Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials
Reagent setup
Procedure
Generation of transgenic mice
Troubleshooting
Timeline
Anticipated results
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(2):91-99. doi:10.7150/ijbs.3.91 This issue Cite
Technical Report
RNAi-based conditional gene knockdown in mice using a U6 promoter driven vector
Vivek Shukla, Xavier Coumoul#, Chu-Xia Deng ![]()
Genetics of Development and Disease Branch, 10/9N105, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland, MD 20892, USA.
# Present address: INSERM UMR-S 747, Unite de Pharmacologie, Toxicologie et Signalisation Cellulaire, Centre Universitaire des Saints-Pères, 45 rue des Saints-Pères, 75270 Paris Cedex 06, FRANCE
Received 2006-12-11; Accepted 2007-1-5; Published 2007-1-5
Abstract
RNA interference (RNAi) is a powerful tool widely used for studying gene function in a number of species. We have previously developed an approach that allows conditional expression of a polymerase III promoter based small hairpin RNA (shRNA) in mice using the Cre-LoxP system. This approach uses a U6 promoter, which is inactive due to the presence of a ploxPneo cassette in the promoter; this promoter can be activated after excision of the neo gene in transgenic mice that express a Cre recombinase transgene. As a proof of principle, we have previously knocked down over 95% of Fgfr2 transcripts in mouse germlines, leading to embryonic lethality, while restricting the knockdown to the progress zone of the limb results in live animals with malformation of digits of both the forelimbs and hindlimbs. We now provide a detailed protocol, including a simplified single-step cloning procedure for vector construction. This method provides a fast yet efficient way to decipher gene functions in vivo in a tissue specific manner.
Keywords: RNAi, shRNA, pol III vectors, transgenic mice, protocol
Introduction
RNA interference (RNAi) is a technique widely used to down-regulate the mRNA level of a specific gene. Small interfering RNAs (siRNAs) or small hairpin RNAs (shRNAs) are composed of a 22 nt-double strand RNA sequence completely homologous to an unique target gene [1-6]. ShRNAs are generally produced by RNA polymerase II or III-based vectors while siRNA can be obtained from biotechnology companies. The siRNA or shRNA-expressing vectors are transfected into cell lines with classical lipotransfectants. The homology of sequence with a specific target gene allows formation of a complex comprising one strand of the shRNA or siRNA hybridized with the mRNA target and the RISC (RNAi-induced silencing complex) proteins in the cytoplasm. RISC then degrades the mRNA, which cannot be translated. The whole process leads to the specific downregulation of the RNA of the corresponding gene within 24-72 hours [2, 3, 7-9]. Because of the dominant feature of RNAi in causing cell lethality, several studies developed inducible regulation of RNAi in mammalian cells to achieved a controllable gene knockdown using either tetracycline, ecdysone, or tamoxifen-inducible systems [8, 10-13].
RNAi based gene knockdown has also been successfully used to study gene functions in mouse models [8, 14-21]. In some of these studies, shRNA constructs were first introduced into mouse embryonic stem (ES) cells through random integration [15, 22-24] or homologous recombination into the Rosa-26 locus [25]. The ES cells with ideal levels of gene knockdown were then injected into blastocysts to obtain mutant mice, following a standard approach for germline transmission used in gene targeting. In some other studies, the mutant mice were generated through a transgenic approach, i.e. injecting shRNA constructs directly into the pronucleus of the oocyte [16, 26, 27]. Mutant mice carrying modification of many genes, including Dnmt1, p53, Fgfr2, RasGAP, and WT1, have been generated using these approaches [22-24, 26, 28-30].
We have recently described a Cre-LoxP mediated shRNA approach that specifically knocks down the Fgfr2 gene with high efficiency (>90% reduction of transcripts) in a tissue specific fashion [6]. This approach has several key features that may contribute to the high efficiency of temporal and spatial control of gene expression. First, the U6 promoter is used to ensure a ubiquitous and high level expression of short transcripts. Secondly, a ploxPneo is inserted into the U6 promoter between its proximal sequence element (PSE) and distal sequence element (DSE). The presence of the neo gene completely blocks transcriptional activity of the U6 promoter, therefore preventing any potentially lethal effect of the shRNA construct during the process of transgenic mice production. Lastly, our construct was introduced through pronuclear injection, making it relatively faster to generate mice than using ES cell based approaches.
This approach has several advantages compared with other in vivo approaches. First, because the neo gene is flanked by loxP sites, it can be deleted by Cre recombinase to restore the activity of the U6 promoter. Therefore, multiple studies investigating gene function in different tissues and different developmental stages can be performed after crossing the mutant mice with transgenic mice that express Cre in different tissues/organs. A recent study indicates that tissue specific gene knockdown can also be achieved by using tissue-specific polymerase-II (polII) promoter, which drives expression of a longer double strand RNA (dsRNA) expression unit [30]. Although effective, this approach is limited to a specific tissue and new transgenic mice carrying dsRNA driven by different tissue specific polII promoters need to be generated for each tissue of interest. Second, we chose to use pronuclear injection to generate mutant mice instead of using ES cells. This avoids the sophisticated ES cell manipulation that is required for germline transmission after the blastocyst injection. Thus, our approach should be technically easy and reliable. Finally, our approach does not require the construction of sophisticated gene-targeting vectors. Indeed, cloning the RNAi oligos into the shRNA expression vector has been simplified into a single step since our initial publications [8, 26], making it relatively simple and fast to generate mutant mice carrying an shRNA construct.
In this article, we present a detailed protocol to generate a U6-based shRNA construct and transgenic mice in order to share our approach with interested readers for their gene knockdown studies.
Materials
Reagents
DNA oligonucleotides
Distilled sterilized water (dH2O)
TE buffer (10 mM Tris-Cl/1 mM EDTA, pH 7.4)
Low-TE buffer (1 mM Tris-Cl/0.1 mM EDTA, pH 7.4)
ApaI, EcoRI, SalI, Not I, Asp 718, Afl III restriction enzymes (Roche)
DNA polymerase I, large fragment (Klenow) (New England Biolabs)
T4 DNA polymerase (New England Biolabs)
MinElute Reaction Cleanup kit (Qiagen)
Qiaprep Spin Miniprep kit (Qiagen)
Dneasy kit (Qiagen)
LB media (Add the following to 800 ml H2O (10 g Bacto-tryptone, 5 g yeast extract, 10 g NaCl). Adjust pH to 7.5 with NaOH. Adjust volume to 1L with dH2O. Sterilize by autoclaving)
LB plates (http://www.bbri.org/faculty/smith/LB_Plates.html)
Ampicillin Stock (100 mg.ml-1) (3 g ampicillin sodium salt (Fisher BP1760-25), add sterile water to raise volume to 30 ml. Filter sterilize through 0.22 µm syringe filter. Aliquot into 1 ml and store at –20°C).
Competent cells (DH5α, Invitrogen)
UltraTMPhenol: Chloroform: Isoamyl Alcohol (Invitrogen, Cat No. 15593-031).
(Caution: these are chemical reagents with harmful effect)
RNA-Stat-60 reagent
Agarose powder (Gibco BRL, Invitrogen)
Ethidium bromide
QIAquick gel extraction kit
Nylon membrane (PROTRAN Plus from Scleicher & Schuell)
Sheets of 3M Whatman Chromatography filter paper and saran wrap
15% (wt/vol) polyacrylamide-8 M urea gel
Loading dye (contains 0.025 % Bromophenol blue, 0.025% Xylene Cyanol, 0.5 mM EDTA, 0.025% SDS)
10X TBE (Quality Biological, Inc.; Cat # 330-001-161)
PERFECTHYBTMPLUS (Sigma, Product no. H 7033)
10X PNK buffer (Roche 1465492)
γ32P ATP (Amersham AA0068 250 μCi). CAUTION: radioactive material. Manipulate under appropriate conditions
Poly Nucleotide Kinase (PNK) (10u/μl, Roche 709557)
G25 column (Amersham 27- 5325-01)
Column 4K (X100)
20X SSPE (Quality Biological, Inc.; Cat # 330-015-161)
20X SSC (Quality Biological, Inc.; Cat # 330-003-161)
20% SDS (Quality Biological, Inc.; Cat # 351-066-101)
dNTP: DNA polymerization mix is available at 20mM/dNTP commercially; take 2 µl of 20mM/dNTP mix and make up the volume 100 µl with dH2O. It will give 400 µM/dNTP solution. CRITICAL: keep on ice during all the preparation of the reactions
Loading buffer: 12% FICOLL 400 by weight or 12 % glycerol by volume ; 60 mM Na2EDTA, pH 8 ; 0.6% by weight SDS ; 0.003% by weight bromphenol blue ; 0.003% by weight xylene cyanol (for high molecular weight bands)
TAE 10X concentrated aqueous solution: Tris-Hcl 48.46 g/L (0,4 M), EDTA-Na,-salt 3.72 g/L (0.01 M), Acetic acid 12.01 g/L (0.2 M)
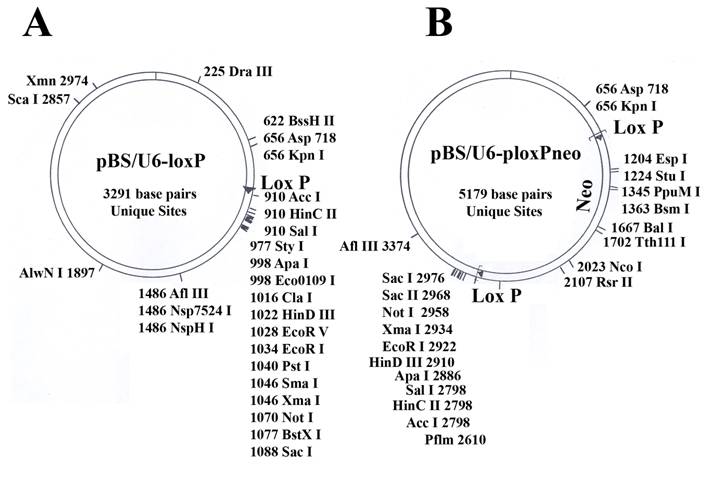
pBS/U6-loxP and pBS/U6-ploxPneo vectors (Fig. 1): The generation of these vectors from the original U6 promoter [31] was as described [8]. Both vectors are available upon request.
Maps of the two vectors used in the study. (A) pBS/U6-LoxP (active). (B) pBS/U6-ploxPneo (inactive, due to the presence of a ploxPneo gene that can be deleted by Cre recombination). The ploxPneo (1936 bp) is derived from the PGKneo [38] after adding loxP sites on both end of the PGK neo. Multiple cloning sites and their locations are indicated.
Lipofectamine (2000) (Invitrogen)
Cells –to-cDNATM II kit (Ambion, Cat # 1722,1723)
Equipment
Computer with internet access
Eppendorf tubes
Eppendorf Centrifuge (5415 D)
Heater
Floater
Beaker
Spectrophotometer (cuves)
Water bath
Shaker for bacteria suspensions
Bacteria plates incubator
Sterilized materials (toothpicks) for transformation procedure
Ultraviolet transilluminator
Gene Amp PCR Systems 9700
Taqman PCR (Gene Choice)
Stratagene UV Crosslinker
Baker
Geiger counter
Cassette and phosphorimager screen
XCell IITM Blot Module
Hybridization oven
Poly Acrylamide Gel Electrophoresis (PAGE) apparatus
Animals
FVB/n mice (Jackson lab) were used for generating transgenic mice.
Reagent setup
1. Designing oligos
With an internet browser, connect to http://www.ncbi.nlm.nih.gov/entrez/query.fcgi. Select in the Search tool bar « Nucleotide » and search for your mRNA or cDNA of interest throughout the Accession Number or name. Within the nucleotide sequence, select a region starting with one or two G (G or GG), followed by 19-20 bases to obtain a total of 20-21 nucleotides (nt) sequence and check that this whole sequence is 45-65% G/C. Finally, use a BLAST2 program (http://www.ebi.ac.uk/blast2/ for example) to check that the sequence is unique and has no significant homology with any other known ones. Additional information regarding nucleotides selection can be found in a recent publication [32].
Oligo 1: the native U6 promoter contains a (GGG), from which transcription is initiated. This motif is part of an ApaI restriction site (GGGCC/C). In the procedure, the ApaI site is cut and blunted, leaving only one G left at its 3' end. The first strand of oligo 1 then contains one or two G at the 5' end which will be cloned at the ApaI site. An example of oligo 1 for Fgfr2 gene is shown in Fig. 2A. In this oligo, we added a HindIII site (AAGCTT), which serves as a 6-base loop of the hairpin, in the oligo following the first 20-21 bp. The last part of the oligo is the complementary sequence of the 20-21 bp followed by three “C” corresponding to the complementary sequence of the first three “G”, five “T” corresponding to the termination site of the RNA polymerase III, and then one G, which will form an EcoRI site after annealing with oligo 2.
An example of used oligos for shRNA vector. (A) Oligo 1 contains a two complementary sequences (in black) that can be formed upon shRNA transcription, a hairpin structure. A Hind III site forms the hairpin structure (see figure 2D in red). (B) Oligo 2 is complementary of oligo 1. Both oligos are designed against mouse Fgfr2. (C) Annealed Oligos 1 and 2. (D) Predicted hairpin structure with the corresponding colors (black: target sequence; red: Hind III site for hairpin).
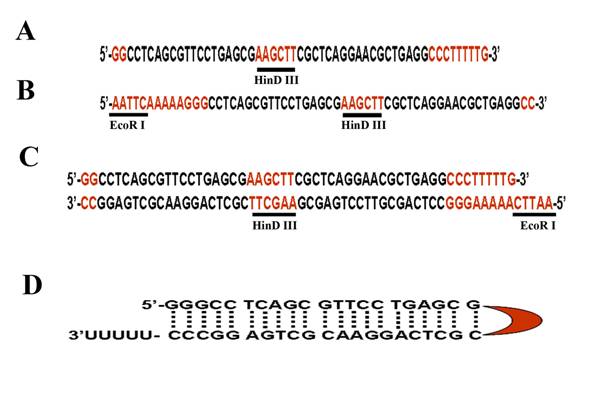
Oligo 2 must be complementary to the Oligo 1 except for 4 nt (AATT) that are added to the former's 5' end (Fig. 2B). This nt forms a digested EcoRI site (Fig. 2C), which will be used for cloning purposes. After transcription by U6 promoter, a hairpin structure will be formed in which the HindIII sequence form the head of the hairpin structure (Fig. 2D).
Note 1: We normally start with at least three candidate regions for the first oligo design so that the best sequence can be determined. Ensure that the designed oligos do not contain a (TTTTT) and a (AAAAA) motifs inside the 20-21 sequence. To avoid multiple inserts, do not order phosphorylated oligos. We ordered oligos from Operon Biotechnologies, although the source may not be critical.
Note 2: A recent study compared hairpin loops formed by several different sequences in cell culture condition. It was shown that the loop formed by “UUCAAGAGA” seems to have a higher efficiency than loops formed by other sequences [33], although its efficiency in vivo remains elusive. In this case, the nucleotide “TTCAAGAGA” can be used instead of “AAGCTT”.
Procedure
2. Annealing oligos
a. Prepare a 0.2 μg/μl stock solution of each Oligo (1 and 2, see Reagent Setup) with TE buffer.
b. Mix the components in an Eppendorf tube as shown in Table 1.
Annealing oligos.
| Component | Amount | Final |
|---|---|---|
| Ligation buffer (10X) | 4 μl | 1X |
| Oligo 1 (0.2 μg/μl) | 18 μl | 0.09 μg/μl |
| Oligo 2 (0.2 μg/μl) | 18 μl | 0.09 μg/μl |
| TOTAL | 40 μl |
c. Heat 1 L of water in a beaker on a heater until the water reaches 70°C.
d. Place the tube on a floater into the beaker and keep the tube at 70°C for 5 minutes.
e. Move the whole beaker onto the bench and let it cool down for 1 hour or until the water reaches room temperature.
Subcloning the double-stranded oligo into the vector.
3. ApaI restriction of the vector
Mix the components in an eppendorf tube as shown in Table 2.
ApaI restriction digestion of U6 vector.
| Component | Amount | Final |
|---|---|---|
| pBS/U6-Neo vector | 5 μg | |
| Buffer A (10X) | 2.5 μl | 1X |
| ApaI | 1 μl | 2 unit/μg of the vector |
| TOTAL (make up with dH2O | 25 μl |
Incubate at 30°C for 2 hours.
Check the digestion of the vector by running 1 μl of the digestion mixture on 1% agarose gel.
Make sure the pBS/U6-Neo vector DNA is produced in a dam– bacteria strain to avoid methylation of the vector (ApaI is methylation-sensitive).
4. Blunt the ApaI-digested DNA with T4 DNA polymerase or Klenow enzyme (which works with lower efficiency than T4 DNA polymerase)
The DNA was then Phenol-Chloroform extracted and resuspend in 10 µl of Low-TE. Measure the DNA concentration. It should contain more than 2.5μg of DNA.
5. EcoRI restriction
Add the reagents to an Eppendorf tube as shown in Table 3.
EcoRI restriction digestion of the vector.
| Component | Amount | Final |
|---|---|---|
| ApaI digested and blunted pBS/U6-neo vector | 2.5μg | |
| Buffer H (10X) | 2.5 μl | 1 X |
| EcoRI | 1 μl (10 unit) | 4 unit/μg of the vector |
| TOTAL (make up with dH2O | 25 μl |
Incubate at 37°C for 2 hours.
Purify the vector by Phenol-Chloroform extraction.
Resuspend the DNA in 20 μl of TE. Measure DNA concentration.
6. Ligation of oligos-1/2 into the pBS/U6-Neo ApaI+EcoRI- digested vector
a. Mix the components in an Eppendorf tube as shown in Table 4.
Ligation of oligos-1/2 into ApaI+EcoRI- digested U6 vector.
| Component | Amount | Final |
|---|---|---|
| ApaI+EcoRI- digested pBS-U6-Neo vector | 100 ng | |
| Annealed oligos | 0.25 μl | |
| Ligation buffer (10X) | 2 μl | 1X |
| T4 DNA ligase | 2 μl | 1-2 unit/reaction |
| dH2O | 14 μl | |
| TOTAL | 20 μl |
b. Centrifuge 10 seconds and incubate overnight at 16°C.
7. Transformation
a. Thaw competent cells (100 µl) on ice.
b. Add 5 to 10µl of the ligation mixture and mix gently by pipetting.
c. Incubate on ice for 30 minutes.
d. Heat shock for 45 seconds at 42°C and chill on ice for 5 minutes.
e. Add 0.9 ml of room temperature LB media and incubate at 37°C for 1 hour in a shaker (225 rpm).
f. Spread 100 to 300 µl of the solution on LB plates containing ampicillin.
g. Incubate overnight at 37°C.
8. Screen for colonies containing insert
a. Pick 18 colonies from the LB plates with sterilized toothpicks and culture each colony in 2 mL of fresh LB media supplemented with ampicillin (100 µg.ml-1) for overnight.
b. Using QIAprep Spin Miniprep Kit, or regular method for miniprep to purify the corresponding pBS/U6-Neo-shRNA vectors and dissolved in 20 μl of TE.
c. Perform the restriction as shown in Table 5.
Digestion of DNA using EcoRI + SalI.
| Component | Amount | Final |
|---|---|---|
| DNA from miniprep | 2.0 μl | |
| EcoRI | 1 μl | 10 unit/reaction |
| SalI | 1 μl | 10 unit/reaction |
| Buffer H | 2.5 μl (10X) | 1X |
| TOTAL | 25 μl |
A master mix can be made for 20 samples (without DNA), and 23 μl aliquots should be divided into 19 Eppendorf tubes. Add 2 μl of DNA extracted from each colony (a total of 18 colonies) into each Eppendorf tube. Also add 100 ng of parent pBS-U6-Neo DNA into the 19th tube, which will be used as a control without insert.
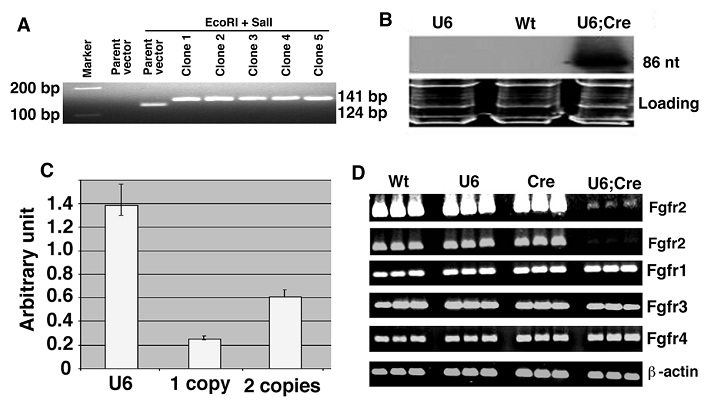
d. Incubate at 37°C for 2 hours. Run the samples on a 3% agarose gel. The parent pBS-U6-Neo vector will release a 124 nt fragment, while the DNA with one insert will release a 141 nt fragment. The difference of 17 nt is visible in the 3% agarose gel (Figure 3 A).
Screening for the U6-Neo-RNAi vector containing the insert and its expression in mouse after deletion of the neo by EIIa-Cre transgene. (A) After double-strand oligo ligation into the pBS/U6-neo vector, check insertion by SalI and EcoRI restriction and migration in a 3% agarose gel. Run two controls (parent and restricted parent vector) and an appropriate molecular weight marker (100, 150 and/or 200 bp). (B) Northern blot showing expression of pBSU6-Neo-RNAi against mouse Fgfr2. Cre mediated deletion of the neo from U6-ploxPneo-Fgfr2 transgene allows expression of Fgfr2 RNAi. (C) Assessment of copy number of U6-ploxPneo-Fgfr2 transgene by realtime PCR using primers for neo gene. Control DNA with 1 copy and 2 copies of neo gene are from Sirt6+/- and Fgfr1-/- ES cells, respectively. (D) RT-PCR analysis of Fgfr1-4 expression in E12.5 embryos of different genotypes. Wt: Wild type; U6: U6-ploxPneo-Fgfr2; Cre: EIIa-Cre; and U6;Cre: U6-Fgfr2;EIIa-Cre. The first row is a longer exposure of the second row. Three embryos for each genotype were shown. RT-PCR was performed using the following primers: Fgfr1-F: 5'-TTCTGGGCTGTGCTGGTCAC-3', and Fgfr1-R: 5'-GCGAACCTTGTAGCCTCCAA-3'. Fgfr2-F: 5'-AAGGTTTACAGCGATGCCCA-3', and Fgfr2-R: 5'-ACCACCATGCAGGCGATTAA-3'. Fgfr3-F: 5'-CTAGTGTTCTGCGTGGCGGT-3', and Fgfr3-R: 5'-TTCTTATCCATTCGCTCCGG-3'. Fgfr4-F: 5'-CTGTTGAGCATCTTTCAGGG-3', and Fgfr4-R: 5'-CGTGGAAGGCCTGTCCATCC-3'.
Critical step
- The insert is 53 nt. Because ApaI+EcoRI digestion cuts away 36 nt from the parent vector, the net gain with one insert is 17 nt.
- In our original study by Coumoul et al. (2004)[8], we used two consecutive steps to clone a total of 4 shorter oligos (oligos 1-4) into the shRNA vector in order to avoid formation of hairpins of the oligos. We now found this is not necessary and only use a one-step protocol to introduce 2 longer oligos (oligos 1 and 2). If you encounter any trouble in using this one-step oligo cloning, a two-step cloning procedure can be used [8, 31].
9. Sequencing the clones
Once DNA vectors containing a single insert is confirmed, we send them to GENEWIZ, INC for sequencing using two primers. One primer (5'– GCTATGACCATGATTACGCCA – 3') is located at 3017-3040, which is 131 bp away from ApaI (located at 2886). Another primer (T3 primer: 5' - ATTAACCCTCACTAAAGGGA - 3') is located at 2992-3011, which is 106 bp away from ApaI. Both primers yield good sequencing.
10. Checking the efficiency of the shRNA vector
Because the presence of the ploxPneo cassette completely blocks the activity of the U6 promoter, it must be deleted by the Cre-loxP mediated recombination in cultured cells using one of the following procedures.
Option A describes a transient transfection procedure to delete the neo and should be used by researchers who consider a fast evaluation of gene knockdown. Option B describes a stable transfection and should be used when the fast evaluation procedure does not yield a clear result. Option C describes a convenient system to get shRNA vector and should be used by the labs, who do not have Cre recombinase system available. In all three procedures, the test should be performed in cells that express the target gene. Alternatively, a plasmid expressing the target gene needs to be co-transfected with shRNA expressing constructs.
Option A
- The U6-Neo-RNAi construct can be transfected into cultured cells bearing constitutively expressed Cre or inducible Cre using lipofectamine 2000 following a recommended procedure (http://www.invitrogen.com/content/sfs/manuals/lipofectamine2000_man.pdf). Parent U6-Neo vector without insert can be used as control.
- After 48-72 hours of transfection, cells will be harvested and total RNA will be prepared using RNA STAT-60 (Tel-Test, Inc., Friendswood, TX) for RT-PCR (Cells-to-cDNA TM II protocol for reverse transcription) analysis of target gene expression. RNAi knockdown is considered successful if about 70% of the mRNA target is degraded.
Option B
- The U6-Neo-RNAi construct can be transfected into regular cells followed by G418 selection.
- Cells with a stable integrated U6-Neo-RNAi construct are then be transfected by Cre-expressing vector to delete the neo gene.
- Alternatively, cells with inducible Cre can also be used for transfection. Cells with a stable integrated U6-Neo-RNAi construct are then placed in a Cre induction condition to delete the neo gene followed by RT-PCR analysis of target gene expression.
Option C
For those laboratories where a Cre expression vector is not available, the shRNA vector can be generated in parallel by cloning the annealed oligo 1/2 (Step 2) into U6-RNAi vector (Fig. 1A). Because this vector does not contain the ploxPneo, the U6 promoter should be active when directly transfected into regular cells.
All above procedures work efficiently in our laboratory [8] and in the literature for assessing knockdown efficiency of vector based siRNAi expression.
Generation of transgenic mice
11. Preparing vectors for pronuclear injection
The pBS/U6-Neo-shRNA vector is digested with Asp718 and AflIII according to the restriction procedure used in the previous paragraphs (see paragraph 4 and 6). After gel electrophoresis, we purify a 2739 bp fragment using the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA). The fragment is resuspended in Low-TE buffer and adjusted to a final concentration of 2ng/μl and injected into oocytes isolated from FVB/N mice through pronuclear injection. All animals are maintained and treated according to the approved NIH guidelines and techniques (http://www.nih.gov/science/models/mouse/sharing/5.html).
12. Identifying G0 transgenic founder mice and screening for germline transmission
a. Cut off 0.5 cm of the tails from weaned pups born to mothers carrying injected blastocysts and carry the tails in Eppendorf tubes. Isolate genomic DNA using Qiagen's Dneasy kit and proceed to genotyping according using PCR with the following pair of primers (5'-CGAAGTTATCTAGAGTCGAC-3', and 5'-AAACAAGGCTTTTCTCCAAGG-3') that amplifies 102 bp from the U6 promoter and the connecting neo gene.
b. Amplify the template as shown in Table 6.
PCR conditions for detecting the U6-neo transgene.
| Cycle number | Denaturation | Annealing | Polymerization |
|---|---|---|---|
| 1 | 2 minutes at 94°C | ||
| 2-34 | 10 seconds at 94°C | 20 seconds at 60°C | 30 seconds at 72°C |
| 35 | 10 seconds at 94°C | 20 seconds at 60°C | 10 minutes at 72°C |
c. Run the PCR product in a 2.5% agarose gel to identify the positive G0 transgenic founder mice.
The PCR positive mice are candidate that carry the U6-Neo-RNAi transgene. These mice are crossed with wild type FVB/N mice to obtain germline transmission. The offspring mice containing the transgene are the F1 generation.
13. Generate U6-ploxPneo-RNAi/Cre mice
Because the presence of the ploxPneo in the RNAi vector that inactivate the U6 promoter activity, the F1 mice should be normal. To study the effect of RNAi mediated knockdown, the ploxPneo gene should be deleted. To achieve this, two independent strains of F1 mice carrying the U6-ploxPneo-RNAi transgene are crossed with mice carrying ubiquitously or tissue specifically expressed Cre transgene to create bigenic mice (U6-Neo-RNAi/Cre) that can be used for phenotypic analysis.
14. Monitoring Cre-mediated recombination
The product generated by Cre-loxP mediated recombination in the U6-ploxPneo-RNAi/Cre bigenic mice can be visualized by using a primer pairs flanking the ploxPneo: 1: 5'-CGCACAGACTTGTGGGAGAA-3'; and 2: 5'-CACAATTACTTTACAGTTAG-3'. 1 μg of DNA extracted from various tissues of bigenic mice can be used in the PCR reaction (as described in step 12).
15. Determining U6-RNAi transgene expression
Because the transcript of the U6-RNAi transgene is very short, it can only be detected by Northern blot protocol that is optimized for short transcripts. A clear and sharp band can be obtained after 24-48 hour exposure (Fig. 3B). The following detailed northern protocol is optimized to detect processed small transcripts [26].
a. Mix 20 μg RNA (volume of 15 μl) with 12 μl of loading dye (contains 0.025% Bromophenol blue, 0.025% Xylene Cyanol, o.5 mM EDTA, 0.025% SDS). Heat samples in 90°C heat block for 5 minutes and then place it on ice. Load the samples onto 15% (wt/vol) polyacrylamide-8 M urea gel.
b. Run gel in 1X TBE running buffer at 200 V approximately 2-3 hrs or until bromophenol blue dye migrates above bottom of the gel.
c. Stain the gel with 4 μg/ml EtBr in 0.5X TBE for 5 min, and look for tRNA and 5S rRNA bands for quality of RNA prep and for loading control. Take digital snapshots of the gel and save images.
d. Set up transfer using XCell IITM Blot Module, (Invitrogen Cat No # EI9051). Cut Nylon Membrane (PROTRAN Plus from Scleicher & Schuell) and 8 sheets of 3M Whatman Chromatography filter paper to the size of the gel. Presoak membrane and filter paper in 0.5X TBE. It is important that both are thoroughly soaked. Place 4 pieces of soaked filter paper on top and 4 on the bottom, with the membrane underneath the gel. Transfer at constant current (12V) for 1-2 hours in 0.5X TBE.
e. While the blot is still moist (leave it on the wet 3MM support), UV crosslink with 1000 mJ of energy (Stratagene UV Crosslinker) RNA side up towards the lamps.
Pause point: Membrane can be wrapped and kept at room temperature for 2-3 days.
f. Prehybridize the membrane with PERFECTHYBTMPLUS (Sigma, Product no. H7033) in a hybridization oven and rotate at 42°C for 2-4 hours.
g. Labeling probe as shown in Table 7.
Label Oligo 1 with γ 32P ATP.
| Component | Amount | Final |
|---|---|---|
| Oligo 1 | 0.6-1 μl | 50-100 ng |
| Polynucleotide kinase buffer (10X) | 2 μl | 1X |
| γ 32P ATP | 6 μl | 50-60 μ Curie |
| Polynucleotide kinase | 1 μl | 10 units |
| TOTAL | 20 μl |
Incubate 30 minutes at 37°C and put on ice.
Pass through G25 column (Amersham 27- 5325-01) and check labeling efficiency (Column 4K (X100); Probe more than 5K (X100).
h. Hybridize the membrane at 42°C overnight with labeled probe in fresh hybridization buffer.
CRITICAL: Hybridization solution was used according to oligo size. For oligo less than 25 mer, 6X SSPE is used, while oligo longer than 25 mer, 5X SSPE should be used. Hybridization solution is comprised of SSPE (5X or 6X), 5X Denharts, 0.5% SDS and carrier DNA 250-300 μl (from 10 mg/ml) for 50 ml.
i. After the hybridization the membrane was washed with 6X SSPE, 0.1% SDS (add 300 ml of 20X SSPE and 5 ml of 20% SDS into 695 ml of dH2O) at 42°C for 10 minutes, and then washed with 2X SSC, 0.1% SDS (add 100 ml of 20X SSC and 5 ml of 20% SDS into 895 ml of dH2O) two times at 42°C for 10 minutes.
j. Drain and wrap in Saran Wrap and expose to X-ray film keeping the blot moist if it is not washed enough and need to be rewashed.
16. Determine the copy number of the transgene
Assessment of copy number of U6-ploxPneo-Fgfr2 transgene by real-time PCR using primers (Neo-U: 5'-AGAGGCTATTCGGCTATGACTG-3', and Neo-D: 5'-GTGGCCAGCCACGATAGC-3') for neo gene. We used DNA extracted from Sirt6+/- and Fgfr1-/- ES cells as controls for 1 copy and 2 copies of neo gene, respectively. However, DNA extracted from any mouse cells carrying one copy and 2 copies of neo gene should be fine for this experiment. Relative copy of copies of transgene can be calculated against the known copy numbers (Fig. 3C).
17. Determining expression levels of the target gene
Expression of the target gene can be evaluated by RT-PCR using RNA isolated from organs/tissues where Cre is expressed. Western blot analysis (Molecular Cloning protocol) can also be used if antibody is available. Efficiency of gene knockdown can be determined through comparing expression levels of the target gene between mutant and control mice. An example of Fgfr2 expression in the Fgfr2-neo-RNAi/EIIa-Cre [34] mice analyzed by RT-PCR was shown in figure 3D.
Sometimes RNAi may generate non-specific toxicity due to off-target effects. Much of the off-target effects can be avoided by conducting a careful homology search in designing the first oligo to select sequences that are unique in the genome (Step 1). In addition, for targeted genes that belong to families containing highly related members, gene expression study should be performed to detect any possible alterations of these members. An example of gene expression of other three members of Fgf receptor family is shown in Fig. 3D.
Troubleshooting
As suggested in Table 8.
Troubleshooting.
| Problems | Solution |
|---|---|
| Step 7, No colony after transformation | The ratio of insert to vector was not adequate. In step 3, the ratio of insert to vector is 40:1. It can be varied from 10-100:1. |
| Step 8, No visible difference of bands | Always use at least 3% agarose gel and run for 2-3 hrs at 180 V. |
| Step 9, Sequencing does not work | Sometimes, it is difficult to read through the hairpin because of its strong secondary structure. We have tried a few companies and found Genewiz Inc. can read through the hairpin structure with good results. |
| Step 10, No decreased expression of the target gene | It can be caused by poor transfection efficiency. Try to find an adequate protocol for transfection. Another possibility is that the RNAi oligo does not work. If this is the case, new RNAi oligo should be used. We normally start with oligos against two different regions of the target gene and rarely found both do not work. |
Timeline
Step 1-9: Two to four weeks.
Step 10: Two to four weeks.
Step 11: One to two months to generate transgenic founder mice.
Step 13-17: After chimera mice are generated, it takes about one to two more months to get F1 mice, and another two months before knockdown phenotypes can be examined from crosses of F1 mice with Cre expressing mice.
Anticipated results
Under our injection conditions (NIDDK Transgenic Core Facility), about 15% of the mice should be chimeras carrying the U6-Neo-RNAi transgene. After crossing with wild type FVB/N mice, more than 50% of the chimeric mice can pass the transgene in the first litter. Based on our expression studies on at least two strains, we show that transgenic mice carrying U6-Neo-Fgfr2RNAi constructs are normal, displaying Fgfr2 transcripts equivalent to those of wild-type controls (Fig. 3D), indicating that the U6 promoter is inactive in vivo due to the presence of the neo in the promoter. We were unable to detect expression of shRNA in those mice (Fig. 3B), suggesting that the presence of the ploxPneo gene inactivated the U6 promoter. Similar results have been recently shown by other studies [25, 35], although a neo gene with of smaller size (about 800 bp) is less tight in silencing U6 promoter activity [35] compared with the ~1900 bp neo cassettes [8, 25, 26]. After excision of the neo through crossing with transgenic mice that express Cre in the mouse germline, the U6 promoter is re-activated, leading to over 95% reduction of Fgfr2 transcripts (Fig. 3D).
Like all other transgenic mice generated by pronuclear injection, the U6-Neo-shRNA construct randomly integrates into mouse genome. The levels of shRNA expression are likely to vary among transgenic lines due to integration sites and copy number. In Step 13, we crossed two independent F1 mice carrying the U6-ploxPneo-Fgfr2RNAi transgene with mice carrying Cre transgene to create bigenic mice (U6-Neo-Fgfr2RNAi/Cre) for assessing knockdown effects, and found that both strains showed similar phenotypes [26]. For those who intend to avoid potential expression variation due to position/copy number effects, the U6-NeoshRNA construct can be targeted into a known open chromosome locus with a single copy shown by Yu and McMahon (2006) [25]. On the other hand, phenotype variation is observed even in mice generated by gene targeting, suggesting that it is an intrinsic nature associated with the loss of function mutation, perhaps due to a different response of each individual to the assault. Actually, this could be beneficial in some circumstances where variable phenotypes may reveal new and unexpected information. For example, we and many others have purposely used sophisticated targeting modifications to generate hypomorphic alleles or isoform knockouts [36, 37]. Thus, for those who intend to find strains with varying levels of target gene expression so that a series of “mutant allele” can be generated for a wild range of gene function analysis, more F1 strains need to be used in Step 13.
Acknowledgements
This work was supported by the intramural Research Program of National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, USA.
References
1. Elbashir SM, Martinez J, Patkaniowska A. et al. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. Embo J. 2001 ;20:6877-88
2. Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001 ;15:188-200
3. Elbashir SM, Harborth J, Lendeckel W. et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001 ;411:494-8
4. Collins RE, Cheng X. Structural domains in RNAi. FEBS Lett. 2005 ;579(26):5841-9
5. Rao M, Sockanathan S. Molecular mechanisms of RNAi: implications for development and disease. Birth Defects Res C Embryo Today. 2005 ;75:28-42
6. Coumoul X, Deng CX. RNAi in mice: a promising approach to decipher gene functions in vivo. Biochimie. 2006 ;88(6):637-43
7. Yu JY, DeRuiter SL, Turner DL. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc Natl Acad Sci U S A. 2002 ;99:6047-52
8. Coumoul X, Li W, Wang RH. et al. Inducible suppression of Fgfr2 and Survivin in ES cells using a combination of the RNA interference (RNAi) and the Cre-LoxP system. Nucleic Acids Res. 2004 ;32:e85
9. Novina CD, Sharp PA. The RNAi revolution. Nature. 2004 ;430:161-4
10. Wang J, Tekle E, Oubrahim H. et al. Stable and controllable RNA interference: Investigating the physiological function of glutathionylated actin. Proc Natl Acad Sci U S A. 2003 ;100:5103-6
11. Czauderna F, Santel A, Hinz M. et al. Inducible shRNA expression for application in a prostate cancer mouse model. Nucleic Acids Res. 2003 ;31:e127
12. Gupta S, Schoer RA, Egan JE. et al. Inducible, reversible, and stable RNA interference in mammalian cells. Proc Natl Acad Sci U S A. 2004 ;101:1927-32
13. Matsukura S, Jones PA, Takai D. Establishment of conditional vectors for hairpin siRNA knockdowns. Nucleic Acids Res. 2003 ;31:e77
14. Hasuwa H, Kaseda K, Einarsdottir T. et al. Small interfering RNA and gene silencing in transgenic mice and rats. FEBS Lett. 2002 ;532:227-30
15. Rubinson DA, Dillon CP, Kwiatkowski AV. et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003 ;33:401-6
16. Tiscornia G, Singer O, Ikawa M. et al. A general method for gene knockdown in mice by using lentiviral vectors expressing small interfering RNA. Proc Natl Acad Sci U S A. 2003 ;100:1844-8
17. Wiznerowicz M, Trono D. Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol. 2003 ;77:8957-61
18. Sarkar SN, Das HK. Regulatory roles of presenilin-1 and nicastrin in neuronal differentiation during in vitro neurogenesis. J Neurochem. 2003 ;87:333-43
19. Miyagishi M, Taira K. U6 promoter-driven siRNAs with four uridine 3' overhangs efficiently suppress targeted gene expression in mammalian cells. Nat Biotechnol. 2002 ;20:497-500
20. Miyagishi M, Taira K. Development and application of siRNA expression vector. Nucleic Acids Res Suppl. 2002 (2):113-4
21. van de Wetering M, Oving I, Muncan V. et al. Specific inhibition of gene expression using a stably integrated, inducible small-interfering-RNA vector. EMBO Rep. 2003 ;4:609-15
22. Kunath T, Gish G, Lickert H. et al. Transgenic RNA interference in ES cell-derived embryos recapitulates a genetic null phenotype. Nat Biotechnol. 2003 ;21:559-61
23. Chang HS, Lin CH, Chen YC. et al. Using siRNA technique to generate transgenic animals with spatiotemporal and conditional gene knockdown. Am J Pathol. 2004 ;165:1535-41
24. Ventura A, Meissner A, Dillon CP. et al. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci U S A. 2004 ;101:10380-5
25. Yu J, McMahon AP. Reproducible and inducible knockdown of gene expression in mice. Genesis. 2006 ;44:252-61
26. Coumoul X, Shukla V, Li C. et al. Conditional knockdown of Fgfr2 in mice using Cre-LoxP induced RNA interference. Nucleic Acids Res. 2005 ;33:e102
27. Peng S, York JP, Zhang P. A transgenic approach for RNA interference-based genetic screening in mice. Proc Natl Acad Sci U S A. 2006 ;103:2252-6
28. Stein P, Svoboda P, Anger M. et al. RNAi: mammalian oocytes do it without RNA-dependent RNA polymerase. Rna. 2003 ;9:187-92
29. Wianny F, Zernicka-Goetz M. Specific interference with gene function by double-stranded RNA in early mouse development. Nat Cell Biol. 2000 ;2:70-5
30. Rao MK, Pham J, Imam JS. et al. Tissue-specific RNAi reveals that WT1 expression in nurse cells controls germ cell survival and spermatogenesis. Genes Dev. 2006 ;20:147-52
31. Sui G, Soohoo C, Affar el B. et al. A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci U S A. 2002 ;99:5515-20
32. Cullen BR. Enhancing and confirming the specificity of RNAi experiments. Nat Methods. 2006 ;3:677-81
33. Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002 ;296:550-3
34. Lakso M, Pichel JG, Gorman JR. et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996 ;93:5860-5
35. Xia X, Zhou H, Huang Y. et al. Allele-specific RNAi selectively silences mutant SOD1 and achieves significant therapeutic benefit in vivo. Neurobiol Dis. 2006 ;23:578-86
36. Xu X, Li C, Takahashi K. et al. Murine fibroblast growth factor receptor 1alpha isoforms mediate node regression and are essential for posterior mesoderm development. Dev Biol. 1999 ;208:293-306
37. Deng CX. Tumor formation in Brca1 conditional mutant mice. Environ Mol Mutagen. 2002 ;39:171-7
38. Tybulewicz VL, Crawford CE, Jackson PK. et al. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991 ;65:1153-63
Author contact
![]() Correspondence to: Chu-Xia Deng, Ph.D., Tel: (301) 402-7225; Fax: (301) 480-1135; Email: chuxiadniddk.nih.gov
Correspondence to: Chu-Xia Deng, Ph.D., Tel: (301) 402-7225; Fax: (301) 480-1135; Email: chuxiadniddk.nih.gov