DNA sequence alignments. Bioinformatics. 2004 ;20:636-643
18. Schwartz S, Zhang Z, Frazer KA, Smit A, Riemer C, Bouck J, Gibbs R, Hardison R, Miller W. PipMaker--a web server for aligning two genomic DNA sequences. Genome Res. 2000 ;10:577-586
19. Ovcharenko I, Loots GG, Giardine BM, Hou M, Ma J, Hardison RC, Stubbs L, Miller W. Mulan: multiple-sequence local alignment and visualization for studying function and evolution. Genome Res. 2005 ;15:184-194
20. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997 ;25:4876-4882
21. Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005 ;21:2933-2942
22. Takebayashi K, Sasai Y, Sakai Y, Watanabe T, Nakanishi S, Kageyama R. Structure, chromosomal locus, and promoter analysis of the gene encoding the mouse helix-loop-helix factor HES-1. Negative autoregulation through the multiple N box elements. J Biol Chem. 1994 ;269:5150-5156
23. Satou Y, Imai KS, Levine M, Kohara Y, Rokhsar D, Satoh N. A genomewide survey of developmentally relevant genes in Ciona intestinalis. I. Genes for bHLH transcription factors. Dev Genes Evol. 2003 ;213:213-21
24. Krzywinski J, Besansky NJ. Frequent intron loss in the white gene: a cautionary tale for phylogeneticists. Mol Biol Evol. 2002 ;19:362-366
25. Cho S, Jin SW, Cohen A, Ellis RE. A phylogeny of caenorhabditis reveals frequent loss of introns during nematode evolution. Genome Res. 2004 ;14:1207-1220
26. Venkatesh B, Ning Y, Brenner S. Late changes in spliceosomal introns define clades in vertebrate evolution. Proc Natl Acad Sci U S A. 1999 ;96:10267-10271
27. Niu DK, Hou WR, Li SW. mRNA-mediated intron losses: evidence from extraordinarily large exons. Mol Biol Evol. 2005 ;22:1475-1481
28. Lewin R. How mammalian RNA returns to its genome. Science. 1983 ;219:1052-1054
29. Robertson HM. The large srh family of chemoreceptor genes in Caenorhabditis nematodes reveals processes of genome evolution involving large duplications and deletions and intron gains and losses. Genome Res. 2000 ;10:192-203
30. Robertson HM. Two large families of chemoreceptor genes in the nematodes Caenorhabditis elegans and Caenorhabditis briggsae reveal extensive gene duplication, diversification, movement, and intron loss. Genome Res. 1998 ;8:449-463
31. Prince VE, Pickett FB. Splitting pairs: the diverging fates of duplicated genes. Nat Rev Genet. 2002 ;3:827-837
32. Zhang Z, Gerstein M. Of mice and men: phylogenetic footprinting aids the discovery of regulatory elements. J Biol. 2003 ;2:11
33. Boffelli D, Nobrega MA, Rubin EM. Comparative genomics at the vertebrate extremes. Nat Rev Genet. 2004 ;5:456-465
34. de la Calle-Mustienes E, Feijoo CG, Manzanares M, Tena JJ, Rodriguez-Seguel E, Letizia A, Allende ML, Gomez-Skarmeta JL. A functional survey of the enhancer activity of conserved non-coding sequences from vertebrate Iroquois cluster gene deserts. Genome Res. 2005 ;15:1061-1072
35. Woolfe A, Goodson M, Goode DK, Snell P, McEwen GK, Vavouri T, Smith SF, North P, Callaway H, Kelly K, Walter K, Abnizova I, Gilks W, Edwards YJ, Cooke JE, Elgar G. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005 ;3:e7
36. McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004 ;32:W20-25
37. Dehal P. et al. The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science. 2002 ;298:2157-2167
38. Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet. 2004 ;5:276-287
39. Tun T, Hamaguchi Y, Matsunami N, Furukawa T, Honjo T, Kawaichi M. Recognition sequence of a highly conserved DNA binding protein RBP-J kappa. Nucleic Acids Res. 1994 ;22:965-971
40. Davis RL, Turner DL. Vertebrate hairy and Enhancer of split related proteins: transcriptional repressors regulating cellular differentiation and embryonic patterning. Oncogene. 2001 ;20:8342-8357
41. Yu JK, Holland ND, Holland LZ. Tissue-specific expression of FoxD reporter constructs in amphioxus embryos. Dev Biol. 2004 ;274:452-461
Author contact
![]() Corresponding address:
Corresponding address: