Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction of HSPs
2. HSPs and PD Pathophysiology
3. PD-Related Gene Mutations and...
4. Protective Role of HSPs in PD
5. Potential Target for the...
6. Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(1):20-26. doi:10.7150/ijbs.3.20 This issue Cite
Review
Are heat shock proteins therapeutic target for Parkinson's disease?
Guang-Rui Luo1, Sheng Chen2, Wei-Dong Le1 ![]()
1. Institute of Health Sciences, Shanghai Institutes of Biological Sciences, Chinese Academy of Sciences, Shanghai, China
2. Institutes of Neurology, Medical School of Jiao Tong University, Shanghai, China
Received 2006-7-28; Accepted 2006-9-27; Published 2006-10-15
Abstract
Heat shock proteins (HSPs), known as molecular chaperone to assist protein folding, have recently become a research focus in Parkinson's disease (PD) because the pathogenesis of this disease is highlighted by the intracellular protein misfolding and inclusion body formation. The present review will focus on the functions of different HSPs and their protective roles in PD. It is postulated that HSPs may serve as protein folding machinery and work together with ubiquitin-proteasome system (UPS) to assist in decomposing aberrant proteins. Failure of UPS is thought to play a key role in the pathogenesis of PD. In addition, HSPs may possess anti-apoptotic effects and keep the homeostasis of dopaminergic neurons against stress conditions. The critical role of HSPs and recent discovery of some novel HSPs inducers suggest that HSPs may be potential therapeutic targets for PD and other neurodegenerative disorders.
Keywords: Parkinson's disease (PD), heat shock proteins (HSPs), ubiquitin-proteasome system (UPS), apoptosis
1. Introduction of HSPs
In the eukaryotic cell, heat shock proteins (HSPs) provide an intrinsic mechanism to defend the cell against external diverse physiological stress that may initiate a cascade of events affecting cell structure and function. The high conservation of HSPs throughout the evolution suggests that these proteins may have a vital role in protecting cells from injury. HSPs are composed of different classes of proteins according to their molecular weight, which include high-molecular-mass HSPs (≥100kD), HSP90 (81 to 99kD), HSP70 (65 to 80kD), HSP60 (55 to 64kD), HSP40 (35 to 54kD) and small HSPs (≤34kD) [1]. Different classes of HSPs play a diversity role in governing proper protein assembly, folding, and translocation [1, 2]. Regulation of these HSPs synthesis creates a unique defense system to maintain cellular protein homeostasis and to ensure cell survival [2].
The current understanding of HSPs' function is based on two main lines of evidence: (1) the clearance of waste proteins requires protein folding machinery called chaperones [1], and (2) HSPs chaperones bind to denatured proteins to promote their degradation [2]. New evidence suggests that HSPs may actively participate in an array of cellular processes, including cytoprotection [3], and HSPs dysfunction may contribute to the pathogenesis of Parkinson's disease (PD), a disease characterized by conformational changes in proteins that result in misfolding, aggregation and intracellular Lewy Body formation [4].
This review provides an update view of the cytoprotective role of HSPs in PD and the potential therapeutic target of HSPs for the treatment of PD.
2. HSPs and PD Pathophysiology
Many neurodegenerative disorders, including PD, Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), Huntington disease (HD) and other polyglutamine expansion disorders, are associated with degeneration and death of specific neuronal populations due to accumulation of certain abnormal polypeptides or proteins [4]. PD is a neurological disorder characterized by movement disturbance that mostly results from progressive degeneration of dopaminergic neurons in substantia nigra pars compacta (SNpc). Numerous studies implicate that at least two components of cellular proteins are associated with PD: the ubiquitin proteasomal system (UPS) and the HSPs [5, 6]. Transcriptional analysis of multiple brain regions in PD indicates the impairment of multiple electron transport chain complexes and the dysfunction of UPS in PD, along with a robust induction of several forms of HSPs [7]. Inclusion bodies called Lewy bodies with aberrant misfolding and aggregative proteins are common pathological hallmark in PD, indicating that abnormality of protein homeostasis may contribute to the pathogenesis of the disease [5]. Hsp70 and Torsin A, a homology to yeast Hsp104 and mutations of the gene causing dystonia, are colocalized with α-synuclein (αSN) containing Lewy bodies [8]. Further, Dedmon et al. [9] found that Hsp70 could inhibit αSN fibril formation through preferential binding to prefibrillar species to change the characteristics of toxic αSN aggregates. This work therefore elucidates a specific role of Hsp70 in the pathogenesis of PD and supports a general concept that chaperone action is a crucial aspect in protecting against the otherwise damaging consequences of protein misfolding [9]. With ageing, the level of HSPs is decreased insufficiently to keep the cellular proteins homeostasis, which may give rise to certain diseases [4, 5]. Since the proportion of patients suffering from PD in our aging society is increasing, it is urgent to find better therapeutic approaches to this devastating disease.
3. PD-Related Gene Mutations and Possible Association with HSPs
During the last decade of discovery of several PD-associated mutant genes a remarkable progress has been made to help our understanding of the biology of PD. So far there are at least 6 genes and several loci that have been identified responsible to PD [10, 11]. It is hypothesized that UPS dysfunction resulted from these defected genes may cause protein misfolding and aggregation, and eventually lead to nigral cell degeneration [12]. Polymorphisms in the 5' promoter regions of Hsp70 gene have been found significantly associated with PD [13].
Alpha-synuclein (αSN)
αSN, which plays a critical role in regulating synaptic vesicle size with particular relevance to dopamine storage, was found to be the main component in the Lewy body. Stress can increase the αSN protein aggregation and inclusion body formation [11, 14]; misfolding αSN can change proteasome composition, impair proteasome-mediated protein degradation, alter protein synthesis, and reduce the ability of cells to withstand stationary phase ageing [11, 15]. Three mutations of αSN, which show toxic gain-of-function, have been found in association with familial PD [10, 11]. Inducible expression of mutant αSN in PC12 cell lines can result in greater sensitivity to proteasomal impairment, leading to mitochondrial abnormalities and neuronal cell death [16]. αSN at nanomolar concentration is able to increase Hsp70 protein level in PC12 cells, which can reduce αSN aggregation and toxicity [17]. In addition, the αSN protein has a tendency to self-aggregate and the protein level of αSN is increased in SNc with ageing [18].
Parkin
Parkin is a member of E3 ligase in the UPS [19]. Parkin mutations are thought to result in the improper targeting of its substrates for proteasomal degradation leading to potentially neurotoxic accumulation [20]. Thus, great emphasis has been placed on the identification of substrates of parkin and their possible role in dopaminergic neuron loss in PD [11]. Kalia et al showed that the bcl-2-associated athanogene 5 (BAG5) can enhance dopaminergic neuron death in a vivo model of PD through inhibiting the E3 ligase activity and the chaperone activity of Hsp70 [21].
Ubiquitin carboxyl-terminal hydrolase L1 (UCH-L1)
UCH-L1, a highly abundant and neuronal specific protein that belongs to a family of deubiquitinating enzymes, is responsible for hydrolyzing polymeric ubiquitin chains to free ubiquitin monomers [10, 11]. UCH-L1 might additionally act as a dimerization-dependent ubiquitin protein ligase [22] and maintain ubiquitin homeostasis by promoting the stability of ubiquitin monomers in vivo [23]. When UCH-L1 mutates, ubiquitin recycling is reduced, which may lead to aggregation of aberrant proteins. It is found that UCH-L1 aggresomes colocalize with Hsp70, chaperone BiP, and other ubiquitinated proteins [24], suggesting that UCH-L1 may interact with HSPs in an attempt to participate in protein degradation.
DJ-1
DJ-1 is a novel oncogene and mutations in this gene can cause familial PD. It is reported that DJ-1 mutations may result in oxidative stress and mitochondrial injury, which may lead to protein aggregation and neuronal cell death [10, 11]. Li et al [25] reported that DJ-1 and its mutants are associated with Hsp70, CHIP and mtHsp70/Grp75, a mitochondria-resident Hsp70; and DJ-1 and its mutants are colocalized with Hsp70 and CHIP in cells. Furthermore, H2O2 treatment in cells enhances DJ-1 interaction with mtHsp70 in mitochondria [25]. These findings suggest that translocation of DJ-1 to mitochondria after oxidative stress is carried out by chaperones.
4. Protective Role of HSPs in PD
It has been reported that Hsp70 is associated with αSN, dopamine transporter (DAT), parkin, proteasome subunits, ubiquitin and UCH-L1 [18]. Hsp70 is believed not only to protect cells from rotenone-mediated cytotoxicity but also to decrease soluble αSN aggregation [26]. Furthermore, Hsp70 can work as a putative anti-apoptotic factor to protect against neuronal cell death in PD [3, 4]. These results highlight the possibility of using Hsp70 as a potential therapy for PD. Recent studies of function and inducer of Hsp90 also indicate its potential therapy for PD [27, 28].
Hsp90
Hsp90 is the main component of the cytosolic molecular chaperone complex that has been implicated in the negative regulation of the heat shock factor 1 (HSF1). HSF1 is responsible for the transcriptional activation of the heat shock genes including Hsp40, Hsp70, and Hsp90 [29], suggesting a regulatory role in Hsp90 synthesis at the transcriptional level. Hsp90 forms a multichaperone complex with Hsp70 and Hsp40 to regulate several regulatory proteins, such as steroid hormone receptors [30] and transcription factors [31], and to modulate the protein translocation from peroxisomal to organelle [32]. The interplay between these chaperones is of crucial importance for cell function and survival. Recently, Uryu et al. demonstrated that Hsp90 was predominantly increased in PD brains, which was in correlation with the elevated level of insoluble αSN. These alterations of Hsp90 in PD brain were recapitulated by neuropathological findings in αSN mutant transgenic mouse model of PD [27]. Furthermore, exposure of cells to proteasome inhibitors resulted in increased levels of Hsp90 [27].
Microglia, which plays a principal role of inflammation in brain [33], express high levels of Hsp90 following excitotoxic lesion in the mouse hippocampus [34]. The protective function of Hsp90 can be very important since inflammation evoked by microglia may increase the risk of PD. Recently, we have demonstrated that (-)-Epigallocatechin gallate EGCG, a major monomer of green tea polyphenols, is a potent inhibitor of microglial activation [35]. EGCG could directly bind to Hsp90 and stabilize the complex of Hsp90 [36]. Thus EGCG could be used to alleviate microglia-mediated dopaminergic neuronal injury in PD.
Hsp70
Auluck et al. [37] reported that application of Hsp70 can prevent dopaminergic neuronal loss in αSN transgenic Drosophila and interference with endogenous chaperone activity can accelerate αSN toxicity. Furthermore, Lewy bodies in human postmortem tissues were usually immunostained positive for molecular chaperones, suggesting that chaperones may play a role in PD progression [37].
It has been reported that Hsp70 can enhance parkin binding and ubiquitinating of expanded polyglutamine protein in vitro, suggesting that Hsp70 may help recruit misfolded proteins as substrates for parkin E3 ubiquitin ligase activity [38]. This finding provides a direct evidence to show the Hsp70 can promote the activity of E3 ligase to degrade aberrant αSN.
It is postulated that Hsp70 itself or cooperating with other factors can protect the neurons from cytotoxicity caused by aberrant proteins. The crosstalk between the Hsp70 and UPS may provide a clue for the intrinsic mechanism of protein aggregation and degradation. Moreover, Hsp70 exerts anti-apoptotic activity by blocking the function of several key proapoptotic factors [3]. Recently, several studies have demonstrated that Hsp70 may play a role in neuroprotection against rotenone-mediated apoptosis in human dopaminergic cell line SH-SY5Y in vitro and against MPTP-induced nigral injury in vivo by inhibiting the proapoptotic factors as well as activating the survival pathway [39, 40].
Small HSPs
Chaperone Hsp25/27(Hsp25 in mice and Hsp27 in humans), is an inhibitor of actin polymerization [41], which has been demonstrated to play a major role in actin filament dynamics in diverse cell types [3].
In human endothelial cells, inhibition of p38-MAPK activation can abolish Hsp27 phosphorylation, actin polymerization, and cell migration [42]. p38-MAPK may act as an upstream activator of stress-inducible Hsp25/27 phosphorylation. It has been demonstrated that Hsp27 could bind to the microtubule associated protein tau and lead to decreased level of hyperphosphorylated tau and therefore enhance cell survival in AD [43]. Another important function of Hsp27 is its protective effects on mitochondria pathway leading to inhibition of apoptosis [44]. It has been found that Hsp27 can block the tBID entering the mitochondria and reduce SMAC and Cytochrome C releasing from mitochondria so as to block the apoptotic process [3].
αB-crystallin Chaperone (Hsp22): Increased expression and abnormal aggregation of small HSPs αB-crystallin has been detected in Lewy bodies and reactive astrocytes in various neurodegenerative diseases [45]. Rekas et al. demonstrated that αB-crystallin was a potent inhibitor of αSN fibrillization in vitro [46]. αB-crystallin may redirect αSN from a fibril-formation pathway towards an amorphous aggregation pathway, thus reducing the amount of physiologically stable amyloid deposits in favor of easily degradable amorphous aggregates [46]. It has been reported that treatment with proteasomal inhibitors MG-132 or lactacystin in cultured rat brain oligodendrocytes can cause apoptotic cell death and induction of heat shock proteins in a time- and concentration-dependent manner [47]. Specifically in this study, αB-crystallin was up-regulated, and ubiquitinated proteins were accumulated. Meanwhile, the tau was dephosphorylated, which enhanced its microtubule-binding capacity [47]. These findings imply that αB-crystallin may work together with other HSPs, ubiquitin and microtubule associated proteins (MAPs) to cope with stressed conditions.
5. Potential Target for the Treatment of PD
Dong et al. [48] reported that Hsp70 gene transferred to dopaminergic neurons by a recombinant adeno-associated virus significantly protected the mouse against MPTP-induced nigral dopaminergic neuron loss and striatal dopamine levels decline [48]. Hsp70 attenuated MPTP induced apoptosis in the SNpc, and increased amphetamine-induced rotation [48]. Collectively, these results demonstrate that increasing chaperone activity may be beneficial for the treatment of PD.
HSPs may exert protective function through two major pathways besides their own chaperon activity: reducing mitochondrial dysfunction and oxidative stress, and preventing UPS impairment.
Anti-apoptotic effects of HSPs in PD
Mitochondrial dysfunction is probably the leading cause of increased oxidative stress and apoptosis in PD. Dopaminergic neurons are more vulnerable to oxidative stress than other neurons because of the special substrate dopamine [49]. In general, apoptotic process can be divided into the three phases: induction (or triggering), transduction of signal, and execution. Theoretically, HSPs may modulate any of these apoptotic phases to rescue the cells [3, 50]. In addition, it has been reported that stable expression of wild-type αB-crystallin protects cancer cells from caspase-3 activation in vitro, indicating that small HSPs αB-crystallin is a novel inhibitor of the activation of apoptosis [51]. (Figure 1) Other gene products linked to monogenic forms of PD also appear to be implicated in mitochondrial dysfunction. Parkin can interact with leucine-rich repeat kinase 2 (Lrrk2) which is part of the mitochondrial outer membrane [52, 53]. Thus Parkin may have an unexpected role in the regulation of normal mitochondrial function in an indirect way [54, 55].
The role of anti-apoptosis of families of HSPs. Most of them function as the inhibitor of the crucial molecule in the apoptosis pathway such as JNK, BAK, Cyto C, caspase-3 and et al. Meanwhile, Hsp27 and Hsp90 can also promote function of AKT to maintain the cell survival.
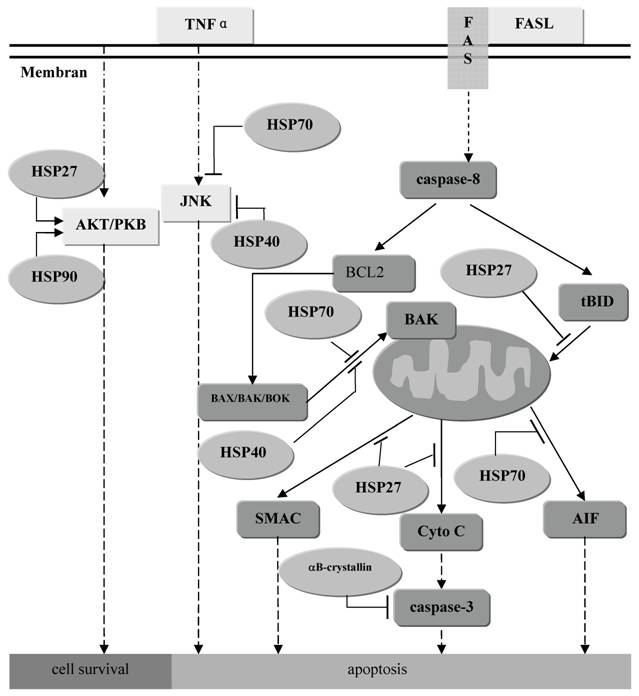
HSPs may promote the UPS in protein degradation
The UPS plays a pivotal role in the degradation of short-lived regulatory proteins which are components of cell cycle regulation, cell surface receptors, ion channels modulation, and antigen presentation [56]. (Figure 2) It is believed that once the disposal system fails to work, the substances, such as regulatory molecules p53, NFκB, and Bax that promote apoptosis, may accumulate to a high level that is harmful to the cell [57]. A hypothesis for the etiology of PD is that subsets of neurons are vulnerable to a failure in proteasome-mediated protein turnover [56]. Accumulation of ubiquitin conjugates has been reported in the pathologic lesions of many chronic neurodegenerative diseases, such as the neurofibrillary tangles in AD and brainstem Lewy bodies in PD [55, 56]. Inhibition of proteasome activity will sensitize dopaminergic neurons to protein alterations and oxidative stress [58].
The Ubiquitin-Proteasome System and the functions of PD associated genes. Parkin works as E3 ligase to help the aberrant protein be poly-ubiquitinated. And UCH-L1 helps hydrolyze the poly-Ub to Ub which can be reused in the next cycle. And when the 26S proteasome is inhibited, HSPs will be induced to compensate the function of protein degradation.
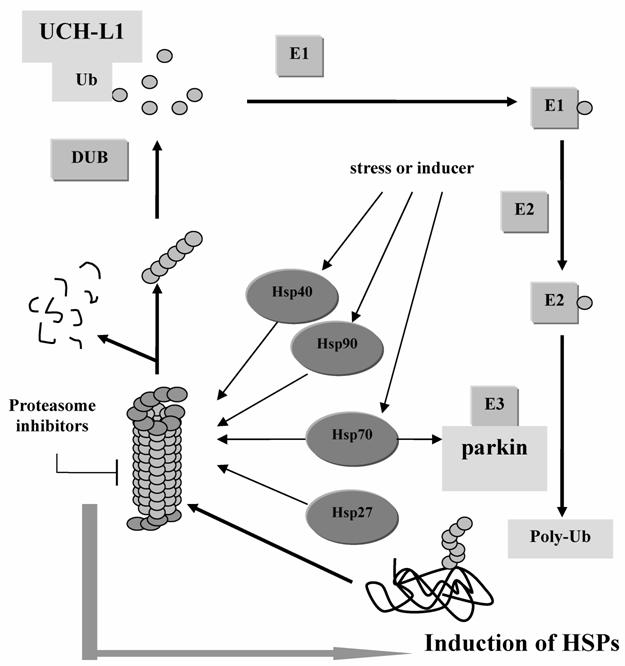
Hsp90, together with Hsc70, Hsp40 and 20S proteasome subunit are the effective components to capture firefly luciferase during thermal inactiveness and to prevent it from undergoing an irreversible off-pathway [59]. The 20S proteasome has been found in tight association with the molecular chaperone Hsp90 [60]. Composed within 26S proteasome subunit, they form a complex involved in a multitude of intracellular processes [56]. In addition, Kim et al has demonstrated that the inhibition of proteasome can increase the expression of Hsp27 and Hsp70 [61], implying that HSPs may act as compensation of UPS or work together to regulate the intracellular protein level. Robertson et al. supported the hypothesis by demonstrating that Hsp70 antisense oligomers enhanced proteasome inhibitor-induced apoptosis [62].
All evidences above implicate that HSPs and UPS are participants in keeping proteins folding correctly. They provide an effective protein quality control system that is essential for cellular functions and survival in many tissues. Dysfunction of these systems leads to protein aggregation and inclusion body formation in dopaminergic neurons.
HSPs inducers and their potential application in PD
It is proposed that up-regulation of protective factors may benefit our cells, but overload of some proteins may be a burden for cells or even cause cancer. So we need to find better way to keep cells in delicate balance with maximal protective effects and minimal side effects.
Cyclopentenone prostaglandin A1 (PGA1), an inducer of HSPs, has been shown to inhibit SH-SY5Y neuron apoptosis. PGA1 can protect against rotenone-induced neuronal degeneration by promoting the expression of HSPs as well as attenuating nuclear translocation of NF-kappaB and caspase-3 activation [63].
Geldanamycin (GA) binds to an ATP site on HSP90 and blocks its interaction with HSF1 to promote HSF1 activation [64]. GA also sensitizes the stress response within normal physiological parameters to enhance chaperone activation and offer protection against αSN neurotoxicity [65]. Furthermore, GA uncouples neuronal toxicity from Lewy body and Lewy neurite formation so that dopaminergic neurons are protected from the effects of αSN expression despite the continued presence of or even increased inclusion pathology [65]. Significantly, GA does not alter the basal level of HSP70, suggesting that GA acts only to elevate chaperone levels in stressed cells and does not alter chaperone activity in neighboring, healthier cells [65]. Because αSN expression leads to a local elevation of inducible HSP70 in dopaminergic neurons [37], these neurons should be preferentially targeted by GA treatment [65]. Its new derivative 17-Allylamino-17-demethoxygeldanamycin 17-AAG shares its important biological activities with less toxicity [28], which gives us a much bright perspective to use GA to induce specific HSPs expression and to attenuate the side effect.
There is feasibility to use Hsp70 as a pretreatment therapy because there are many nontoxic or low toxic Hsp70 inducers available, such as paeoniflorin [66], bimoclomol (co-inducer to increase the activity) [67, 68], radicicol, and valproic acid (VPA) [39]. These Hsp70 inducers can up-regulate Hsp70 effectively for reconfirmation of the cellular homeostasis. Thus, it is hope that modulates the stress response by inducers can be a promising target for treatment of PD.
6. Conclusion
HSPs have two main cellular functions aimed at promoting the UPS function and inhibiting the apoptotic activity. However, the detailed molecular mechanisms underlying their biological functions are still unclear. It is believed that HSPs, UPS, mitochondria and other organelles may work coordinately to keep the cell in a stable and well-operated state. HSPs are particularly important in PD and other neurodegenerative disorders because aberrant protein aggregation and neuron degeneration are the common pathophysiology of these disorders. Numerous studies in vitro and in vivo model of PD have demonstrated that increase in the expression of HSPs especially Hsp70 by gene transfer or HSPs inducers can reduce the aberrant protein misfolding and inhibit the proapoptotic pathway to attenuate dopaminergic neuron degeneration. Thus such intervention provides a promising therapy for PD. Advance in the research of HSPs targets will shed a light on the feasibility of clinical application of HSPs in PD. The future study will focus on finding the mechanisms of aberrant protein aggregation and searching for the selective HSPs inducers for the treatment of PD.
Acknowledgements
This review was in part supported by grants from Science and Technology Commission of Shanghai Municipality (03D214021 and 04DZ14902) and E-Institutes of Shanghai Municipal Education Commission (iE03003).
Conflict of interest
The author has declared that no conflict of interest exists.
References
1. Hartl F. Molecular chaperones in cellular protein folding. Nature. 1996 ;381(6583):571-9
2. Hightower LE. Heat shock, stress proteins, chaperones, and proteotoxicity. Cell. 1991 ;66(2):191-7
3. Benn SC, Woolf CJ. Adult neuron survival strategies—slamming on the brakes. Nat Rev Neurosci. 2004 ;5(9):686-700
4. Meriin AB, Sherman MY. Role of molecular chaperones in neurodegenerative disorders. Int J Hyperthermia. 2005 ;21(5):403-19
5. Berke SJ, Paulson HL. Protein aggregation and the ubiquitin proteasome pathway: gaining the upper hand on neurodegeneration. Curr Opin Genet Dev. 2003 ;13(3):253-61
6. Grunblatt E, Mandel S, Jacob-Hirsch J. et al. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J Neural Transm. 2004 ;111(12):1543-73
7. Zhang Y, James M, Middleton FA, Davis RL. Transcriptional analysis of multiple brain regions in Parkinson's disease supports the involvement of specific protein processing, energy metabolism, and signaling pathways, and suggests novel disease mechanisms. Am J Med Genet B Neuropsychiatr Genet. 2005 ;137(1):5-16
8. McLean PJ, Kawamata H, Shariff S. et al. TorsinA and heat shock proteins act as molecular chapersones: suppression of alpha-synuclein aggregation. J Neurochem. 2002 ;83(4):846-54
9. Dedmon MM, Christodoulou J, Wilson MR, Dobson CM. Heat shock protein 70 inhibits alpha-synuclein fibril formation via preferential binding to prefibrillar species. J Biol Chem. 2005 ;280(15):14733-40
10. Le W, Appel SH. Mutant genes responsible for Parkinson's disease. Curr Opin Pharmacol. 2004 ;4:79-84
11. Moore DJ, West AB Dawson VL, Dawson TM . Molecular pathophysiology of Parkinson's disease. Annu. Rev. Neurosci. 2005 ;28:57-87
12. McNaught KS, Perl DP, Brownell AL, Olanow CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson's disease. Ann Neurol. 2004 ;56(1):149-62
13. Wu YR, Wang CK, Chen CM. et al. Analysis of heat-shock protein 70 gene polymorphisms and the risk of Parkinson's disease. Hum Genet. 2004 ;114(3):236-41
14. Macario AJ, Conway de Macario E. Sick chaperones, cell stress, and disease. N Engl J Med. 2005 ;353(14):1489-501
15. Chen Q, Thorpe J, Keller JN. Alpha-synuclein alters proteasome function, protein synthesis, and stationary phase viability. J Biol Chem. 2005 ;280(34):30009-17
16. Tanaka Y, Engelender S, Igarashi S. et al. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001 ;10(9):919-26
17. Kluchen J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 reduces alpha-synuclein aggregation and toxicity. J Biol Chem. 2004 ;279(24):25497-502
18. Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004 ;305(5688):1292-5
19. Shimura H, Hattori N, Kubo S. et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000 ;25(3):302-5
20. Kim SJ, Sung JY, Um JW. et al. Parkin cleaves intracellular alpha-synuclein inclusions via the activation of calpain. J Biol Chem. 2003 ;278(43):41890-9
21. Kalia SK, Lee S, Smith PD, Liu L, Crocker SJ, Thorarinsdottir TE, Glover JR, Fon EA, Parkin DS, Lozano AM. BAG5 inhibits parkin and enhances dopaminergic neuron degeneration. Neuron. 2004 ;44:931-45
22. Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT Jr. The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility. Cell. 2002 ;111(2):209-18
23. Osaka H, Wang YL, Takada K. et al. Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum. Mol. Genet. 2003 ;12(16):1945-58
24. Ardley HC, Scott GB, Rose SA, Tan NG, Robinson PA. UCH-L1 aggresome formation in response to proteasome impairment indicates a role in inclusion formation in Parkinson's disease. J Neurochem. 2004 ;90(2):379-91
25. Li HM, Niki T, Taira T, Iguchi-Ariga SM, Ariga H. Association of DJ-1 with chaperones and enhanced association and colocalization with mitochondrial Hsp70 by oxidative stree. Free Radic Res. 2005 ;39(10):1091-9
26. Zhou Y, Gu G, Goodlett DR. et al. Analysis of alpha-synuclein-associated proteins by quantitative proteomics. J Biol Chem. 2004 ;279(37):39155-64
27. Uryu K, Richter-Landsberg C, Welch W. et al. Convergence of heat shock protein 90 with ubiquitin in filamentous alpha-synuclein inclusions of alpha-synucleinopathies. Am J Pathol. 2006 ;168(3):947-61
28. Waza M, Adachi H, Katsuno M. et al. Modulation of Hsp90 function in neurodegenerative disorders: a molecular-targeted therapy against disease-causing protein. J Mol Med. 2006 ;84(8):635-46
29. Bharadwaj S, Ali A, Ovsenek N. Multiple components of the HSP90 chaperone complex function in regulation of heat shock factor 1 in vivo. Mol Cell Biol. 1999 ;19(12):8033-41
30. Sabbah M, Radanyi C, Redeuilh G, Balieu E. The 90 kDA heat-shock protein (hsp90) modulates the binding of the estrogen receptor to its cognate DNA. Biochem J. 1996 ;314(pt 1):205-213
31. Zhang X, Clark AF, Yorio T. Heat shock protein 90 is an essential molecular chaperone for nuclear transport of glucocorticoid receptor beta. Invest Ophthalmol Vis Sci. 2006 ;47(2):700-8
32. Crookes WJ, Olsen LJ. The effects of chaperones and the influence of protein assembly on peroxisomal protein import. J Biol Chem. 1998 ;273(27):17236-42
33. Mor G, Nilsen J, Horvath T. et al. Estrogen and microglia: A regulatory system that affects the brain. J Neurobiol. 1999 ;40(4):484-96
34. Jeon GS, Park SW, Kim DW. et al. Glial expression of the 90-kDa heat shock protein (HSP90) and the 94-kDa glucose-regulated protein (GRP94) following an excitotoxic lesion in the mouse hippocampus. Glia. 2004 ;48(3):250-8
35. Li R, Huang YG, Fang D, Le WD. (-)-Epigallocatechin gallate inhibits lipopolysaccharide-induced microglial activation and protects against inflammation-mediated dopaminergic neuronal injury. J Neurosci Res. 2004 ;78(5):723-31
36. Palermo CM, Westlake CA, Gasiewicz TA. Epigallocatechin gallate inhibits aryl hydrocarbon receptor gene transcription through an indirect mechanism involving binding to a 90 kDa heat shock protein. Biochemistry. 2005 ;44(13):5041-52
37. Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002 ;295(5556):865-8
38. Tsai YC, Fishman PS, Thakor NV, Oyler GA. Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J Biol Chem. 2003 ;278(24):22044-55
39. Pan TH, Li XQ, Xie WJ, Jankovic J, Le WD. Valproic acid-mediated Hsp70 induction and anti-apoptotic neuroprotection in SH-SY5Y cells. FEBS Lett. 2005 ;579(30):6716-20
40. Shen HY, He JC, Wang Y, Huang QY, Chen JF. Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J Biol Chem. 2005 ;280(48):39962-9
41. Miron T, Vancompernolle K, Vandekerckhove J, Wilchek M, Geiger B. A 25-kD inhibitor of actin polymerization is a low molecular mass heat shock protein. J Cell Biol. 1991 ;114(2):255-61
42. Huot J, Houle F, Marceau F, Landry J. Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ Res. 1997 ;80(3):383-392
43. Shimura H, Miura-Shimura Y, Kosik KS. Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J Biol Chem. 2004 ;279(17):17957-62
44. Concannon CG, Gorman AM, Samali A. On the role of Hsp27 in regulating apoptosis. Apoptosis. 2003 ;8(1):61-70
45. Yun SJ, Hahm DH, Lee EH. Immobilization stress induces the expression of alphaB-crystallin in rat hippocampus: implications of glial activation in stress-mediated hippocampal degeneration. Neurosci Lett. 2002 ;324(1):45-8
46. Rekas A, Adda CG, Andrew Aquilina J. et al. Interaction of the molecular chaperone alphaB-crystallin with alpha-synuclein: effects on amyloid fibril formation and chaperone activity. J Mol Biol. 2004 ;340(5):1167-83
47. Goldbaum O, Richter-Landsberg C. Proteolytic stress causes heat shock protein induction, tau ubiquitination, and the recruitment of ubiquitin to tau-positive aggregates in oligodendrocytes in culture. J Neurosci. 2004 ;24(25):5748-57
48. Dong Z, Wolfer DP, Lipp HP, Bueler H. Hsp70 gene transfer by adeno-associated virus inhibits MPTP-induced nigrostriatal degeneration in the mouse model of Parkinson's disease. Mol Ther. 2005 ;11(1):80-8
49. Jenner P. Oxidative stress in Parkinson's disease. Ann Neurol. 2003 ;53(Suppl 3):S26-36
50. Beere HM, Green DR. Stress management - heat shock protein-70 and the regulation of apoptosis. Trends Cell Biol. 2001 ;11(1):6-10
51. Kamradt MC, Lu M, Werner ME. et al. The small heat shock protein alpha B-crystallin is a novel inhibitor of TRAIL-induced apoptosis that suppresses the activation of caspase-3. J Biol Chem. 2005 ;280(12):11059-66
52. Smith WW, Pei Z, Jiang H. et al. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci U S A. 2005 ;102(51):18676-81
53. West AB, Moore DJ, Biskup S. et al. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005Nov15 ;102(46):16842-7
54. Palacino JJ, Sagi D, Goldberg MS. et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004 ;279(18):18614-22
55. Winklhofer KF, Henn IH, Kay-Jackson PC, Heller U, Tatzelt J. Inactivation of parkin by oxidative stress and C-terminal truncations: a protective role of molecular chaperones. J Biol Chem. 2003 ;278(47):47199-208
56. Schwartz AL. The Ubiquitin-proteasome pathway and pathogenesis of human disease. Annu Rev Med. 1999 ;50:57-74
57. Hernandez F, Diaz-Hernandez M, Avila J, Lucas JJ. Testing the ubiquitin-proteasome hypothesis of neurodegeneration in vivo. Trends Neurosci. 2004 ;27(2):66-9
58. Mytilineou C, McNaught KS, Shashidharan P. et al. Inhibition of proteasome activity sensitizes dopamine neurons to protein alterations and oxidative stress. J Neural Transm. 2004 ;111(10-11):1237-51
59. Minami Y, Kawasaki H, Minami M. et al. A critical role for the proteasome activator PA28 in the Hsp90-dependent protein refolding. J Biol Chem. 2000 ;275(12):9055-61
60. Whittier JE, Xiong Y, Rechsteiner MC, Squier TC. Hsp90 enhances degradation of oxidized calmodulin by the 20 S proteasome. J Biol Chem. 2004 ;279(44):46135-42
61. Kim D, Kim SH, Li GC. Proteasome inhibitors MG132 and lactacystin hyperphosphorylate HSF1 and induce hsp70 and hsp27 expression. Biochem Biophys Res Commun. 1999 ;254(1):264-8
62. Robertson JD, Datta K, Biswal SS, Kehrer JP. Heat-shock protein 70 antisense oligomers enhance proteasome inhibitor-induced apoptosis. Biochem J. 1999 ;344(2):477-85
63. Wang X, Qin ZH, Leng Y. et al. Prostaglandin A1 inhibits rotenone-induced apoptosis in SH-SY5Y cells. J Neurochem. 2002 ;83(5):1094-102
64. Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998 ;94(4):471-80
65. Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Mechanisms of Suppression of {alpha}-Synuclein Neurotoxicity by Geldanamycin in Drosophila. J Biol Chem. 2005 ;280(4):2873-8
66. Yan D, Saito K, Ohmi Y. et al. A novel heat shock protein-inducing compound. Cell Stress Chaperones. 2004 ;9(4):378-89
67. Nanasi PP, Jednakovits A. Multilateral in vivo and in vitro protective effects of the novel heat shock protein coinducer, bimoclomol: results of preclinical studies. Cardiovasc Drug Rev. 2001 ;19(2):133-51
68. Hargitai J, Lewis H, Boros I. et al. A heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1. Biochem Biophys Res Commun. 2003 ;307(3):689-95
Author contact
![]() Correspondence to: Dr. Weidong Le, Professor and Principal Investigator, Institute of Health Sciences, Shanghai Institutes of Biological Sciences, Chinese Academy of Sciences, 225# South Chongqing Road, 200025, Shanghai, China. Tel: +86-021-64370045-611201, Fax: 001-713-798-9307, Email: Weidongltmc.edu
Correspondence to: Dr. Weidong Le, Professor and Principal Investigator, Institute of Health Sciences, Shanghai Institutes of Biological Sciences, Chinese Academy of Sciences, 225# South Chongqing Road, 200025, Shanghai, China. Tel: +86-021-64370045-611201, Fax: 001-713-798-9307, Email: Weidongltmc.edu