Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(1):1-7. doi:10.7150/ijbs.3.1 This issue Cite
Research Paper
Effect of Reparation of Repeat Sequences in the Human α-Synuclein on Fibrillation Ability
Koji Sode, Sayaka Ochiai, Natsuki Kobayashi, Eri Usuzaka ![]()
Department of Biotechnology, Graduate School of Engineering, Tokyo University of Agriculture & Technology, 2-2-4-16, Nakacho, Koganei, Tokyo 184-8588, Japan
Received 2006-7-6; Accepted 2006-9-29; Published 2006-10-2
Abstract
The aggregation and fibrillation of α-synuclein has been implicated as a causative factor in the Parkinson's disease. The hexamer motif KTKEGV is found in each of the seven imperfect repeat sequences in the N-terminal half of α-synuclein. The motif is not fully conserved in the sixth and seventh repeats. We created mutants in which the motif was repaired to be fully conserved in either (Rep6 and Rep7) or both (Rep67) of these two repeats. The Rep6 and Rep67 mutants showed a greatly reduced propensity to aggregate and fibrillate while all three mutants showed greater resistance to HFIP-induced formation of the α-helix intermediate. Resistance to formation in the partially folded intermediate may repress the folding of α-synuclein, consequently interfering with the aggregation and fibril formation. These results demonstrated that KTKEGV repeats may have a significant role in keeping native unfolded status of α-synuclein.
Keywords: α-synuclein, aggregation and fibrillation, site-directed mutagenesis, natively unfolded protein, NAC region, 11-residue repeat sequences
1. Introduction
Several neurodegenerative disorders have been characterized by the deposition and accumulation of abnormal protein components in cells, leading to selective synaptic damage. Such disorders are often described according to the type of misfolded and deposited protein, although it is not fully understood what causes the abnormal protein folding and accumulation as well as how they become cytotoxic. Alzheimer's disease (AD) and Lewy body (LB) disease, represented by Parkinson's disease (PD), are the most common neurodegenerative diseases, which are characterized by the accumulation of the misfolded proteins β-amyloid and α-synuclein, respectively. That α-synuclein is the major fibrillar protein component of LB in both sporadic and familial PD, and that two different α-synuclein missense mutations (A53T [1, 2] and A30P [3]) cause autosomal-dominant PD, strongly support a causative role for α-synuclein in PD pathogenesis.
α-Synuclein is a relatively abundant brain protein of unknown function that is a type of natively unfolded proteins (disordered proteins) [4], which have little or no ordered structure under physiological conditions in vitro. The protein's 140-amino acid amino acid sequence consists of an N-terminal region (residues 1-95) containing seven 11-residue repeats (XKTKEGVXXXX) that include a highly conserved KTKEGV hexameric motif, and an acidic C-terminal region (residues 96-140). Similarities in these 11-residue repeats with those found in apolipoproteins have suggested a possible role in the association of α-synuclein with lipid through formation of α-helical secondary structure [5, 6]. Furthermore, portions of the α-synuclein N-terminal region have been reported to adopt an α-helical structure and bind SDS micelles [7-9] and lipid vesicles [10]. α-Synucleins binding to lipid vesicles have been reported not to fibrillate in vitro [11]. These repeat sequences also have an effect of decreasing the tendency of α-synuclein to form β-sheet structures, such as fibrils [12]. The central region of α-synuclein (residues 61-95), known as the non-amyloid β component of Alzheimer's disease amyloid (NAC), is recognized as the core region responsible for the formation of fibrils [13], which have a β-sheet-rich structure [14].
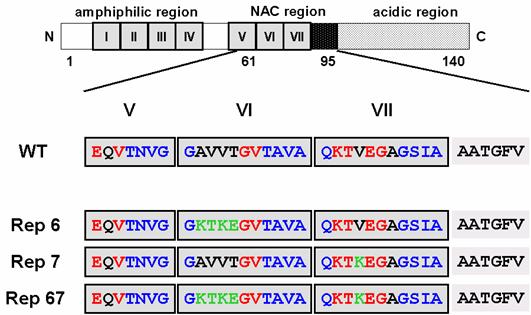
However, none has ever reported on the role of KTKEGV repeated sequence in α-synuclein structure, particularly in the protein folded process and/or keeping its random structure. The lack in the information about the role of this one of the characteristic primary sequence in α-synuclein, KTKEGV, inspired us to initiate the reparation of imperfect sequence in this molecule. Interestingly, among the seven 11-amino acid imperfect repeats, the characteristic hexameric motifs in the two repeats located within the NAC region are not conserved (Figure 1). In order to investigate the role of these two non-conserved hexameric motifs on the aggregation and fibril formation ability of α-synuclein, we carried out a site-directed mutagenesis study on this region. We constructed three mutant α-synucleins in which either or both of the nonconserved hexameric motifs in the 6th and 7th repeats were substituted to the conserved KTKEGV sequence (Figure 1). We investigated the propensity of the mutant α-synucleins to form fibrils and aggregates, as well as their propensity to form α-helices. Our findings suggest that the two 11-residue repeat sequences in the NAC region have a strong effect on α-synuclein folding and the 6th repeat sequence has a particularly strong influence on α-synuclein's propensity to aggregate and fibrillate.
α-Synuclein mutants introduced amino acid substitution into the incomplete repeat sequences in NAC region. The boxed regions indicate incomplete repeat regions. Red letters indicate conserved amino acids in the characteristic hexamer motif (KTKEGV). Green letters represent the mutated residues.
2. Materials and Methods
Chemicals
Thioflavin T (TfT) was obtained from Sigma. 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP) was obtained from Wako.
Recombinant technology
PCR amplification of the α-synuclein structural gene was carried out using a human bone marrow cDNA library as the template and the following oligonucleotide primer pair: 5'-CCAAGCTTCATATGGATGTATTCATGAAAGGACTTTCAAAGG-3' (forward) and 5'-GGTACCCTTGGCAAAGCAGGCTTCAGGTTCGTAGTCTTGATA-3' (reverse). The forward primer was designed with an NdeI immediately following a HindIII site, while the reverse primer has a KpnI site at its 5' end (all sites are underlined). Amplification was carried out using Ampli-Taq Gold DNA polymerase (Roche) with 35 cycles of 95 oC for 1 min, 55 oC for 1 min, and 72 oC for 1 min. The amplified fragment was cloned into the pGEM-T TA cloning vector (Promega) and its sequence was confirmed by automated DNA sequencing (Perkin Elmer ABI Model 310).
The HindIII-KpnI fragment of the pGEM-T clone, containing the amplified α-synuclein gene, was inserted into the vector pKF19k to carry out site-directed mutagenesis using Mutan®-Super Express Km (Takara, Japan). Mutations were introduced with the following oligonucleotides: 5'-GTGACAAATGTTGGAGGAAAAACAAAAGAAGGTGTGACAGCAGTAGCC-3' (Rep6 (Ala69Lys/Val70Thr/Val71Lys/Thr72Glu)), 5'-CCCAGAAGACAAAAGAGGGAGCAGG-3' (Rep7 (Val82Lys)). The mutant Rep67 (Ala69Lys/Val70Thr/Val71Lys/Thr72Glu/Val82Lys) was created using the two primers for Rep6 and Rep7 simultaneously. After confirming the mutations by DNA sequencing, the wild type and mutated genes were digested with NdeI and KpnI and inserted into pTYB1 (New England Biolabs) expression vectors.
Wild type and mutant α-synuclein genes in pTYB1 were expressed in E. coli with C-terminal intein/chitin binding domain tags and purified using the IMPACT T7 one-step protein purification system (New England Biolabs). This system allowed simple purification of α-synucleins by binding of the fusion proteins to a chitin column, followed by addition of a thiol agent (dithiothreitol) to induce the self-cleavage reaction of the intein and release the α-synucleins. The resultant proteins were highly homogeneous by SDS-PAGE and their concentrations were determined by the DC protein assay kit (BioRad).
Circular dichroism measurements
CD spectra of 0.2 mg/ml α-synucleins at 25 oC were recorded from 190 to 250 nm in a 0.1 cm cell, a 1.0 nm bandwidth, a 4 s response time, and a scan speed of 20 nm/min using a J-720 spectropolarimeter (JASCO). Each spectrum was generated from the average of five scans. The CD spectrum of 10 mM Tris buffer, pH 7.4, was used as background and subtracted from the protein spectra. The data were presented as the mean residue molar ellipticity (deg∙cm2∙dmol-1).
Fibril formation analysis
All protein samples were concentrated to approx. 10 mg/ml by using Amicon Ultra-15 filters (Millipore) in 10 mM Tris-HCl, pH 7.4, and were centrifuged at 150,000g for 60 min to remove insoluble aggregates. α-Synucleins were incubated at concentrations of 2.0 mg/ml in 0.5 ml of the same buffer with 0.02% NaN3 at 37 oC with shaking in 1.5 ml tubes. Fibril formation was monitored by Thioflavin T (TfT) fluorescence by transferring 10 μl aliquots from the incubated sample to 1.0 ml of 25 μM TfT in 10 mM Tris buffer, pH 7.4. Fluorescence measurements were performed in a FP6500 spectrofluorometer (JASCO) with a 0.5 cm excitation light path. TfT fluorescence was recorded immediately after addition of protein aliquots to the TfT mixture at 1 nm increments from 460 to 560 nm, with an excitation of 450 nm, response time of 1 s, scan speed of 100 nm/min, and slits of 5 nm for both excitation and emission. For each sample, a blank measurement recorded prior to addition of α-synuclein to the TfT solution was subtracted from the final TfT intensity at 482 nm. All data were processed using Spectra Manager (JASCO) software. Rayleigh light scattering at 330 nm was used to monitor the total aggregation.
1,1,1,3,3,3-Hexafluoro-2-propanol titration
HFIP was added to a 0.6 mg/ml α-synuclein solution in 10 mM Tris-HCl, pH 7.4, and diluted with the same buffer to obtain final concentrations of 0 to 10% (v/v) HFIP and 0.2 mg/ml α-synuclein. Samples were analyzed by CD spectroscopy after incubation at room temperature for 24 hours.
3. Results and Discussion
Aggregation and fibrillation of wild type and mutant a-synuclein
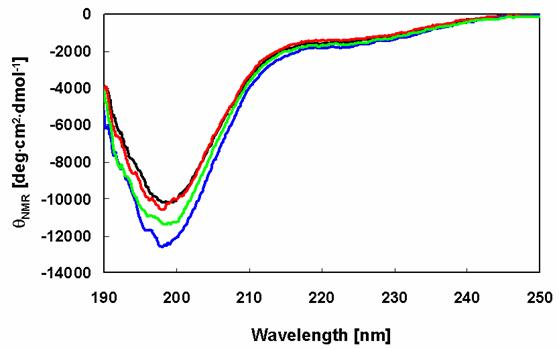
The far-UV CD spectrum of the prepared wild-type human recombinant α-synuclein sample is almost indistinguishable from those of the Rep6, Rep7, and Rep67 α-synuclein mutants (Figure 2). The CD spectra of these samples are typical of an essentially unfolded polypeptide chain, including the characteristic minima in the vicinity of 198 nm. These results show that the amino acid substitutions had no impact on the native state of α-synuclein.
Secondary structures of α-synuclein and its mutants. CD spectra of α-synucleins determined after purification: WT (black), Rep6 (blue), Rep7 (red), Rep67 (green).
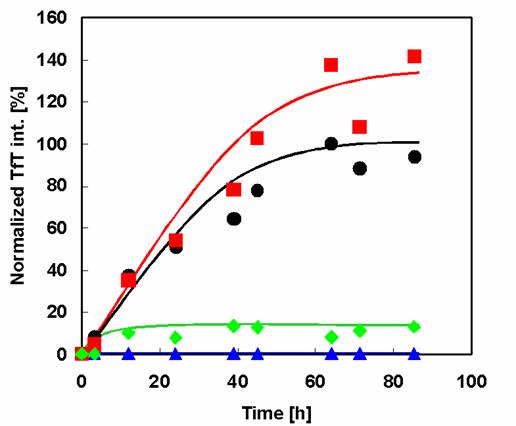
The prepared wild type and mutant α-synuclein samples were investigated for their aggregation and fibril formation abilities using the fluorescent dye TfT. Because TfT appears to embed itself within the clearance of pleats of β-sheets, it interacts quite specifically with fibrils but not with most native proteins or amorphous aggregates. Binding of TfT to protein fibrils is accompanied by a characteristic increase in the fluorescence intensity in the vicinity of 482 nm [15]. Comparison of the fibrillation patterns of wild type and mutant α-synucleins (Figure 3) shows that Rep7 has a similar fibrillation rate and higher maximal fluorescence intensity than wild type. Therefore, reparation of the 7th repeat sequence, mutating it to contain the characteristic KTKEGV hexameric motif, did not affect the propensity of fibril formation. In contrast, Rep6 and Rep67 had much lower fibril formation rates than both wild type and Rep7, with maximal fibril formation values less than 10% of the wild-type value. Therefore, both Rep6 and Rep67 achieved great reductions in fibril formation ability by reparation of the 6th repeat sequence to contain the characteristic hexamer motif.
Effect of amino acid substitution on amyloid fibril formation of α-synuclein. Time course of fibril formation of α-synuclein and its mutants as determined by TfT fluorescence assay analysis: WT (black circles), Rep6 (blue triangles), Rep7 (red squares), Rep67 (green diamonds).
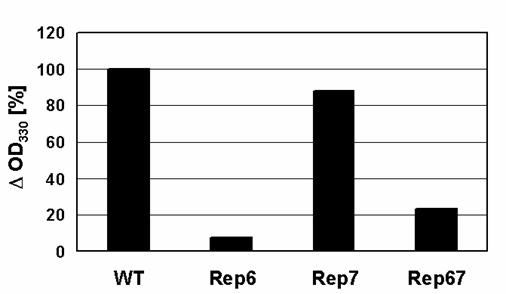
Light scattering from protein solutions is recognized as an association-dependent parameter reflecting the formation of large particles. Unlike TfT, the increase in static light scattering reflects the formation of all large particles, both fibrillar and amorphous, including large soluble oligomers. Total α-synuclein aggregation levels were determined by light scattering and compared (Figure 4). While the aggregation of Rep7 after 120 hours of incubation was almost identical to that of wild type, Rep6 and Rep67 were much less prone to aggregate. These results suggest that reparation of the 6th conserved characteristic hexamer motif has an especially strong effect on α-synuclein aggregation and fibrillation.
Changes in α-synuclein light-scattering. The OD330 values recorded after incubation of the samples were subtracted from those before incubation to determine their decrease in light-scattering (ΔOD330).
The primary structure at the NAC region strongly characterizes the preference of the formation of fibril. As we and other researcher reported [16, 17], the amino acid substitution at NAC region may result in the formation of less amyloid forming prone mutant α-synuclein, only if the introduced residues are positive toward the resistance in fibril formation. However, simultaneous substitution of 4 residues without changing their random and water soluble structures, is hard to be achieved. To design such simultaneous multi residues substitution, the precise prediction of possible secondary structure at the target region and the information about the interaction of residues under the folded condition are necessary, although few structural information in the folded structure of α-synuclein is available. Our results indicated that the introduction of KTKEGV sequence at the NAC region resulted in drastic decrease in the fibril formation ability without losing its primary random structure and its water solubility. This fact indicated that KTKEGV is a sequence which may have a crucial role in preventing amyloid fibril formation, keeping its random structure in a water soluble form. Considering that the replacement of NAC region itself may result in negative effect of amyloid forming ability, Rep6 showed less prone in amyloid forming than Rep7.
α-Helix formation by wild type and mutant α-synucleins in the presence of HFIP
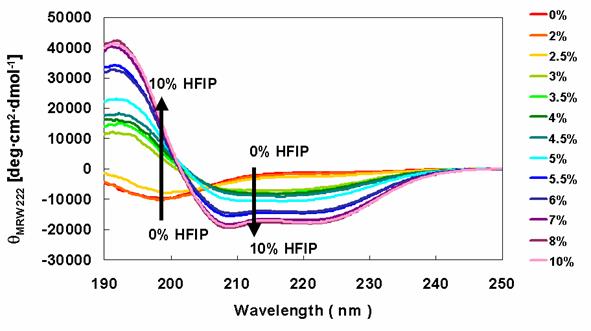
HFIP was used to induce the conversion of α-synucleins to an α-helical structure, which was monitored by far-UV CD spectra. Solutions containing the wild type protein (data not shown) and Rep6 (Figure 5) showed increased secondary structure with increasing HFIP. The minima at 198 nm, representing random structure, were converted to peaks with large positive ellipticity. This was accompanied by the appearance of minima around 208 nm and 222 nm, indicating the formation of α-helical structure with increasing HFIP. Similar HFIP concentration-dependent CD spectral changes were observed in solutions containing Rep7 or Rep67 (data not shown). Consistent with earlier studies, these observations indicate that the transition from the unfolded state to a partially folded intermediate conformation concludes with the formation of α-helix rich species as evidenced by the two minima at 208 nm and 222 nm.
Secondary structures of α-synuclein Rep6 in the presence of various concentrations of HFIP. CD spectra of α-synuclein Rep6 were determined in the presence of various concentration of HFIP.
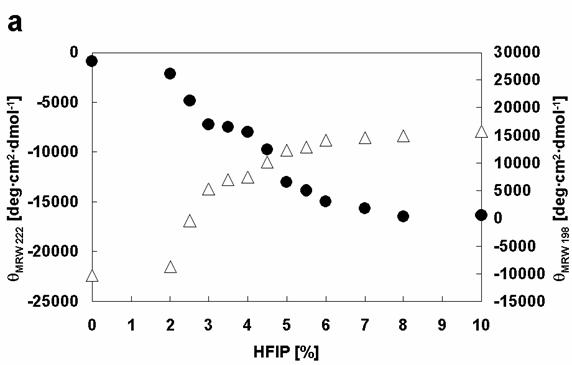
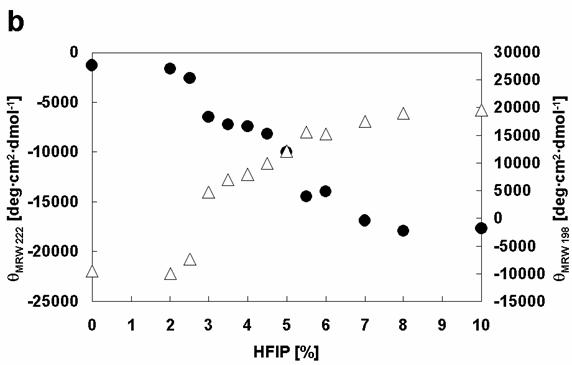
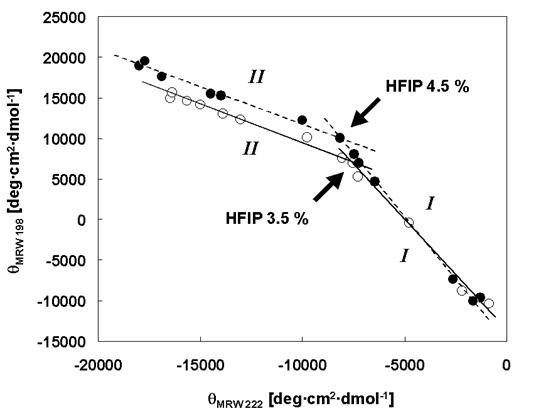
Closer analysis of the changes in [θ]λ (λ=198 and λ=222) of wild type and mutant α-synucleins with increasing HFIP concentration indicates that the formation of α-helical structure appears to proceed through two transitions (Figure 6). To examine the transitions in greater detail, we produced phase diagrams (Figures 7) showing the relationship between [θ]λ198 and [θ]λ222. A linear relationship reflects an all-or-none transition between two different conformations, while a nonlinear relation represents multiple sequential transformations. For the wild type α-synuclein, the structural changes with increasing HFIP clearly proceeds through two transitions (Figure 7): (I) from natively unfolded to partially folded (0-3.5% HFIP), and (II) from a partially folded intermediate to an α-helical conformation (3.5-10% HFIP). These observations are also consistent with earlier work [18]. Although the Rep6 (Figure 7), Rep7, and Rep67 (data not shown) mutants also go through two transitions, the break-point HFIP concentration has shifted higher than with the wild type protein. Whereas the breaking point between the two transitions occurred near 3.5% HFIP for wild-type α-synuclein, all three mutants had breaking points near 4.5% HFIP (Figure 7). These greater HFIP requirements indicate that the reparation of the characteristic hexamer motif in the 6th or 7th repeat sequence help decrease the propensity of α-synuclein to form a partially folded intermediate.
Comparison of the effects of HFIP on the conformations of α-synuclein WT with Rep6. Dependencies of [θ]MRW 222 (black circles) and [θ]MRW 198 (white triangles) on HFIP concentration: (a) WT, (b) Rep6.
Phase diagrams for the structural changes induced in α-synuclein WT and Rep6 by HFIP. The effects of HFIP on the conformation of α-synucleins: WT (white circles and solid lines), Rep6 (black circles and dashed lines). The roman numbers, I and II indicate the phases of transition: (I) from natively unfolded to partially folded intermediate, (II) from partially folded intermediate to α-helical conformation. The arrows indicate the breaking points. The break-point HFIP concentrations are 3.5% (WT) and 4.5% (Rep6) respectively.
The first critical step of aggregation and fibrillation of α-synuclein is considered to be the folding from a natively unfolded state to a partially folded intermediate [19]. Munishkina et al. [18] reported the enhancement of fibril formation by low (1-2.5%) concentrations of HFIP in the presence of alcohol. Such low concentrations of HFIP were sufficient due to the ability of alcohol to stabilize the partially folded α-synuclein intermediate.
Our results clearly indicated that all mutants showed the resistant toward formation of partially folded structure compared with wild type. Therefore, KTKEGV repeats may have significant role in keeping native unfolded status of α-synuclein. Since the partial folded status is critical step in amyloid fibril forming process, the resistance toward the formation of partial folded structure revealed that KTKEGV repeats may give a positive contribution in preventing fibril formation.
As expected, Rep6 and Rep67 also showed lower propensities to aggregate and fibrillate than wild type α-synuclein. Although Rep7 also showed difficulty in intermediate formation, it appears to have a similar propensity to aggregate and fibrillate to wild type α-synuclein. Considering that only one amino acid was substituted to create Rep7, the NAC region remains relatively intact, and the interaction of the NAC region may dominate during the fibril or aggregation formation.
Acknowledgements
The authors thank Dr. Stefano Ferri for kindly revising the manuscript.
Conflict of interests
The authors have declared that no conflict of interest exists.
References
1. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997 ;276:2045-7
2. Munoz E, Oliva R, Obach V, Marti MJ, Pastor P, Ballesta F, Tolosa E. Identification of Spanish familial Parkinson's disease and screening for the Ala53Thr mutation of the alpha-synuclein gene in early onset patients. Neurosci Lett. 1997 ;235:57-60
3. Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998 ;18:106-8
4. Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PTJr. NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996 ;35:13709-15
5. George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995 ;15:361-72
6. Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998 ;273:9443-9
7. Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem. 2005 ;280:9595-603
8. Bussell RJr, Eliezer D. A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins. J Mol Biol. 2003 ;329:763-78
9. Chandra S, Chen X, Rizo J, Jahn R, Sudhof TC. A broken alpha -helix in folded alpha -Synuclein. J Biol Chem. 2003 ;278:15313-8
10. Jao CC, Der-Sarkissian A, Chen J, Langen R. Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc Natl Acad Sci U S A. 2004 ;101:8331-6
11. Zhu M, Fink AL. Lipid binding inhibits alpha-synuclein fibril formation. J Biol Chem. 2003 ;278:16873-7
12. Kessler JC, Rochet JC, Lansbury PTJr. The N-terminal repeat domain of alpha-synuclein inhibits beta-sheet and amyloid fibril formation. Biochemistry. 2003 ;42:672-8
13. Giasson BI, Murray IV, Trojanowski JQ, Lee VM. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem. 2001 ;276:2380-6
14. Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A. 2000 ;97:4897-902
15. LeVine H 3rd. Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999 ;309:274-84
16. Sode K, Usuzaka E, Kobayashi N, Ochiai S. Engineered alpha-synuclein prevents wild type and familial Parkin variant fibril formation. Biochem Biophys Res Commun. 2005 ;335:432-6
17. Du HN, Tang L, Luo XY, Li HT, Hu J, Zhou JW, Hu HY. A peptide motif consisting of glycine, alanine, and valine is required for the fibrillization and cytotoxicity of human alpha-synuclein. Biochemistry. 2003 ;42:8870-8
18. Munishkina LA, Phelan C, Uversky VN, Fink AL. Conformational behavior and aggregation of alpha-synuclein in organic solvents: modeling the effects of membranes. Biochemistry. 2003 ;42:2720-30
19. Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001 ;276:10737-44
Author contact
![]() Correspondence to: Prof. Koji Sode, Tel/Fax: +81-42-388-7027, E-mail: sodetuat.ac.jp.
Correspondence to: Prof. Koji Sode, Tel/Fax: +81-42-388-7027, E-mail: sodetuat.ac.jp.