International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(1):64-70. doi:10.7150/ijbs.3.64 This issue Cite
Research Paper
Urokinase Separation from Cell Culture Broth of a Human Kidney Cell Line
Vibha Bansal1, Pradip K. Roychoudhury1, Ashok Kumar2 ![]()
1. Department of Biochemical Engineering and Biotechnology, Indian Institute of Technology, Hauz-Khas, New Delhi-110016, INDIA
2. Department of Biological Sciences and Bioengineering, Indian Institute of Technology, Kanpur-208016, INDIA
Received 2006-9-25; Accepted 2006-11-15; Published 2006-11-22
Citation:
Bansal V, Roychoudhury PK, Kumar A. Urokinase Separation from Cell Culture Broth of a Human Kidney Cell Line. Int J Biol Sci 2007; 3(1):64-70. doi:10.7150/ijbs.3.64. https://www.ijbs.com/v03p0064.htm
Other stylesAbstract
A single step ion-exchange chromatography on a sulfo-propyl (SP)- Sepharose column was performed to separate both the high molecular weight (HMW)- and low molecular weight (LMW)- forms of enzymatically active urokinase type plasminogen activator from human kidney (HT1080) cell culture media. The level of urokinase secreted by the cell line reached to about 145 Plough units/ml culture broth within 48 h of cultivation. The conditioned cell culture media was applied directly to the column without any prior concentration steps. Polyacrylamide gel electrophoresis of the column eluates in the presence of sodium dodecyl sulphate showed that the cell line secretes three forms of two-chain high molecular weight (HMW) urokinase of molecular weights (Mr) 64,000, 60,900 and 55,000. In addition, two low molecular weight (LMW) forms of Mr 22,000 and 20,000; proteolytic cleavage products of HMW, were also found. The HMW and LMW forms had intrinsic plasminogen dependent proteolytic activity as judged by zymographic analysis. The specific activity of the pooled peak fractions increased (approximately 93-fold) to values as high as 1481 Plough units/ mg protein. Both HMW as well as LMW forms were obtained in significantly high yields.
Keywords: Urokinase, Kidney cell culture (HT-1080), SP-Sepharose, Ion-exchange chromatography, Tissue plasminogen activator, Single step purification
1. Introduction
Urokinase (UK; EC 3.4.21.73), a plasminogen activating proteolytic enzyme, was first described in 1951 as occurring in trace quantities in mammalian urine [1]. The urokinase detected in urine is produced by kidney cells. It has been reported that the urokinase secreted by kidney cells is antigenically similar to the one isolated from urine [2]. Urokinase catalyzes the conversion of plasminogen to plasmin by cleaving the Arg-Val linkage in the Pro-Gly-Arg-Val sequence of the former. As the resulting plasmin dissolves clots of fibrin in blood vessels, urokinase is intravenously administered for treatment of thromboembolic disease [3]. There are two kinds of this enzyme, the low molecular weight (LMW-) and the high molecular weight (HMW-) urokinase. The former being an autocatalytic fragmentation product of the latter [4] and both these forms are glycoproteins [5].
All characterized commercial preparations of urokinase contain one or both of these enzymatically active forms. The major source of commercially available urokinase is human urine [6]. However, extremely low concentrations of urokinase (10-15 μg/ml) in human urine create a major problem in isolation and purification of this enzyme [7]. Human kidney cells cultured in vitro, are a recognized source of urokinase. These cells are known to secrete biologically active and heavily glycosylated urokinase in the culture medium [2]. The yields of this enzyme are relatively low. In order to enhance the urokinase production by these cells it is required to design operational strategies and perform media optimizations for the growth of the cells and enzyme production. Moreover, urokinase production per unit confluent area and per unit time must be increased. This potential for increased secretion of urokinase by cells is an important consideration for using cell culture for urokinase production. Attempts are being made to have an insight for separation of urokinase from cell culture broth. In view of this therapeutic importance of obtaining a plasminogen activator from a human source, we have been working on the urokinase production using human kidney cell line HT1080. The cell lines, however, must be fed with a complex growth medium. This need for nutrients such as sugars, amino acids and growth factors, is supplied from preparations of animal serum in the growth medium.
The separation strategies devised till date involve a series of concentration steps followed by conventional chromatographic separations [8]. In one such reported work, Hou and Zaniewski [6] purified urinary urokinase by an SP cation exchanger followed by a zinc-chelated affinity chromatographic cartridge. This report demonstrated the combined use of a cation exchanger with zinc-chelated chromatographic cartridges in purifying urokinase on a relatively large scale with an 18-fold increase in urokinase specific activity and approximately 70 % yield. Multi-step chromatographic methods have also been reported for the separation of urokinase from conditioned cell culture media of different human cell lines [9, 10]. Similarities in the structures of known synthetic substrates and inhibitors of urokinase have led to the formulation of suitable competitive inhibitors for affinity chromatography ligands such as BAPA (α-benzylsulfonyl-p-aminophenylalanine) [11], aminobenzamidine [12], homoarginine benzyl ester [13] or agmatine [14, 15] coupled to a gel matrix. However, these adsorbents do not exhibit specificity to yield highly purified urokinase. Immunoaffinity chromatography offers a powerful approach for the separation of urokinase from cell culture broth [16]. However, because of the high costs and low operational stability of the antibody columns, such approach suffers some limitations. Most of the isolation methods for urokinase use multi-step approach which lead to lower yields coupled with high capital and operating costs, thus making cost-effective and high-fold purification of urokinase from crude sources a major challenge. Thus the new developments for efficient and cost effective separation strategies for urokinsae are highly desired.
The present work describes a single-step purification process for the separation of urokinase from the conditioned media obtained from the human kidney (HT1080) cell cultures.
2. Materials and Methods
Materials
Urokinase from human kidney cells (activity 0.37 Sigma units/mg protein), plasminogen (bovine plasma) and urokinase substrate (pyro-Glu-Gly-Arg-p-nitroanilide-HCl) were obtained from Sigma (St Louis, MO, USA). Casein for zymography was purchased from ICN Biomedicals Inc. (Aurora, USA). Medium range protein molecular weight marker was bought from Bangalore Genei Private Ltd. (Banglore, India). Centriplus YM-30 and Centriplus YM-10 were purchased from Amicon.
Cell culture medium, Dulbecco's Modified Eagles medium (DMEM) and New Born Calf Serum (NBCS) were from Life Technologies (Gibco BRL). Sulfo Propyl (SP)-Sepharose® Fast Flow from Pharmacia Fine Chemicals (Uppsala, Sweden) was a kind gift from Dr. Panda (NII, India). All other chemicals were of analytical reagent grade and used without further purification.
Cell line
Human kidney cell line HT1080 (NCCS, Pune, India) was cultured in DMEM containing 10 % NBCS. Conditioned media samples were withdrawn from the T-flasks at regular intervals to monitor urokinase production. The media withdrawn between 48 and 72 hours of cell cultivation was pooled and used as the source material for urokinase purification.
Ammonium sulphate precipitation
Powdered ammonium sulphate was added (in small aliquots) to the conditioned media kept in ice to approximately 50 % saturation with constant stirring. The precipitate was collected by centrifugation at 8,000 g for 30 min. The pellet was then resuspended in 500 μl of 0.05 M Tris-HCl buffer, pH 8.0. Resuspended pellet samples were dialyzed overnight against the suspension buffer (50 mM Tris-HCl, pH 8.0) and assayed for protein concentration and urokinase activity.
Centrifugal filtration
Centrifugal filtration modules, Centriplus YM-30 and Centriplus YM-10 were used to concentrate the urokinase activity in conditioned cell culture media. The conditioned media was filtered through these modules at 4,000 g for 1 hour at 4ºC.
Ion-exchange chromatography
SP-Sepharose (3.0 ml) was washed in 0.5 M NaOH according to the manufacturer's instructions. Thereafter, the gel was washed with distilled water and packed in a 1.0 x 10-cm column (Pharmacia Fine Chemicals) to a length of about 3.0 cm. The gel was then equilibrated with 50 mM sodium acetate buffer, pH 4.5 containing 5 mM EDTA, at a flow rate of 0.1 ml/min. Three milliliters of the cell culture broth, predialysed against equilibration buffer were applied to the column. The column was then washed using five column volumes of pre-elution buffer (0.2 M NaCl, 50 mM sodium acetate, 5mM EDTA, pH 4.5) to remove unbound or loosely proteins. The bound urokinase was subsequently eluted with 40 ml of linear gradient of 0.2 M – 2.0 M NaCl in 50 mM sodium acetate buffer, pH 4.5 containing 5 mM EDTA at a flow rate of 0.1 ml/min. Fractions of 1.0 ml each were collected and those containing the protein peak were assayed for urokinase activity and desalted by dialysis.
Analytical methods
Enzyme assay: Amidolytic activity of urokinase was determined by the method of Tait et al. [17], using pyro-Glu-Gly-Arg-p-nitroanilide-HCl as urokinase substrate. The activity units were expressed as Plough units/mg protein and were determined by using urokinase from human kidney cells (0.37 Sigma units/mg protein; 1 Sigma unit = 75 000 Plough units) as standard. One Sigma unit corresponds to the amount of enzyme that hydrolyzes 1 μmol pyro-Glu-Gly-Arg-p-nitroanilide-HCl per minute at 37 ºC and at pH 8.8.
Protein measurements: The amount of protein was determined by Folin´s method according to Lowry et al. [18], using bovine serum albumin (BSA) as a standard.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE): The electrophoresis was performed according to the method of Laemmli [19], using 4-10 % gradient gel. Reduction of protein was accomplished with 10 % β-mercaptoethanol (v/v) in the sample buffer. Gels were stained with 0.1 % Coomasie blue in 50 % methanol and 10 % acetic acid for 1 hour followed by destaining in 7 % methanol and 7 % acetic acid till the background reduced to the extent of convenient visibility of the protein bands on the gel. The molecular weights of samples were determined by SDS-PAGE using medium range protein molecular weight marker (PMW-M).
Zymography: The Zymography for urokinase activity was performed by modification of the method by Heussen and Dowdle [20]. Samples (mixed with sample buffer containing 10 % SDS and 40 % glycerol (v/v) but without β-mercaptoethanol) were loaded on to 10 % SDS-PAGE gels containing 0.1 % casein with 7.5 μg/ml of plasminogen at 4ºC. The gels were then treated with 100 ml of 50 mM Tris-HCl (pH 7.5) containing 2.5 % Triton X-100 for 1 hour at room temperature with gentle shaking. The gels were then washed in 100 ml deionized water for 10 min, and incubated in 0.1 M glycine-NaOH (pH 8.3) overnight at 37ºC with gentle shaking. Finally, these were stained with Coomasie brilliant blue. Urokinase (Sigma) was used as a positive control.
3. Results
Concentration of urokinase activity in starting material
Human kidney cell line HT 1080 was cultured in DMEM containing 10% NBCS. The cells adhered to the tissue culture dish and full confluence was obtained within 48 h of cell cultivation (Fig. 1). The conditioned media samples withdrawn at regular intervals were assayed for urokinase activity. The cells secreted about 145 PU/ml culture broth at 48 h of cell cultivation (Fig. 2). The slight decreases in activity observed latter are because of the secretion of urokinase inhibitors by the cell line.
The specific activity of urokinase in the resuspended and subsequently dialyzed pellet obtained from 50 % ammonium sulphate saturation of the conditioned medium from cell culture showed an approximately 4-fold increase. However, the recovery of urokinase was only about 20 %.
Centrifugal filtration was then tried as an alternative to ammonium sulphate precipitation. The conditioned cell culture media was concentrated by means of centrifugal filtration through 10 kD and 30 kD cut off centriplus modules. Urokinase activity was detected in both the retentate as well as permeate. The centriplus was consequently treated with BSA prior to the filtration process. However, no considerable increase in recovery could be achieved. Thus both ammonium sulphate precipitation and ultrafiltration were not successful for the concentration of the enzyme either because of the low recoveries or loss of the enzymatic activity.
The dialyzed conditioned media from the cell cultures was finally applied directly to the ion-exchange column, without any prior concentration step, to eliminate any possible loss in enzymatic activity.
Figure 1
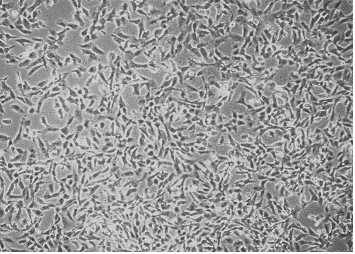
Growth of human kidney (HT 1080) cell line in a T-flask in DMEM containing 10 % NBCS.
Figure 2
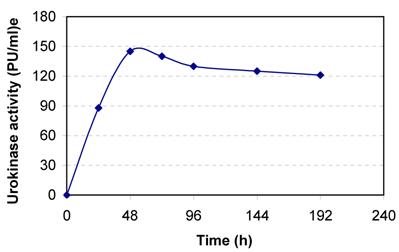
Secretion of urokinase by human kidney (HT 1080) cell line. The cells are grown in 25cm2 T-flask in DMEM containing 10 % NBCS.
Ion-exchange chromatography
BSA is one of the main constituents of conditioned media and must be removed. The pI of BSA is 4.7, compared to a pI in the range of 7-9 for urokinase [6]. Based on this difference in pI, SP-Sepharose cation exchanger was used for binding and elution under acidic conditions. The use of SP-Sepharose ion-exchange column led to as high as 93-fold purification of urokinase (Table 1).
Table 1
Separation of urokinase from conditioned media of human kidney (HT1080) cell culture
| Fraction | Vol. (ml) | Total protein (mg) | Urokinase activity (PU/ml) | Total urokinase (PU) | Specific activity (PU/mg) | Fold purification | Yield (%) |
|---|---|---|---|---|---|---|---|
| Conditioned cell culture media load | 3.0 | 26.25 | 140 | 420 | 16 | 1 | 100 |
| Breakthrough + wash | 30.0 | 24.06 | 4.92 | 147.67 | 2.16 | 0.13 | 35 |
| Eluate fractions | |||||||
| P1* | 4.0 | 1.97 | 26 | 103.66 | 52.62 | 3.3 | 25 |
| P2** | 3.0 | 0.21 | 104 | 311 | 1481 | 92.56 | 74 |
* Peak 1 pooled fractions
** Peak 2 pooled fractions
Urokinase activity of 420 Plough units (PU) contained in 3.0 ml conditioned media from cell culture was applied to SP-Sepharose matrix packed in a glass column. Subsequently, the column was washed with pre-elution buffer to remove nonspecifically bound material. Most of the protein passed through the column unabsorbed. Approximately 35 % of the original urokinase activity appeared in the flow through and washing. Urokinase bound to the column was then eluted by the application of linear sodium chloride gradient (0.2 – 2. 0 M NaCl). Two protein peaks were observed in the resulting elution profile. When the fractions constituting the protein peaks were analyzed for urokinase activity, major enzyme activity was observed in peak (P2). (Fig. 3, Table 1). The fractions constituting each of the peaks were pooled independently. In the first protein peak (P1) pool comprising fractions 6-9, about 1.97 mg of the applied protein constituting 103.66 PU of urokinase activity was recovered. This constituted 25.0 % of the enzyme activity loaded and resulted in a 3.3-fold increase in specific enzyme activity. In the second pool (P2) comprising fractions 13-15, 0.064 mg protein constituting about 311 PU was recovered. This constituted 74.0 % of the total activity loaded and resulted in about 93-fold increase in urokinase specific activity in a single step. The productivity and purification of the enzyme thus seems to be suitable for commercial production. However, it may be useful to consider the production of the enzyme on the large scale and further separation of LMW and HMW urokinase may be desirable.
Figure 3
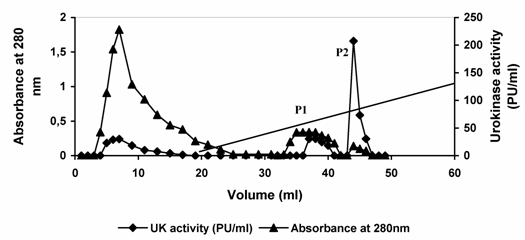
Elution profile of urokinase type-plasminogen activator from sulfo-propyl Sepharose. Dialyzed conditioned culture media from HT1080 cells was applied to a column of SP-Sepharose equilibrated in 0.05 M acetate buffer, 5mM EDTA (pH 4.5). After collecting the flow-through of the column, the column was washed with pre-elution buffer. The bound enzyme was eluted by applying 40 ml of linear gradient of 0.2 M – 2.0 M NaCl in 50 mM sodium acetate buffer, pH 4.5 containing 5 mM EDTA at a flow rate of 0.1 ml/min. Diagonal line between 20 ml to 60 ml in the graph is the start and end of the elution gradient. Fractions of 1 ml were collected and assayed for protein concentration and enzyme activity). P1 and P2 refer to eluted protein peaks 1 and 2, respectively. For more details see materials and methods.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
The pooled fractions P1 and P2 constituting the chromatographic peaks 1 and 2 respectively, were analyzed by SDS-PAGE under reducing (Fig. 4) and non-reducing conditions (picture not shown). The pooled fractions P1 (Fig. 4, lane 3) showed presence of multiple bands on SDS-PAGE. The P2 pool showed fewer bands (Fig. 4, lane 4), as also reflected in the higher specific activity of urokinase so obtained by amidolytic assay. This lane showed a significant reduction in the intensity of serum albumin band that was distinctly visible in the band pattern obtained with the starting material on the gel (Fig. 4, lane 5). Here, the triplet bands of Mr 64,400, 60,900 and 55,000 were not distinct and appeared faint. The two bands of Mr 22,000 and 20,000 seen in lane 3 were not observed for P2 pool. Under non-reducing conditions, both P1 and P 2 pooled fractions showed the presence of an extra band of Mr 97,000.
The urokinase (Sigma, U5004) shown in lane 2 (Fig. 4) was used as the reference sample for comparison with ion-exchange column eluates. Since Sigma specifications indicate 90-95 % of the total protein in the vial to be human albumin, therefore the major band (Mr 66,000) corresponded to albumin, while the minor band (Mr 55,000) was that of HMW urokinase. The LMW urokinase bands could not be visualized.
Zymography
To test the plasminogen activating potential of the urokinase eluted in the two peak pools, zymographic analysis of both the peak materials was performed (Fig. 5) using reference urokinase as the positive control. The P1 pool failed to yield any detectable zones of lysis on the zymogram (Fig. 5, lane 2). This may be attributed to the extremely low specific activity of urokinase P1 eluate. However, the P2 pooled fractions (Fig. 5, lane 1) produced discrete triplet bands of lysis in the Mr range of 64,000-55,000, which corresponded accurately to the triplet bands observed in the SDS-PAGE pattern. Two clear zones of Mr 22,000 and 20,000 were observed confirming the presence of plasminogen activating potential in the polypeptides eluted in P2 pool. Thus, the activity of urokinase was found to be well preserved in 1.0 % SDS. Clear bands of lysis failed to appear on a gel containing casein but no plasminogen thus ruling out the presence of any directly caseinolytic protein in the column eluates. Although the Mr 22,000 and 20,000 bands were not observed on SDS-PAGE for either reference urokinase or the P2 pool, clear zones corresponding to LMW urokinase, appeared for both in the zymogram. This indicated a lower concentration of LMW as compared to HMW in the preparations but higher plasminogen activating capability. Thus, the clear zones produced by peak (P2) pool yielded a pattern almost identical to that obtained with the reference urokinase (Fig. 5, lane 3).
Figure 4
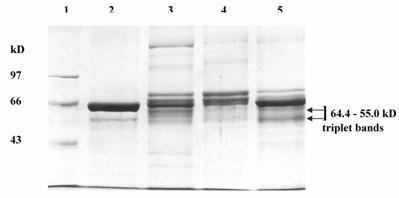
SDS-PAGE of pooled peak fractions eluted from the sulfo-propyl Sepharose column. The electrophoresis was carried out at 20 mA. The protein samples (15 μg protein in each) were applied in each lane. Lanes: (1) molecular weight protein marker (M); (2) urokinase (Sigma); (3) protein peak 1 pool (P1); (4) protein peak 2 pool (P2); and (5) conditioned media from HT1080 cell culture.
4. Discussion
Despite the immense clinical application of urokinase, no satisfactory cost efficient methods have yet been described for its purification from cell cultures. Conditioned cell culture medium contains relatively higher concentrations of urokinase (65 μg/liter) [9], as compared to urine (10-15 μg/liter). Hence, it offers a more enriched source for urokinase separation.
Figure 5

Zymogram of pooled peak fractions eluted from the sulfo-propyl Sepharose column. Protein (20 μg) was loaded in each well and electrophoresis carried out at 20 mA at 4 ºC. Lanes: (1) protein peak 2 pool (P2); (2) protein peak 1 pool (P1); and (3) urokinase (Sigma). Clear zones of lysis indicate bands of urokinase.
In this study, conventional methods were initially tried for concentration and partial purification of the enzyme, like ammonium sulphate precipitation or centrifugal filtration, however, these methods were not successful for the purpose. We have subsequently been able to achieve highly purified urokinase (1481 PU/mg protein) directly from the conditioned media of HT1080 cell cultures in a single ion-exchange chromatography step. The total enzyme activity mass balance obtained were surprisingly high (>100 %, together in breakthrough and eluate). This could be due to the presence of some enzyme-inhibitor complexes in the conditioned cell culture media being used as the starting material. Subsequently, a variation in pH or exposure to some nucleophilic agent during purification process might have led to the partial dissociation of inhibitor from urokinase. Consequently the percentage of free and active urokinase in the eluate increased. This accounts for nearly 100 % recovery from the ion-exchange column. Wun et al. [21] have also reported that HT-1080 cell cultures allow enhanced secretion of plasminogen activator inhibitor-1 (PAI-1), which forms enzymatically inactive urokinase-PAI-1 complexes with the urokinase. It has also been reported previously that a urokinase-related protein with M.W. 95000, representing a complex of two-chain urokinase with an inhibitor accounts for about 70 % of the total urokinase-related antigen in urine [9]. Nucleophilic agents dissociate the complex into active two-chain (tc) urokinase and a protein with M.W. 54,000. This highly purified UK was obtained with a yield of 5 μg/l of urine. It was concluded that UK-related proteins in human urine consist of about 25% of single chain (sc)-urokinase (10-20 μg/l) and of about 75% tc-urokinase (40-50 μg/l), the bulk of which is complexed to an inhibitor.
SDS-PAGE analysis of the eluates from ion-exchange column revealed urokinase bands in both the high (55-64 kD) as well as low (22-20 kD) molecular weight ranges (as confirmed by zymography). The Mr 64,000, 60,900 and 55,000 bands are apparently glycosylation variants of HMW urokinase as cultured cells are known to secrete differently glycosylated urokinase in the media [22]. The presence of these bands is in agreement with the findings of previous researchers wherein different HMW forms of urokinase (58.5±5.5 kD) have been reported. These forms of urokinase, upon reduction, produced two bands of Mr 22.0±3.5 and 20.0±3.5 as reported earlier [23 - 25]. This explains the presence of the 22,000 and 20,000 LMW bands, which are probably cleavage products of the HMW forms of urokinase. This is probably also the reason that urokinase activity was detected in the permeate in centrifugal filtration using 10 kD or 30 kD cut-off filters whereas the cleavage products of HMW forms of urokinase can be permeable.
The HMW forms of urokinase are initially secreted in the media as single-chain forms. Autocatalytic activity of urokinase and other proteolytic activities present in the serum result in cleavage of the single chain HMW urokinase. This leads to the formation of two-chain HMW form in which two polypeptide chains are held together by disulphide bridges and also the LMW form [26]. However, addition of aprotinin, a proteolytic inhibitor, in the serum-supplemented media, did not lead to any change in urokinase composition of the conditioned media. Both LMW as well as HMW urokinase activities could still be detected in the media (data not shown). This observation suggested the presence of aprotinin-resistant proteolytic activities in the media. Andreasen et al. [27] have previously reported similar findings.
The presence of a Mr 97,000 band in the native PAGE (absent in SDS-PAGE) is in agreement with the minor 95,000 form of urokinase reported by Booyse et al. [10]. Disappearance of this band from SDS-PAGE suggests it to be an enzyme-inhibitor complex, which could be dissociated into Mr 55,000 urokinase and a 46,000-inhibitor protein on treatment with nucleophilic agents. Wun et al. [21], also showed that SDS treatment caused partial inactivation of fully or moderately active PAI-1 [7, 10].
Prior to the ion-exchange chromatography step, attempts were made to concentrate urokinase activity in the conditioned media. For this purpose, classical techniques of ammonium sulfate precipitation, ethanol precipitation and centrifugal filtration were tried. However, neither of the precipitation techniques could successfully precipitate urokinase from the conditioned media into a single pellet. Centrifugal filtration also failed to yield any appreciable results. Appearance of activity in retentate as well as permeate was due to the presence of active forms of urokinase over a wide Mr range that was later confirmed through SDS-PAGE and zymography. The activities in the permeate and retentate, on being pooled together could not account for the total activity initially loaded on to the centriplus. This implied that urokinase in the medium (approximately 25 %) was somehow binding to the polysulfone membranes. Similar losses were also reported by others [28]. The binding of urokinase to polysulfone membrane might be attributed to the glycoprotein nature of urokinase thus leading to increased hydrophobic interaction between protein and the membrane. Even using BSA pretreated filtration modules or increasing the ionic strength of the starting material could not eliminate these losses. A detailed study of the glycosylation patterns of the purified urokinase can lead to a better understanding of the losses incurred during centrifugal filtration.
It can be concluded that HT1080 cells secrete three forms of HMW urokinase (Mr 64,000, 60,900 and 55,000) that are expected to be glycosylated to different degrees. Some proteolytic activity in the media, in addition to the autocatalytic activity of HMW urokinase, leads to fragmentation of the HMW form. This results in the generation of a LMW form in the range of Mr 22000 to 20, 000. All the forms so observed have been found to be enzymatically active.
Also, as can be seen in the SDS-PAGE and zymography results here, the LMW and HMW forms of urokinase have eluted together into the same fraction. It has been suggested previously that it might be impossible to separate completely the HMW urokinase from the LMW form [29]. The LMW and HMW forms of urokinase are known to have different efficiencies as regards their plasminogen activating roles. Experiments are being conducted to achieve the separation of these different forms of urokinase, thus ensuring more homogeneous preparations of purified urokinase. This will be communicated shortly.
Acknowledgements
The authors gratefully acknowledge the financial support from Department of Biotechnology and Council of Scientific and Industrial Research, India and Swedish Research Council/SIDA, Sweden- research link project.
Conflict of interest
Declared none.
References
1. Christman JK, Acs G. Purification and characterization of a cellular fibrinolytic factor associated with oncogenic transformation: The plasminogen activator from SV-40-transformed hamster cells. Biochim. Biophys. Acta. 1974 ;340:339-347
2. Bernik MB, Kwaan HC. Plasminogen activator activity in cultures from human tissues. An immunological and histochemical study. J Clin Invest. 1969 ;48:1740-1753
3. Lopez-Sendon J, de Lopez SE, Bobadilla JF, Rubio R, Bermejo J, Delcan JL. Cardiovascular pharmacology (XIII). The efficacy of different thrombolytic drugs in the treatment of acute myocardial infarct. Rev Esp Cardiol. 1995 ;48:407-439
4. White WF, Barlow GH, Mozen MM. The isolation and characterization of plasminogen activators (urokinase) from human urine. Biochemistry. 1966 ;5:2160-2169
5. McLellan WL, Vetterlein D, Roblin R. The glycoprotein nature of human plasminogen activators. FEBS Lett. 1980 ;115:181-184
6. Hou KC, Zaniewski R. Purification of urokinase by combined cation exchanger and affinity chromatographic cartridges. J. Chromatogr. B. 1990 ;525:297-306
7. Stump DC, Thienpont M, Collen D. Urokinase- related proteins in human urine. J. Biol. Chem. 1986 ;261:1267-1273
8. Herion P, Bollen A. Purification of urokinase by monoclonal antibody affinity chromatography. Biosci. Rep. 1983 ;3:373-379
9. Stump DC, Lijnen HR, Collen D. Purification and characterization of single-chain urokinase type plasminogen activator from human cell cultures. J. Biol. Chem. 1986 ;26:1274-1278
10. Booyse FM, Scheinbuks J, Lin PH, Traylor M, Bruce R. Isolation and interrelationships of the multiple molecular tissue-type and urokinase-type plasminogen activator forms produced by cultured human umbilical vein endothelial cells. J. Biol. Chem. 1988 ;263:15129-15138
11. Maciag T, Weibel MK, Pye EK. Urokinase. Jakoby WB, and Wilchek M, eds. Methods Enzymol New York: Plenum 1974: 451-459
12. Holmberg L, Bladh B, Astedt B. Purification of urokinase by affinity chromatography. Biochim. Biophys. Acta. 1976 ;445:215-222
13. Tamura Y, Fujii S. Purification of human plasma kallikrein and urokinase by affinity chromatography. J. Biochem. 1976 ;80:507-511
14. Soberano ME, Ong EB, Johnson AJ, Levy M, Schoellmann G. Purification and characterization of two forms of urokinase. Biochim. Biophys. Acta. 1976 ;445:763-773
15. Astedt B, Holmberg L, Wagner G, Richter P, Plough J. Purification of urokinase by a beta-naphthamidine affinity column. Thromb. Haemostasis. 1979 ;42:924-928
16. Vetterlein D, Calton GJ. Purification of urokinase from complex mixtures using immobilized monoclonal antibody against urokinase light chain. Thromb. Haemostasis. 1983 ;49:24-27
17. Tait JF, Engelhardt S, Smith C, Fujikawa K. Prourokinase-annexin V chimeras. J. Biol. Chem. 1995 ;37:21594-21599
18. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951 ;193:265-275
19. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970 ;227:680-685
20. Heussen C, Dowdle EB. Electrophoretic analysis of plasminogen activators in polyacrylamide gels containing sodium dodecyl sulfate and copolymerized substrates. Anal. Biochem. 1980 ;102:196-202
21. Wun TC, Palmier MO, Siegel NR, Smith CE. Affinity purification of active plasminogen activator inhibitor-1 (PAI-1) using immobilized anhydrourokinase. J Biol Chem. 1989 ;264:7862-7868
22. Rasmussen JR. Glycosylation of recombinant proteins. Biol. Carbohydrates. 1991 ;3:179-285
23. Reagan ME, Robb M, Bornstein I, Niday E G. Immunoaffinity purification of tissue plasminogen activator from serum-supplemented conditioned media using monoclonal antibody. Thromb. Res. 1985 ;40:1-9
24. Park K-W, Choi S-H, Song X-X, Funahashi H, Niwa K. Production of plasminogen activators (PAs) in bovine cumulus-oocyte complexes during maturation in vitro: effects of epidermal growth factor on production of PAs in oocyte and cumulus cells. Biol. Reprod. 1999 ;61:298-304
25. Husain SS, Gurewich V, Lipinski B. Purification and partial characterization of a single-chain high molecular-weight form of urokinase from human urine. Arch. Biochem. Biophys. 1983 ;220:31-38
26. Astedt B. Purification and characterization of human vascular plasminogen activator. Biochim Biophys. Acta. 1980 ;621:241-254
27. Andreasen PA, Neilsen LS, Grondahl-Hansen J, Skriver L, Zeuthen J, Stephens RW, Dano K. Inactive proenzyme to tissue-type plasminogen activator from human melanoma cells, identified after affinity purification of a monoclonal antibody. Embo J. 1984 ;3:51-56
28. Male KB, Nguyen AL, Luong JHT. Isolation of urokinase by affinity ultrafiltration. Biotechnol. Bioeng. 1990 ;35:87-93
29. Takahashi R, Akiba K, Koike M, Noguchi T, Ezure Y. Affinity chromatography for purification of two urokinases from human urine. J. Chromatogr. B. 2000 ;742:71-78
Author contact
![]() Correspondence to: Dr. Ashok Kumar, Department of Biological Sciences and Bioengineering, Indian Institute of Technology, Kanpur-208016, INDIA. Tel. +91-512-2594051; Fax. +91-512-2594010; e-mail: ashokkumac.in
Correspondence to: Dr. Ashok Kumar, Department of Biological Sciences and Bioengineering, Indian Institute of Technology, Kanpur-208016, INDIA. Tel. +91-512-2594051; Fax. +91-512-2594010; e-mail: ashokkumac.in
Citation styles
APA
Bansal, V., Roychoudhury, P.K., Kumar, A. (2007). Urokinase Separation from Cell Culture Broth of a Human Kidney Cell Line. International Journal of Biological Sciences, 3(1), 64-70. https://doi.org/10.7150/ijbs.3.64.
ACS
Bansal, V.; Roychoudhury, P.K.; Kumar, A. Urokinase Separation from Cell Culture Broth of a Human Kidney Cell Line. Int. J. Biol. Sci. 2007, 3 (1), 64-70. DOI: 10.7150/ijbs.3.64.
NLM
Bansal V, Roychoudhury PK, Kumar A. Urokinase Separation from Cell Culture Broth of a Human Kidney Cell Line. Int J Biol Sci 2007; 3(1):64-70. doi:10.7150/ijbs.3.64. https://www.ijbs.com/v03p0064.htm
CSE
Bansal V, Roychoudhury PK, Kumar A. 2007. Urokinase Separation from Cell Culture Broth of a Human Kidney Cell Line. Int J Biol Sci. 3(1):64-70.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.