Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2006; 2(4):161-170. doi:10.7150/ijbs.2.161 This issue Cite
Short Research Communication
Murine Wee1 Plays a Critical Role in Cell Cycle Regulation and Pre-Implantation Stages of Embryonic Development
Yohei Tominaga, Cuiling Li, Rui-Hong Wang, Chu-Xia Deng ![]()
National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, 10/9N105, NIH, Bethesda, MD 20892, USA
Received 2006-3-29; Accepted 2006-5-15; Published 2006-5-18
Abstract
Wee1 kinase regulates the G2/M cell cycle checkpoint by phosphorylating and inactivating the mitotic cyclin-dependent kinase 1 (Cdk1). Loss of Wee1 in many systems, including yeast and drosophila, leads to premature mitotic entry. However, the developmental role of Wee1 in mammals remains unclear. In this study, we established Wee1 knockout mice by gene targeting. We found that Wee-/- embryos were defective in the G2/M cell cycle checkpoint induced by γ-irradiation and died of apoptosis before embryonic (E) day 3.5. To study the function of Wee1 further, we have developed MEF cells in which Wee1 is disrupted by a tamoxifen inducible Cre-LoxP approach. We found that acute deletion of Wee1 resulted in profound growth defects and cell death. Wee1 deficient cells displayed chromosome aneuploidy and DNA damage as revealed by γ-H2AX foci formation and Chk2 activation. Further studies revealed a conserved mechanism of Wee1 in regulating mitotic entry and the G2/M checkpoint compared with other lower organisms. These data provide in vivo evidence that mammalian Wee1 plays a critical role in maintaining genome integrity and is essential for embryonic survival at the pre-implantation stage of mouse development.
Keywords: Cdk1, Chk2, G2/M, apoptosis, genetic instability
1. Results and Discussion
Cells treated with DNA damaging agents, such as γ-radiation, coordinately arrest their cell cycle progression at varying phases to allow time for repairing the DNA damage. This is achieved by the activation of cell cycle checkpoints, which monitor DNA status and ensure the completion of the previous phase in the cell cycle before advancing to the next phase [4-7]. It was shown that Wee1 and Cdc25 cooperatively regulate G2/M boundary by inhibiting Cdk1 activity [1, 8, 9]. Wee1 and Cdk1 also regulate each other and play a critical role in controlling the transition into mitosis [10]. However, the function of Wee1 in mammalian development is unknown. To investigate this, we have established a Wee1-deficient mouse strain by gene targeting and studied its role in early mouse embryos and cultured mouse embryonic fibroblast (MEF) cells.
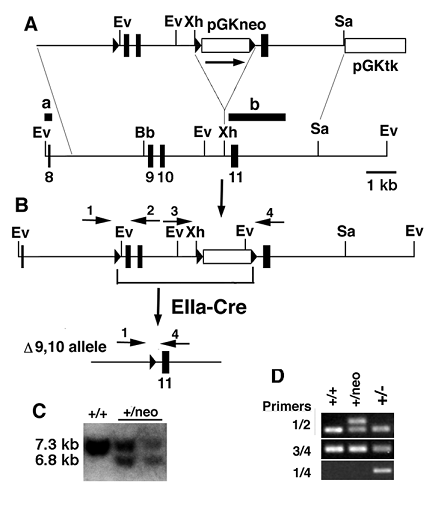
Wee1 was mutated by a replacement type gene-targeting vector, ploxPneo-Wee1, which introduced a ploxPneo [11] into intron 10 and the third loxP into intron 8 of the Wee1 locus, respectively (Fig. 1A). The correct targeting of the vector followed by Cre-loxP mediated recombination would delete exons 9 and 10, which encode the kinase domain of Wee1. Three out of 114 G418r/FIAUr ES clones had both the ploxPneo and the loxP introduced into the Wee1 locus based on Southern blot analysis (Fig. 1C). After germline transmission, we crossed heterozygous mutant (Wee1+/neo) mice with EIIa-Cre transgenic mice to delete exons 9 and 10 from mouse germline (Fig. 1B). Our PCR analysis on genomic DNA extracted from mice with various genotypes confirmed the predicted targeting and deletion in the Wee1 locus (Fig. 1D).
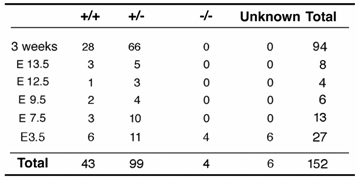
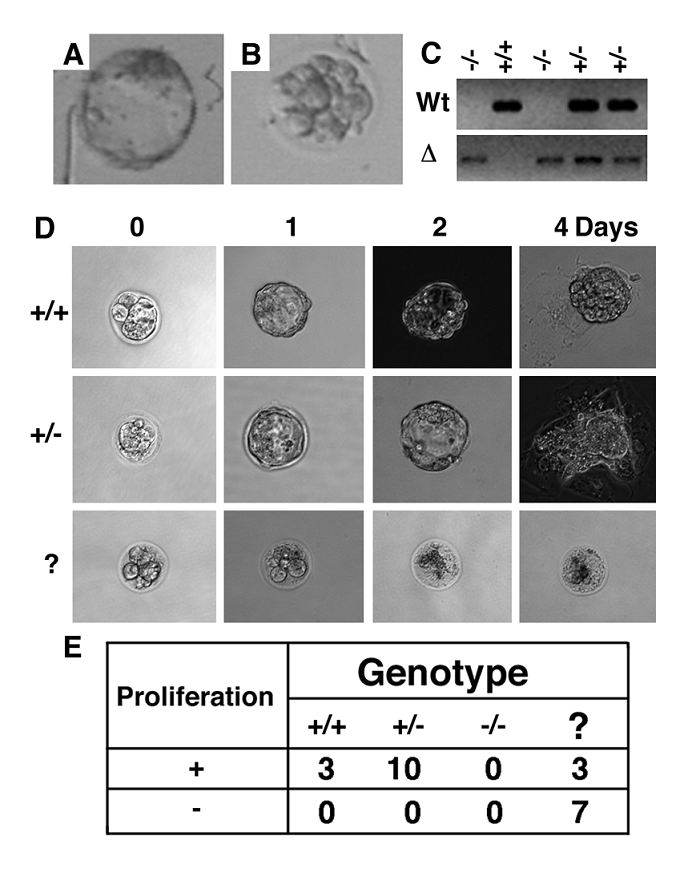
Adult Wee1+/- mice showed no obvious abnormality and were fertile. After interbreeding the Wee1+/- mice, we failed to obtain Wee1-/- mice from 94 mice genotyped at weaning while the wild type and Wee1+/- mice were presented at a ratio of 1:2.3 (Table 1). As no dead Wee1-/- mice were found after birth, we suspected that Wee1-/- mice died at embryonic development stages. Our analysis of embryos at embryonic (E) 13.5 to E7.5 also did not detect any homozygotes and abnormal deciduas (Table 1), suggesting that the Wee1-/- embryos died at stages prior to implantation. To verify this, we isolated E3.5 embryos derived from Wee1+/- interbreeding. We found 4 Wee1-/- embryos out of 27 embryos analyzed although genotypes of 6 embryos could not be reliably determined due to a technical difficulty in manipulating small embryos. The majority of the Wee1+/+ and Wee1+/- embryos were at the blastocyst stage, and possessed a distinct inner cell mass (ICM) and an outer layer of trophectoderm cells. The Wee1-/- embryos were comprised of fewer cells than their Wee1+/+ and Wee1+/- littermates and remained at the morulae stage (Fig. 2A-C). The mutant cells were disorganized without an obvious compaction. Those observations suggest that the development of Wee1-/- embryos was interrupted before reaching the blastocyst stage.
Because Wee1-/- embryos were retarded with abnormal morphology at the blastocyst stage, we investigated whether these embryos would grow if extra time was given. To monitor this process, we inoculated blastocytsts isolated from Wee1+/- female mice, which were mated with Wee1+/- males, into 24-well culture dishes. Our analysis of 23 blastocysts indicated that all embryos showing obvious cell proliferation were either Wee1+/+ or Wee1+/- (Fig. 2D,E). The remaining 7 embryos, which were abnormal initially, did not show any signs of growth and all cells degraded during culture (Fig. 2E). Genotype of these embryos could not be determined as we failed to extract DNA from them. We suspected that most of these embryos would be homozygous for Wee1 mutation based on our earlier observations that Wee1-/- embryos were presented at E3.5 with abnormal morphology (Fig. 2B,C). Thus, Wee1-/- embryos failed to grow even when more time is given.
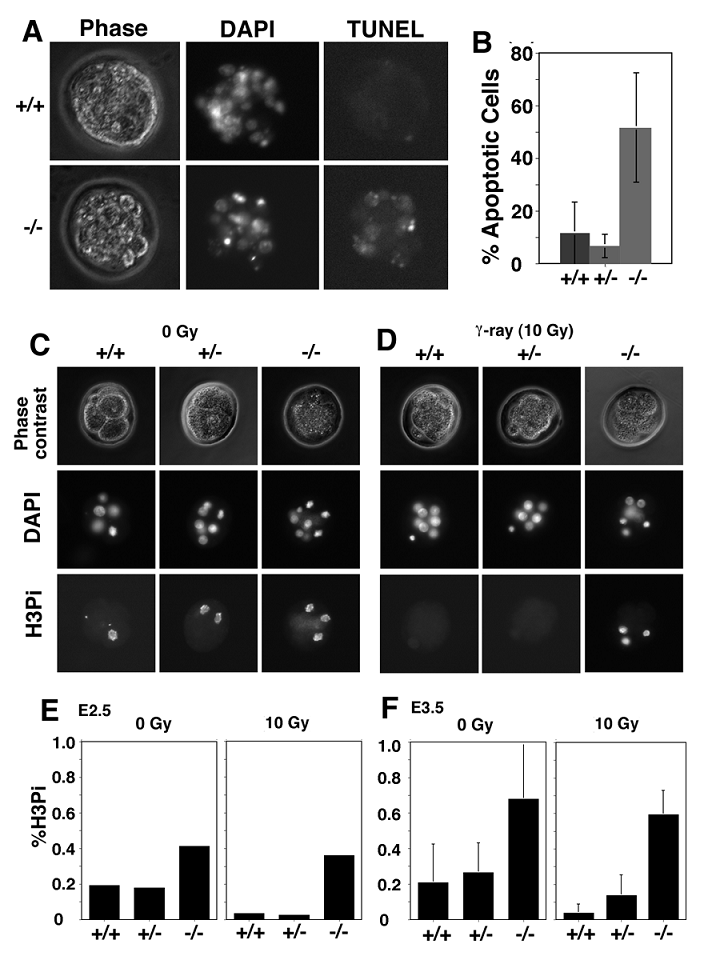
Next, we determined potential causes for the death of Wee1-/- embryos by TUNEL assay. Our analysis revealed that about 50% of cells in Wee1-/- embryos underwent apoptosis while about 5-10% cells in wild type and Wee1+/- embryos were TUNEL positive (Fig. 3A). There was no statistic difference between wild type and Wee1+/- embryos, however the differences between wild type and Wee1-/- embryos (p<0.001), and between Wee1+/- and Wee1-/- embryos (p<0.001) were significant (Fig. 3B). This observation suggests that Wee1-/- embryos die at 16-32 cell stages due to apoptosis.
Next, we studied possible causes for the increased apoptosis observed in Wee1-/- embryos. In vitro studies revealed that Wee1 plays an essential role in the G2/M cell cycle checkpoint [1, 8, 9]. Therefore, we investigated whether Wee1 had a similar role in vivo. We isolated embryos at E2.5 and E3.5 from Wee1+/- females after they were mated with Wee1+/- male mice. The mitotic index was determined in these embryos prior to, and after they were irradiated with γ-radiation (10 Gy) using an antibody against phosphorylated histone H3. A significant decrease in the number of mitotic cells was observed in both E2.5 Wee1+/+ and Wee1+/- embryos after irradiation while no obvious change was seen in Wee1-/- embryos (Fig. 3C-E). This observation suggests that Wee1-/- embryos were defective in activating the G2/M cell cycle checkpoint induced by DNA damage. The mitotic index of E3.5 embryos declined from 20% to 4% (p=0.048), from 27% to 13% (p=0.056) in Wee1+/- embryos, and from 65% to 60% (p=0.594) in Wee1-/- embryos (Fig. 3C). Thus, compared with Wee1+/+ embryos, absence of one wild type copy of Wee1 in E3.5 embryos might result in a partial loss of the G2/M checkpoint induced by γ-irradiation. Of note, Wee1-/- embryos had much more mitotic cells than Wee1+/+ and Wee1+/- embryos at both time points prior to irradiation. Thus, although the Wee1-/- embryos appeared morphologically normal at E2.5, some fundamental alterations at cellular level had already started, which might account for the developmental abnormality shown in the blastocyst stage.
To study cellular defects associated with Wee1 deficiency, we established MEF cells allowing for the acute mutation of Wee1 using a 4-hydroxytamaxifen (4-HT) inducible Cre-LoxP system. These cells (Wee1Co/-;Tm-Cre) carried a conditional Wee1 allele with its exons 9 and 10 flanked by loxP sites (Fig. 4A), a mutant allele missing exons 9 and 10, and a tamoxifen inducible Cre gene (Tm-Cre) [12]. Treatment with 4-HT in these cells causes nuclear translocalization of Cre, leading to the deletion of the conditional allele (Fig. 4B). Our MTT assay revealed significantly reduced cell quantity after the Wee1Co/-;Tm-Cre cells were exposed to 4-HT compared with untreated or vehicle (ethanol) treated cells (Fig. 5A). In contrast, only slight reduction in cell quantity was observed in control cells (Wee1+/Co;Tm-Cre, Wee1+/+;Tm-Cre, and Wee1+/-;Tm-Cre) after they were treated with 4-HT compared to untreated or vehicle treated cells (data not shown). Microscopic analysis revealed that many of the 4-HT treated Wee1Co/-;Tm-Cre cells showed nuclear fragmentation and membrane blebbing, and were rounded up detaching from the culture plate (Fig. 6A). These abnormalities were not seen in untreated cells (Fig. 6B). Staining with annexin-V, we detected increased apoptotic cells 24 hours upon the addition of 4-HT (Fig. 5B), while the fraction of BrdU positive cells maintained unaltered (not shown).
Because our analysis of E2.5-3.5 embryos indicated that the absence of Wee1 impaired the G2/M cell cycle checkpoint, we next studied this cell cycle checkpoint in the cultured MEF cells. Our analysis revealed a sharp reduction of the mitotic index in the ethanol treated Wee1Co/-;Tm-Cre cells one hour after γ-irradiation, while only a slight reduction of mitotic fraction was observed in the in 4-HT treated cells (Fig. 5C), suggesting a defective G2/M checkpoint in these cells. Based on this observation, we conclude that Wee1 plays a critical role in G2/M cell cycle checkpoint in mouse cells both in vitro and in vivo.
Of note, the growth defects observed in Wee1 deficient cells are much more severe than those observed in many other mutant cells that are defective in the G2/M cell cycle checkpoint, such as BRCA1 [13] or ATM mutant [14] cells, suggesting the phenotypes associated with Wee1 deficiency could also be contributed to other defects in addition to the defective G2/M cell cycle checkpoint. It was shown previously in budding yeast and Drosophila that the absence of Wee1 results in premature entry of mitosis [2, 3]. To investigate whether Wee1 deficiency impairs this function in the mouse, we analyzed the Wee1Co/-;Tm-Cre cells for a possible defect in this checkpoint in the absence or presence of 4-HT. Using double immunofluorescent staining with antibodies against BrdU and phosphorylated histone H3, we found about 40% of histone H3 positive Wee1Co/-;Tm-Cre cells were also positive for BrdU when analyzed 24 hours after 4-HT exposure, while only less than 5% of ethanol treated cells were double positive (Fig. 5D-F). To provide independent confirmation, we had also stained Wee1Co/-;Tm-Cre cells with PI and an antibody against phosphorylated histone H3 followed by flow cytometry analysis. We found that about 50% of phosphorylated H3 positive cells had a DNA content less than 4n while this was not found in the untreated population (Fig. 5G). Thus, like the yeast and Drosophila, the absence of Weel triggered premature entry into mitosis.
Because Wee1 is a negative regulator of Cdk1, the absence of Wee1 is predicted to increase Ckd1 activity. To investigate if the premature mitotic entry observed in the Wee1 mutant cells is indeed associated with the increased Cdk1 activity, we first examined the level of Cdk1-Tyr15, which is specifically phosphorylated by Wee1 [1, 8, 9]. Our data revealed a significant reduction of Cdk1 phosphorylation at Tyr15 24 hours after 4-HT treatment (Fig. 5H), indicating that Cdk1 is activated in the absence of Wee1. To test whether the activation of Cdk1 is responsible for premature mitotic entry, we treated Wee1Co/-;Tm-Cre cells with 4-ht in the presence or absence of purralanol A that specifically inhibits Cdk1 activity. Our data indicated that while purralanol A reduced slightly the percentage of BrdU positive cells (Fig. 5I), it decreased significantly the fraction of mitotic cells (Fig. 5J), leading to the blocking of premature mitotic entry of 4-HT treated Wee1Co/-;Tm-Cre cells (Fig. 5K)
Western blot analysis also revealed increased phosphorylation of Chk2 upon acute deletion of Wee1 (Fig. 5H). Because Chk2 is phosphorylated by ATM upon DNA damage [15], we checked DNA damage in the 4-HT treated Wee1Co/-;Tm-Cre cells using an antibody against phosphorylated form of H2AX (γ-H2AX), which relocates to double strand breaks (DSBs) upon DNA damage [16]. This analysis detected a robust increase of γ-H2AX nuclear foci in the Wee1Co/-;Tm-Cre cells 24 hours after 4-HT treatment but not in the ethanol treated cells (Fig. 7A,B). Correlating with DNA damage, the 4-HT treated Wee1Co/-;Tm-Cre cells 24 hours exhibited increased chromosome aneuploidy compared with ethanol treated cells (Fig. 7C-E). Because Wee1 does not play a direct role in DNA damage repair, we believe that the premature mitotic entry due to Wee1 deficiency could be a cause for DSBs when mutant cells try to initiate mitosis prior to the completion of semi-conservative DNA replication. The DSBs and genetic instability in Wee1 deficient cells, in turn, trigger the DNA damage response, leading to the increased cell death.
In conclusion, our analysis on Wee1-null embryos and Weel-conditional mutant MEFs indicates that Wee1 plays an essential role in maintaining genetic stability and embryonic survival at the pre-implantation stage of mouse development. Our data revealed that similar to lower organisms [2, 3], murine Wee1 controls mitotic entry and the G2/M cell cycle checkpoint through the regulation of Cdk1 activity. Recent studies reveal that Wee1 serves as a downstream mediator of the breast cancer associated gene 1 (BRCA1) in activating the G2/M cell cycle checkpoint upon DNA damage [17, 18]. Future studies should be directed to the relationship between BRCA1 and Wee1 in breast cancer formation and genomic integrity. In addition, phenotypes associated with Wee1-deficiency, in terms of embryonic lethality, premature mitotic entry and defective G2/M cell cycle checkpoint, are similar to those caused by Chk1-null mutation [19, 20]. Because activity of Chk1 is also found to be regulated by BRCA1 [17, 18], the genetic interplay between Chk1 and Wee1 should also be addressed in future studies. A recent investigation indicates that cell division in many mammalian tissues may be associated with specific times of day and that regulation of Wee1 expression by the molecular components of the circadian clockwork may play a key role in this process [21]. Conditional mutations of Wee1 using a Cre-loxP approach [22-24] to overcome early embryonic lethality would facilitate functional study of Wee1 in all phases of murine development.
2. Experimental Procedures
Establishment of Wee1 Knockout Mice
To make a targeting vector for Wee1, we isolated mouse Wee1 genomic DNA from a mouse 129SVEV genomic library using a 904 bp Wee1 cDNA fragment (from 895 to 1799bp) as a probe. A 5.4 kb fragment, which is 5' to the exon 11 starting from a XhoI site in intron 10 of Wee1 gene, was inserted into the pLoxPneo plasmid [25] that was digested with NotI and XhoI. The resulting construct was digested with SalI followed by insertion of a 3 kb fragment, which starts from the XhoI site in the intron 10 to a SalI site 3' to exon 11. A loxP sequence [11] was inserted into a BbeI site in 5' arm (Fig. 1A). After electroporation of the targeting vector into mouse TC-1 ES cells [26], we analyzed G418 and FIAU double resistant clones for homologous recombination in the Wee1 locus. Genomic DNAs from these clones were digested with EcoRV followed by Southern transfer analysis using a 5' flanking probe and then a 3' internal probes. The 5' flanking probe is a 338 bp fragment obtained by PCR using the following primers: 5' GAT ATCAAG CCA GGG CTT TG 3' and 5' GAG AAA GGC TAT ACA ACT TTG AAG 3'. The 3' internal probe is a 1,908 bp fragment obtained by PCR using the following primers: 5' GGC TTC TTA GAC TCT TTC CA 3' and 5' GTT GGG TTG GAT GTT TAG AG 3'. Two ES clones containing correct target event were injected into blastocysts derived from C57B6 mice to generate chimeric mice, and germline transmissions following a mating procedure as previously described [11].
To establish the mice carrying Wee1 mutant allele, we mated F1 mutant mouse (Wee1+/neo) with the EIIa-Cre transgenic female mice [27]. Genomic DNA was extracted from the tail of offspring followed by PCR genotyping. Heterozygous mice containing predicted deletion of both exons 9 and 10 were named as Wee1+/- and were further interbred to generate Wee1-/- mice.
PCR Genotyping for Blastocysts
Blastocysts were transferred into eppendorf tubes containing 20 μl of 5X PCR buffer. They were incubated at 60°C for overnight followed by 100°C for 10 min. We performed PCR amplification using these samples with these primers (35 cycles of 94°C for 30 sec., 60°C for 30 sec. and 72°C for 2 min. 30 sec. and following 72°C for 10 min.). We used the following pair of primers (5' TGT CTA CAA GTT GTC TTG TCA TGA 3' and 5' ACT GTG GAG AGC TCT CAA TG 3') to amply Wee1 wild type allele (369 bp) and another set of primer (5' TGT CTA CAA GTT GTC TTG TCA TGA 3' and 5' CTG CAG GCA AAC CTG TTT C 3') for Wee1 mutant allele (253 bp). PCR products were analyzed running into 2% agarose gel.
TUNEL Assay for Blastocysts
Blastocysts were washed with 1% BSA in PBS 2 times at 4°C, and fixed with 4% paraformaldehyde in PBS at room temperature for 30 min. They were then incubated with 0.1% sodium citrate, 0.1% Triton-X 100, 1% BSA in PBS for 2 min at 4°C, washed with 1% BSA in PBS 2 times at 4°C, and incubated in TUNEL reaction mixture (Roche) at 37°C for 60 min in dark. After washing with 1% BSA in PBS 2 times at 4°C and incubated with DAPI in PBS, they were observed with an inverted fluorescent microscope (Model IX81F, Olympus) and photographed by a cooled CCD color camera, CoolSNAP FX (Photometrics) and IPLab Scientific Image Processing 3.6.3 (Scanalytics, Inc) as the software.
Analysis of G2/M Cell Checkpoint in Blastocysts
Blastocysts in Brinster's BMOC-3 medium (Gibco) were treated with γ-ray (10 Gy) and incubated for three hours at 37°C in CO2 incubator. After incubating them with growth medium containing nocodazole (0.1 ng/ml) for another 3 hours, they were fixed with 4% paraformaldehyde in PBS at room temperature for 30 min followed by the treatment of 0.15% triton-X and 1.5% BSA in PBS for 20 min at room temperature. The embryos were then incubated with a polyclonal antibody to phosphorylated histone H3 at Ser10 (1:300 dilution, Upstate biotechnology) in 1.5% BSA in PBS for 20 min at room temperature and followed by a 2nd antibody (1:200 dilution, Alexa Fluor 488 gout anti-rabbit IgG (H+L) Molecular probes) with DAPI (100 ng/ml) for 1 hour at room temperature before imaging analysis.
MEF Cells and Treatment
MEF cells were derived from E14.5 embryos generated from intercrosses of Wee1+/Co;Tm-Cre mice. For MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay, we plated 2X104 cells into each well of a 24-well plate and 4-HT at 1mM was added twenty-four hours later. At different time points after further incubation, we removed the medium, added 0.5 ml of 0.1% MTT solution dissolved by PBS in each well. 30 min after in 37°C CO2 incubator, we removed the MTT solution and added 0.4 ml of 2-propanol. After 30 min shaking, we measured OD560 by a plate reader. Analysis for the irradiation induced G2/M cell cycle checkpoint and annexin-V-FITC positive Cells were performed as described by [13] and [28], respectively. For analysis of premature mitotic entry, cells grown on glass slides were paused labeled with BrdU half hours before they were fixed with 70% ethanol for 30 min. The cells were washed with PBS, suspended in 1 ml of 2N HCl, 0.5% Triton X-100 and incubated for 30 min, and in 0.5% Triton X-100 in PBS incubated for 10 min. The cells were stained with antibodies to BrdU (Becton-Dickinson) phosphorylated histone H3 (Upstate biotechnology) followed by secondary antibody.
Immunoblot Analysis
Western blot analysis was accomplished according to standard procedures using ECL detection (Amersham). The following primary antibodies were used: Chk2 (Transduction Laboratories), Wee1 (H-3 Santa Cruz), Cdk1, (SC-54 Santa Cruz), and Phospho-Cdk1 (Tyr-15) (#9111, Cell Signaling), and β-actin (Sigma). Horseradish peroxidase-conjugated anti-rabbit, sheep, and anti-mouse antibodies (Amersham) were used as secondary antibodies.
Acknowledgements
We thank members of Deng lab for critical discussion and reading of the manuscript. This work was supported by the intramural Research Program of National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, USA.
Conflict of interests
The authors have declared that no conflict of interest exists.
References
1. Kellogg DR. Wee1-dependent mechanisms required for coordination of cell growth and cell division. J Cell Sci. 2003 ;116:4883-90
2. Stumpff J, Duncan T, Homola E. et al. Drosophila Wee1 kinase regulates Cdk1 and mitotic entry during embryogenesis. Curr Biol. 2004 ;14:2143-8
3. Harvey SL, Kellogg DR. Conservation of mechanisms controlling entry into mitosis: budding yeast wee1 delays entry into mitosis and is required for cell size control. Curr Biol. 2003 ;13:264-75
4. Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996 ;274:1664-72
5. Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004 ;432:316-23
6. Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005 ;5:773-85
7. Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response, and cancer evolution. Nucleic Acids Res. 2006 ;34:1416-26
8. Dunphy WG, Kumagai A. The cdc25 protein contains an intrinsic phosphatase activity. Cell. 1991 ;67:189-96
9. Strausfeld U, Labbe JC, Fesquet D. et al. Dephosphorylation and activation of a p34cdc2/cyclin B complex in vitro by human CDC25 protein. Nature. 1991 ;351:242-5
10. Harvey SL, Charlet A, Haas W. et al. Cdk1-dependent regulation of the mitotic inhibitor Wee1. Cell. 2005 ;122:407-20
11. Deng CX, Xu X. Generation and analysis of Brca1 conditional knockout mice. Methods Mol Biol. 2004 ;280:185-200
12. Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002 ;244:305-18
13. Xu X, Weaver Z, Linke SP. et al. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell. 1999 ;3:389-95
14. Chen P, Gatei M, O'Connell MJ. et al. Chk1 complements the G2/M checkpoint defect and radiosensitivity of ataxia-telangiectasia cells. Oncogene. 1999 ;18:249-56
15. Falck J, Mailand N, Syljuasen RG. et al. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001 ;410:842-7
16. Paull TT, Rogakou EP, Yamazaki V. et al. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000 ;10:886-95
17. Yarden RI, Pardo-Reoyo S, Sgagias M. et al. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet. 2002 ;30:285-9
18. Yan Y, Spieker RS, Kim M. et al. BRCA1-mediated G2/M cell cycle arrest requires ERK1/2 kinase activation. Oncogene. 2005 ;24:3285-96
19. Liu Q, Guntuku S, Cui XS. et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000 ;14:1448-59
20. Takai H, Tominaga K, Motoyama N. et al. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev. 2000 ;14:1439-47
21. Matsuo T, Yamaguchi S, Mitsui S. et al. Control mechanism of the circadian clock for timing of cell division in vivo. Science. 2003 ;302:255-9
22. Xu X, Wagner KU, Larson D. et al. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999 ;22:37-43
23. Sauer B. Inducible gene targeting in mice using the Cre/lox system. Methods. 1998 ;14:381-92
24. Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000 ;26:99-109
25. Yang X, Li C, Xu X. et al. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci U S A. 1998 ;95:3667-72
26. Deng C, Wynshaw-Boris A, Zhou F. et al. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996 ;84:911-21
27. Lakso M, Pichel JG, Gorman JR. et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996 ;93:5860-5
28. Wang RH, Yu H, Deng CX. A requirement for breast-cancer-associated gene 1 (BRCA1) in the spindle checkpoint. Proc Natl Acad Sci U S A. 2004 ;101:17108-13
Figures and Tables
Genotypes of offspring of Wee1+/- interbreeding
Generation of Wee1 knockout mice. (A) Structure of Wee1 targeting vector, ploxPneoWee1 (upper) and map of a part of normal mouse Wee1 gene (lower). Be: BbeI, Ev: EcoRV, S: SalI, and Xh: XhoI. Probes used were as indicated. Probes “a”, which is 5' flanking the targeting vector, and probe “b”, which is located in the 3' arm of the targeting vector, were used in Southern blot analysis. (B) Structure of targeted allele (upper), and exons 9/10 deleted allele (lower). (C) Southern blot showed targeted allele (lanes 2 and 3) after EcoRV digestion and probed with probe b. (F) PCR based genotype of offspring obtained from crosses between Wee1+/neo and EIIa-Cre mice. Primer 1: 5' TGT CTA CAA GTT GTC TTG TCA TGA 3', Primer 2: 5' ACT GTG GAG AGC TCT CAA TG 3', Primer 3: 5' CTC CAG GTG TGT CAT ATA CC 3', Primer 4: 5' CCG TTC CTC AGC TGC AAC TT 3'.
Abnormal appearance of Wee1-/- embryos at pre-implantation stages. (A,B) Images of Wee1+/+ (A) and Wee1-/- (B) embryos. (C) Genotyping of E3.5 embryos by PCR. (D,E) Morphology (D) of blastocysts after they were cultured after different times as indicated. All embryos showing proliferation were either Wee1+/+ or Wee1+/- embryos. Genotypes (E) were determined by PCR 4 days after culture.
Apoptosis and impaired G2/M cell cycle checkpoint in Wee1-/- embryos. (A, B) TUNEL assay revealed increased apoptosis in Wee1-/- embryos. (C,D) Mitotic cells in E2.5 embryos. (E,F) Mitotic index of E2.5 and E3.5 embryos. Each graph bar summarizes data obtained from at least 4 embryos.
Generation of tamoxifen inducible Wee1 knockout MEF cells. (A) Structure of conditional allele (upper) and exons 9/10 deleted allele (lower). (B) PCR based genotyping of Wee1Co/+ and Wee1Co/-;Tm-Cre MEFs in the presence (+) or absence (-) of 4-ht. The same primers were used for genotyping as shown in Fig. 1.
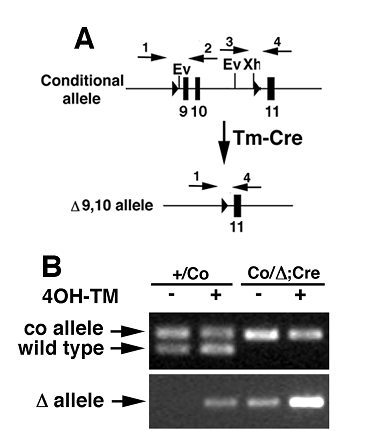
Increased cell death and impaired cell cycle regulation of Wee1-deficient cells (A) MTT assay of Wee1Co/-;TM-cre MEF cells that were treated with ethanol or no treatment (upper curve), or 1μM of 4-HT. (B) Apoptotic fraction of Wee1Co/-;TM-cre MEF cells after 4-ht treatment revealed by annexin V and EtBr staining. (C) Defective G2/M cell cycle checkpoint of Wee1Co/-;TM-cre MEF cells. Cells were treated with 1 μM 4-HT for 24 hours and fixed at 1-4 hours after they were irradiated with 10 Gy γ-ray. (D,E) Premature entry into mitosis revealed by BrdU and histone H3 double staining. Wee1Co/-;TM-cre MEFs were incubated in the absence (D) or presence (E) of 4-HT for 24 hours prior to the staining. (F) Percentage of BrdU and histone H3 double positive cell. The percentage is calculated by the following formula [(BrdU++H3Pi+)/H3Pi+]. (G) Flow cytometry analysis of mitotic cells in ethanol or 4-ht treated Wee1Co/-;TM-cre MEFs by PI and H3Pi staining. (H) Western blotting of Wee1Co/-;TM-cre MEFs. The arrow points to the phosphorylated form of Chk2 after lighter expossure of the gel. (I,J,K) Effect of purralanol A (10 μM) on mitotic entry of Wee1Co/-;Tm-Cre cells in the absence (-) and presence (+) of 4-HT. Percentages of BrdU (I), H3Pi (J), and double (K) positive cells were shown.
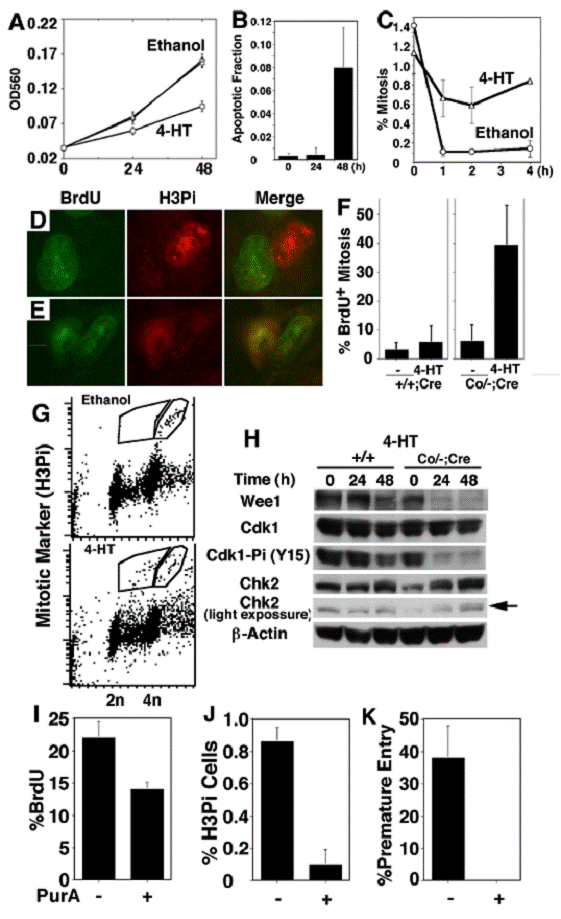
Morphology of Wee1Co/-;Tm-Cre MEFs in the presence (A) and absence (B) of 4-HT for 48 hours.
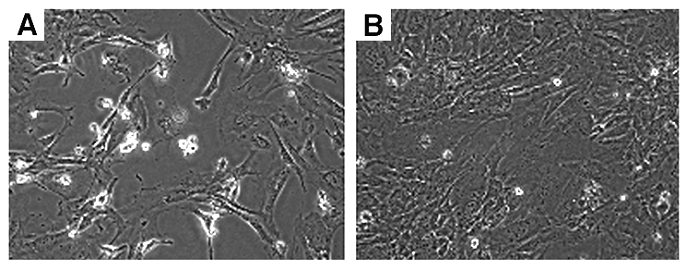
DNA damage and chromosome instability of Wee1Co/-;TM-cre MEF cells. (A,B) Immunofluorescent staining of Wee1Co/-;TM-cre MEF cells using an antibody to γ-H2AX in the absence (A) or presence (B) of 1 μM 4-ht for 24 hours. (C,D) Chromosome spread of Wee1Co/-;TM-cre MEF cells after they were grown in the absence (C) or presence (D) of 1 μM 4-ht for 24 hours. (E) A summary of chromosomal spreading data from more than 60 cells in the absence (black bar) and presence (open bar) of 4-HT.
Author contact
![]() Correspondence to: Dr. Chu-Xia Deng, Tel: (301) 402-7225; Fax: (301) 480-1135; Email: chuxiadniddk.nih.gov
Correspondence to: Dr. Chu-Xia Deng, Tel: (301) 402-7225; Fax: (301) 480-1135; Email: chuxiadniddk.nih.gov