International Journal of Biological Sciences 10
Impact Factor
ISSN: 1449-2288
Impact Factor
ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(2):100-107. doi:10.7150/ijbs.3.100 This issue Cite
Research Paper
Mouse 24p3 Protein Has an Effect on L929 Cell Viability
Pei-Tzu Li2, Ying-Chu Lee1, Namasivayam Elangovan2, Sin-Tak Chu1 2 ![]()
1. Institute of Biological Chemistry, Academia Sinica, Taipei, Taiwan
2. Institute of Biochemical Science, College of Life Science, National Taiwan University, Taipei, Taiwan.
Received 2006-8-28; Accepted 2007-1-12; Published 2007-1-18
Citation:
Li PT, Lee YC, Elangovan N, Chu ST. Mouse 24p3 Protein Has an Effect on L929 Cell Viability. Int J Biol Sci 2007; 3(2):100-107. doi:10.7150/ijbs.3.100. https://www.ijbs.com/v03p0100.htm
Other stylesAbstract
It is well known that mouse uterine 24p3 protein, is an acute phase protein, secreted from the L929 cell line, and that it will be induced by the dexamethasone stimulation of the cell. We investigated the possible effects of 24p3 protein on the L929 cell line, by observing its morphological change, ROS increase and viability decrease, by the process of culturing in a 24p3 protein-supplemented medium. Following the L929 cells′ exposure to the 24p3 protein supplement for a period of 72 hours, S-phase cells accumulated to a significant degree, suggesting that the entry into the G2/M phase from the S phase, in the cell cycle progression, was blocked. There was a significant decrease in cell numbers and increased DNA damage within the cells in the presence of 24p3 protein within the medium for 96 hours, implying that they have undergone pathway of cell death. After 96h incubation in low concentration of 24p3 protein, the result of PI/annexin V double staining showed cell death obviously. These results suggest that 24p3 protein-induced S phase arrest in the cell cycle, would cause DNA damage, followed by cell death in the L929 cells.
Keywords: Cell death, Lipocalin, S phase arrest.
1. Introduction
Lipocalin is a functionally diverse family of proteins that generally bind small hydrophobic ligands [1] and interact with cell surface-receptors [2]. All of these family proteins have limited amino acid sequence similarity, but do have a common tertiary structure, and it is also proposed that they are variously involved in the transport, modulation of cell growth and metabolism, as well as regulation of tissue development [1]. The importance of the lipocalin family has been recently noted. The murine 24p3 protein was identified as a protein induced in response to various proliferative signals, and is highly expressed in uterine luminal fluids, epithelial cells and epididymal caput fluid [3-5]. This protein has also been implicated as an apoptotic factor [6] which has also been proven to play a role in kidney cell differentiation [7]. Garay-Rojas et al. [4] found that the addition of dexamethasone to mouse L cell cultures (L929, from mouse connective tissue) led to an increase in 24p3 mRNA transcription and 24p3 protein synthesis, resulting in the accumulation of 24p3 protein in the culture media. They suggested that the 24p3 protein may be a mediator in the processes of growth, differentiation and inflammation. They also mentioned about the autocrine mechanism of 24p3 protein might exist in L929 cells. It suggests that the 24p3 protein plays a biological function in cells. Devireddy et al. [6] concluded that IL-3 deprivation activates 24p3 transcription, leading to both the synthesis and secretion of 24p3 protein, which in turn suggests the induction of apoptosis via an autocrine pathway. Liu et al. [8] provided evidence to show that 24p3 protein is highly induced during the acute phase response in vivo and can be regulated in cultured 3T3 cells by TNF-α, which is a major mediator of the acute phase response. A strong proapoptotic agent, MK886, has also been shown to act as an inducer of 24p3 gene expression, and it has been suggested that 24p3 protein may play an important role in MK886-induced apoptosis [9]. Recently, Bong et al. [10] have provided further evidence that the over-expression of the 24p3 gene may be related to apoptosis in mammary epithelial cells. Although it has been suggested that 24p3 protein may play a role in cell death, more direct evidence and further study are required to demonstrate the possible pathway by which 24p3 protein induces cell death. Show as a result according to the research of our laboratory, 24p3 protein is internalized and routed back of the L929 cell [11]. It suggests the protein plays a function in the cells. Based on the above, we aim to look for the possibility of affecting cell death by treatment with 24p3 protein. In this study, we use connective tissue cell line (L929) to investigate the directed effect of 24p3 protein on L929 cell viability. L929 cells do not synthesize 24p3 protein in the absence of glucocorticoid in the culture medium [4]. We purified the 24p3 protein from mouse uterine fluid then identified via MS spectrum and ready for use. This study allows us to explore the effect of 24p3 protein on L929 cells without the endogenous 24p3 protein. An extracellular fatty acid binding protein (Ex-FABP) is a lipocalin; it proposes that represents a stress protein in tissue [12]. We estimated the possibility of 24p3 protein as a stress protein via ROS measurement. As using PI/annexin V double staining, we observed the cell death as the supplement of 24p3 protein in culture medium. We found that 24p3 protein caused S phase arrest after 72 h incubation, and increased DNA damage after 96 h incubation. This suggested that cell cycle arrest occurs after 24p3 protein has been present for a significant time period, and then is followed by cell death.
2. Materials and Methods
Chemicals and biochemicals
Anti-mouse IgG FITC-conjugated antibody and anti-BrdU antibody (clone BU33) fluorescein isothiocyanate (FITC) conjugate were obtained from Sigma (St. Louis, MO. USA). The 24p3 protein was purified from DES-stimulated female mice as previously described [3]. All chemicals were of reagent grade quality.
Cell culture
The L929 cells were maintained in DMEM: F12 supplemented with 10% fetal calf serum. Cells were seeded into 24-well plates and left to acclimatize for at least 24 h before being treated. To induce death, cells were incubated with different amounts of 24p3 protein. After various time intervals, media containing non-adherent (floating) cells were removed, while the remaining adherent cells were incubated with trypsin/EDTA for 1 min at 37oC to remove them from the plastic. Both populations of cells were pooled before staining. After purification of the 24p3 protein from HPLC chromatography, we evaporated the excess acetonitrile in the sample and washed three times with water to avoid the contamination of the organic solvent.
Measurement of intracellular reactive oxygen species (ROS) production
The intracellular ROS were measured essentially as described by Halliwell and Whiteman [13]. Cells (2×104) were cultured on 96 well plates for 48 h and prepared for use. For the measurement of hydrogen peroxide, following incubation in 1.0 or 10 μM 24p3 protein containing medium for various time intervals, L929 cells were treated with 20 μM 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; Molecular Probes, Eugene, OR) for 15 min. The dichlorodihydrofluorescein (DCFH), the deacetylated product of DCFH-DA by intracellular esterases, reacts with H2O2 to form dichlorofluorescein (DCF), which is an oxidized fluorescent compound. The amount of intracellular hydrogen peroxide can be quantified by detection of DCF using a microfluorometer (Berthold Twinkle LB970) with excitation and emission wavelengths set at 485 nm and 535 nm, respectively.
The Trypan blue exclusion method for cell viability
Trypan blue is a dye that cannot enter cells through an intact membrane and therefore stains only those cells that are undergoing membrane disruption. L929 cells were cultured in medium, either with or without the presence of 24p3 protein for 72 h. The cells were then washed twice with PBS and stained with 0.025% Trypan blue in PBS for 3 min. Blue cells were then counted and these counts were used to calculate the relative proportion of the dead cells.
Assessment of apoptosis by PI/annexin V double staining
After incubation with 0.5 or 1.0 μM 24p3 protein, the cell death is measured using annexin V-FITC conjugate [14]. Cells were incubated in serum-free DMEM containing 10 ng/ml annexin V-FITC and 10 ng/ml PI for 15 min. The fluorescences of annexin V-FITC and PI were monitored by using a COULTER EPICS XL-MCL™ flow cytometer with excitation wavelength of 488 nm and emission wavelength of 525 and 625 nm, respectively. For each sample, 10,000 cells were analyzed. The necrotic cells lost cell membrane integrity that permits PI entry; PI+/annexin V− (upper left quadrant in the plot). Viable cells exhibit PI−/annexin V− (control cells are represented on the low left hand quadrant in the plot); early apoptotic cells exhibit PI−/annexin V+ (lower right quadrant in the plot); late apoptotic cells or necrotic cells exhibit PI+/annexin V+ (the upper right quadrant in the plot).
Fig 1
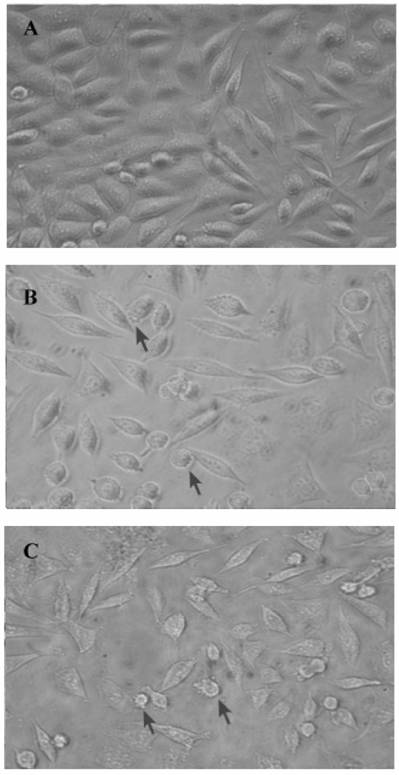
Comparative morphology of L929 cells under 24p3 protein incubation. L929 cells were incubated in medium in the presence or absence of 24p3 protein for 72 h. (A) control; (B) 1 μM 24p3 protein incubation; (C) 10 μM 24p3 protein incubation. The morphology of L929 cells was observed under bright field microscopy and the magnification was X 400. The cells with blebs are indicated by arrows.
Flow cytometry and fluorescence microscopy
Flow cytometry data was collected using a COULTER EPICS XL flow cytometer (Beckman-Coulter, FL, USA). Fluorescence was initiated by excitation at a wavelength of 488 nm and measured via a 625 nm filter. The fluorescence intensity of propidium iodide (PI) was quantified for 10,000 individual cells. For the examination of nuclear morphology, cells were stained with 2 μg/ml of 4', 6-diamidino-2-phenylindole (DAPI) to stain the DNA, and observed under a fluorescence microscope (AH3-RFCA, Olympus, Tokyo, Japan).
Immunodetection of incorporation of BrdU
L929 cells were cultured in the absence or presence of 24p3 protein (5 and 10 μM) for 69 h, and were then pulse-labeled (3 h) with 10 μM 5'-bromo-2'- deoxyuridine (BrdU). After labeling, the cells were washed twice with PBS and fixed in methanol. Fixed cells were washed with PBS to remove the organic solvent. FITC-labeled anti-BrdU antibody, diluted with PBS buffer containing 0.1% Triton X-100, was used to measure BrdU incorporation by fluorescence microscopy.
Statistical analysis
Data were analyzed, with the use of ANOVA by GraphPad software, and p<0.05 indicated significance. Post-test comparisons were made, with the use of the Student-Newman-Keuls test.
3. Results
24p3 protein changes the morphology of L929 cells
Because of the 24p3 protein had about 5 to 10 μM in mature mouse uterine fluid (data not shown); using the dosage of the protein in L929 cells should be reasonable near the physiological concentration. In our previous study, we have confirmed that 24p3 protein is specifically bound to the L929 cell surface and internalized into the cells via receptor-mediated endocytosis [11]. We attempt to know what kind of response can cause by 24p3 protein on L929 cells. L929 cells appeared spindle-shaped, adhered to the surface of the culture plate and were growth confluent after 72 h incubation (Fig. 1A). After confluent growth, some unattached but live round cells appeared in the crowded culture plate (Fig. 1A). After co-incubation with 1 μM 24p3 protein (Fig. 1B) or 10 μM 24p3 protein (Fig. 1C), prominent blebs on cell surface could be observed (indicated by arrows) and the cells had started rounding up, thus gradually detaching from the culture plate after 72 h in proportion to the concentrations of 24p3 protein treatment. Lesser cell numbers were observed in the 24p3 protein supplemented culture medium, in comparison to the unsupplemented medium, even using 1.0 μM 24p3 protein. These results suggest that changes may have occurred at the level of the plasma membrane. Trypan blue staining was used to assess whether the plasma membranes of cells treated with 24p3 protein were undergoing permeability changes. Trypan blue staining was performed on cells treated with either 1.0 or 10 μM 24p3 protein for 72 h. There was an increase in the death of L929 cells with the increasing concentration of 24p3 protein. After exposure to 10 µM 24p3 protein for 72 h, there was 60~70% decrease in cell viability compared to the control cells (data not shown). These results implied that the 24p3 protein has an effect on the cell proliferation during 24p3 protein treatment for 72 h.
Induction of ROS by 24p3 protein
Increasing intracellular ROS may lead to stress and resultant induction of cell death. Hence, we attempted to evaluate the ROS in L929 cells. For this purpose, the cultured cells were incubated with the membrane permeable probe DCFH/DA. The oxidation of DCFH to fluorescent DCF within the cells resulted in high cellular fluorescence intensities. We tested ROS production in L929 at various time intervals, in 24p3 protein supplement medium. The data is shown in figure 2. After exposure to 24p3 protein for 2 h, although no distinct morphological changes were observed, ROS increased almost up to 2-fold at 1.0 μM (Fig.2 ●) and 4.5-fold at 10 μM 24p3 protein treatment (Fig.2 ■) comparing to the control cells (Fig.2 ▲). We found that 24p3 protein could induce the increase of intracellular ROS significantly within a short time, upon the protein interaction with the cells in a dose-dependent manner. The intracellular ROS would maintain high levels for a period of time (data not shown). It suggests decrease the cell viability. It indicates 24p3 protein has the stress effect on L929 cells and causing cell morphological change.
Fig 2
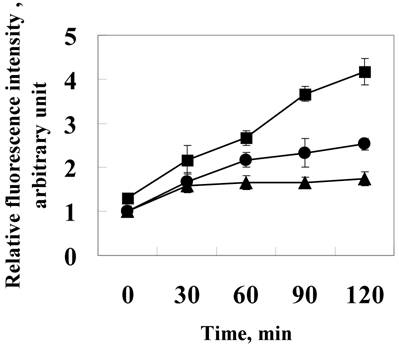
Induction of ROS by 24p3 protein treatment. L929 cells were treated with 1.0 mM (●) , 10 μM (■)24p3 protein or without 24p3 protein (▲) for various time intervals, followed by incubation with 20 μM DCFH-DA for 15 min. The amount of intracellular ROS can be quanitified by detection via a microfluorometer with excitation and emission wavelengths at 485nm and 535nm, respectively. The results were confirmed in multiple experiments and presented as the mean±SD, **p < 0.01, n=5.
24p3 protein treatment causes S-phase cell cycle arrest in L929 cells
Environmental stress affecting the cell progression, to demonstrate the effect of 24p3 protein on L929 cells progression, L929 cells were investigated by flow cytometry. Figure 3 shows the analyzed results of L929 cells using the fluorescent probe, propidium iodide (PI), in which PI binding with DNA produced a red fluorescence with 70% alcohol fixed cells. After treatment with 10 μM 24p3 protein for 24 h (Fig. 3A) (blank graph), the cell progression was similar to the control cells (solid graph). The percentage of cells in each phase is shown in the lower table. After treatment for 72 h (Figure 3B), the percentage of the cells in S phase increased significantly to 61.2%, compared to control cells with 6.0%. The G0/G1 cells decreased to 24.2%, compared to the control cells with 87.6%. After 72 h, the proliferation rate would have slowed down and hence more control cells occurred in the G1/G0 phase. These results agree well with those found for decreased cell viability in Figure 1. Cells are not able to proliferate normally under S phase cell cycle arrest. Upon the cell cycle arresting, cell death may be triggered for further incubation. After 72h incubation, the S-phase arrested may initiate the cell death and the morphological change would be observed at this moment.
Fig 3
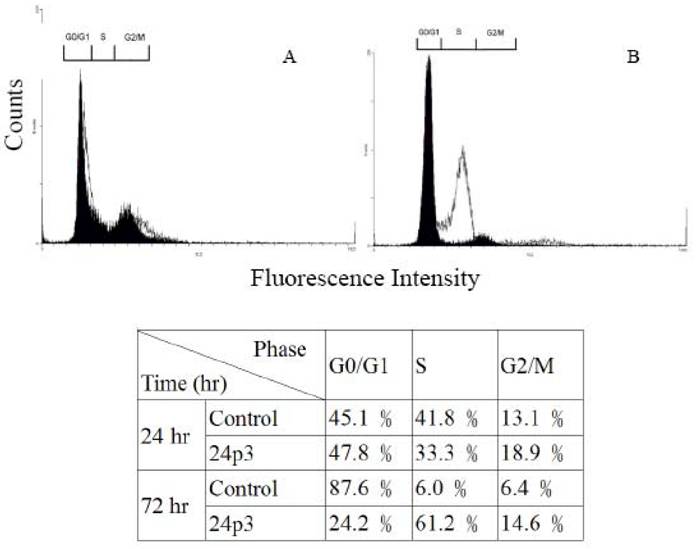
Cell cycle analysis of L929 cells treated with 24p3 protein. L929 cells were incubated in medium in the presence or absence of 10μM 24p3 protein for 24 and 72 h, and this was followed by incubation with propedium iodide as described in “Material and Methods”. The results of two independent experiments are shown. Solid graph, control cells; blank graph, 10 μM 24p3 protein-treated cells. The lower table shows the percentage of cells in each phase.
BrdU incorporation assay
After cell progression blocking, the cells may have further damage by 24p3 protein treatment. To confirm the effect of 24p3 protein on cell response, the incorporation of BrdU into DNA was measured using a DNA strand break assay. L929 cells were treated with 5.0 and 10 μM 24p3 protein for 69 h, and this was followed by BrdU labeling for 3 h. Immunodetection by FITC-labeled BrdU antibody was then carried out. An increase in DNA breakage could be detected by the presence of more FITC-labeled anti-BrdU antibody, as shown in figure 4 e and f. The percentage of BrdU incorporated into the cells increased from 2.1 ± 2.1% to 42.5 ± 9.6% under 5.0 μM 24p3 protein treatment, and from 2.1 ± 2.1% to 72.4 ± 5.6% under 10 μM 24p3 protein treatment (Fig. 4, lower table). The figures highlighted the accumulation of DNA breakage in S-phase arrested cells which leads to cell death.
Analysis of DNA damage by fluorescence microscopy
According to the above results, it showed the DNA damage after 24p3 protein treatment because of the S-phase arrest causing by the stress. To demonstrate the cell death after S phase arrest, we investigate the DNA status of the cell by DNA staining method. Using DAPI (4', 6-diamidino-2-phenylindole) staining, we analyzed changes in nuclear morphology of L929 cells after incubation with the 24p3 protein. Concentration dependent decrease in viability was observed in response to 24p3 protein application. After further (96 h) incubation with protein supplemented medium, fewer changes in the nuclei were observed in the control cells (Fig. 5, a & d) whereas, 1.0µM of 24p3 induced nuclei change in L929 cells (Fig.5, b & e). Thus, a lower concentration of 24p3 (1.0µM) affected L929 cells with DNA condensation in cell nuclei which is seen to be associated with nuclei membrane (indicated by arrows in Fig.5, e). On the other hand, a high concentration of 24p3 protein (10µM) treated cells showed a majority of dead cells (Fig.5, c). Smeared DNA is an indicator of DNA undergoing breakdown and condensed DNA appearing in dead cells (indicated by arrows in Fig. 5, f). It suggested the DNA fragmentation. These results emphasized that 24p3 protein is involved in DNA breakdown, leading to cell death. This result was coincided with the BrdU labeling assay (Fig. 4).
Fig 4
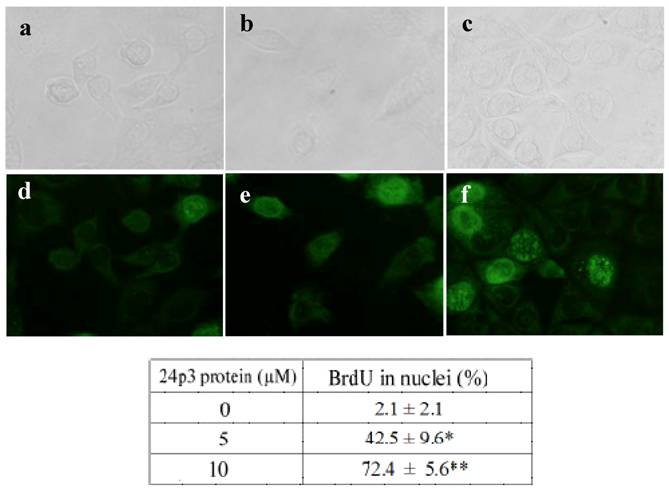
S phase arrest detected by BrdU incorporation analysis. L929 cells were cultured in the presence or absence of 24p3 protein (5 and 10 μM) for 69 h, and then were pulse-labeled (3 h) with 10 μM BrdU. After removing the BrdU, the cells were washed, and fixed for measurement of BrdU incorporation using FITC-labeled anti-BrdU antibody (1000 X dilution). Upper panel showed the L929 cells under bright field microscope, lower panel showed the L929 cells under fluorescence microscope. a and d, control cells; b and e, 5 μM 24p3 protein treated cells; c and f, 10 μM 24p3 protein treated cells. The percentage of BrdU incorporated cells is shown in the table. The data are expressed as mean ± SD, n=3. *, p<0.05; **, p<0.01.
Flow cytometry analysis of DNA damage
Flow cytometric analysis of 24p3 protein treated L929 cells showed severe dose dependent manner of DNA damage and fragmentation. A representative example of L929 cells, relative to the amounts of 24p3 protein treatment during 96 h of incubation, is given in Table 1. To determine the population of cells undergoing cell death, we used a cell cycle analysis technique. Cells were treated with 1 and 10 μM of 24p3 protein for 96 h and harvested at the indicated time interval. PI-labeled cells were analyzed with flow cytometry, demonstrating the decreased number of cells in the G1/G0 phase and the development of a sub-G1 dead cell population. The cell cycle progression of 10 μM 24p3 protein treated-L929 cells was shown in Fig. 6. After 24p3 protein treatment, the sub-G1 cells increased and the G1/Go cells decreased obviously. Upon the 96 h incubation with 24p3 protein, the S cells decreased, comparing to the 72 h incubation (Fig.3B). It indicated that the S-phase arrested cells would undergo the cell death after further incubation with the 24p3 protein. This data are coincided with Fig.5, the DNA breakdown in 10 μM 24p3 protein treatment for 96h. The distribution of cells in the sub-G1 and G1/G0 phase of the cell cycle is indicated in Table 1. As shown in Table 1, 57.6±6.5% of the L929 control cells were found in the G1/Go phase of the cell cycle, whereas about 24.0±5.9% and 12.7±3.7% cells were observed in the G1/Go phase with 1 and 10µM of 24p3 treated cells respectively, after 96 h. The data shown in Table 1, suggests that a low concentration of 24p3 protein (1.0 μM) could prolong the time of S phase arrest to 96h and showing 61.8±6.2% cells in S phase. Interestingly, there was an increase in sub-G1 cells to 42.7±6.7%, with an increase in 24p3 protein concentration, when compared to the control cells. However, the number of cells in the S phase of 10 μM 24p3 protein treated for 96 h would decrease to 39.5±5.5%, indicating cell death after S phase arrest. Thus, damaged cell nuclei might undergo DNA fragmentation, and accordingly, the number of dead cells will increase.
Table 1
Flow cytometry analysis of 24p3 protein on the L929 cell cycle after 96 h incubation.
| Sub-G1 | G1/G0 | S | G2/M | |
|---|---|---|---|---|
| Control cells | 4.2±3.5% | 57.6±6.5% | 28.8±4.6% | 9.5±3.5% |
| 1.0 μM 24p3 protein treatment | 6.7±4.9% | 24.0±5.9% | 61.8±6.2% | 8.3±3.8% |
| 10 μM 24p3 protein treatment | 42.7±6.7% | 12.7±3.7% | 39.5±5.5% | 5.1±3.3% |
Fig 5
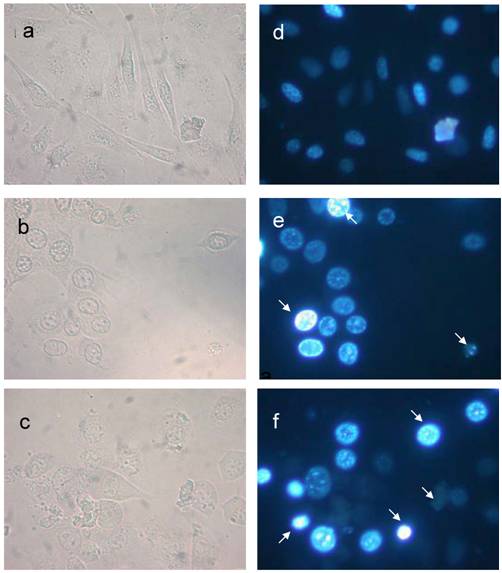
The nuclear morphological changes of 24p3 treated L929 cell. The assessment of nuclear damage in L929 cell using nuclear stain DAPI. The nuclear stained cells observed in bright field (a, b & c) and epifluorescence (d, e & f) microscopy. One micro molar 24p3 treated cell (b & e) showed aggregation of DNA (arrow) and more number of cellular DNA, smeared, aggregated and condensed (arrow) observed in 10 µM 24p3 treated cell (c & f).
Fig 6
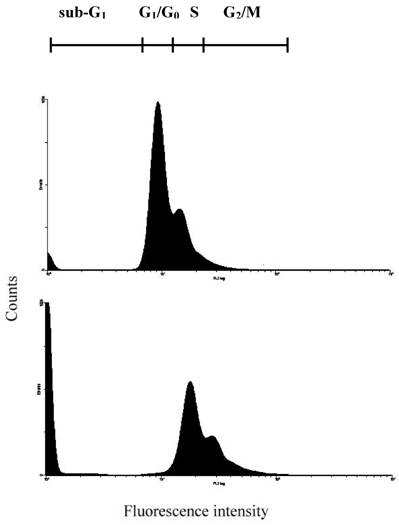
Detection of 24p3 protein induced L929 cells DNA damage. Cells were cultured in cover slips with 10 μM 24p3 protein supplement for 96h; the formation of DNA damage can be observed by cell cycle measurement. Upper panel, Control; Lower panel, 10 μM 24p3 protein treated cells. After treatment with 24p3 protein, cells were detached by trypsinization and stained with PI then determined cell cycle by flow cytometry.
Cell viability assay
To confirm the effect of 24p3 protein on the increase in number of sub-G1 cells, which indicates the decrease in cell viability, we used Trypan blue staining method for detecting cell viability. The data showed that cells were suppressive and that this effect was 24p3 concentration dependent (Fig. 7A). At 1.0µM of 24p3 protein, the viable cells were reduced up to 10% compared with the 24p3 protein-free control. It coincided with that more S-arrest cells but less sub-G1 cells appeared in 1.0 μM 24p3 protein-treated cells in Table 1. However, at a higher concentration of 10 µM of 24p3 protein, a greater reduction in the viability of L929 cells was observed. The difference in the level of absorbance, when compared with the control, was statistically significant at 10 µM 24p3 protein treated cells. The percentage of viable cells was reduced to 20% when compared with the control group, when the cells were treated with 10 µM of 24p3 protein (Fig. 7A). These results exhibited that induction of 24p3 protein might be involved in the cell death of L929. However, this method does not distinguish between apoptotic and necrotic cell death. We attempt to confirm the cell death via annexin V labeling method. Cells were double-labeled with annexin V-FITC and PI, and analyzed by flow cytometry (Fig.7B). Upon 96 hours 0.5 μM and 1.0 μM 24p3 protein treatment (Fig. 7B middle panel and left panel), the apoptotic cells (PI−/annexin V+) had yet appeared compared to the control population. The upper left would contain cells that take up PI but do not bind annexin V. These cells would most likely be necrotic. There were 11.2% and 6.85% PI+/annexin V− cells after 96 h 0.5 and 1.0 μM 24p3 protein treatment. The PI+/annexin V+ cells may be late apoptotic cells that are binding annexin V and taking up PI. There were 0.42% and 28.1% after 0.5 and 1.0 μM 24p3 protein treatment. Although, we did not detect the early apoptotic cells in this moment, but we observed the late apoptotic cells and sub-G1 cells in PI assay via flow cytometry. According to the results, 24p3 protein treated L929 cells may undergo the cell death via apoptosis.
Fig 7
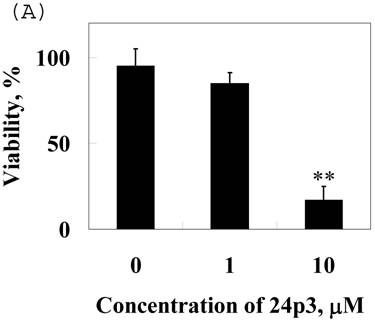
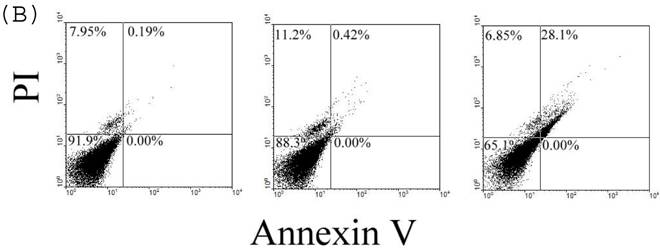
Effect of 24p3 protein treatment on plasma membrane integrity assessed by Trypan blue exclusion and PI/annexin V double staining. (A) L929 cells were washed twice with PBS and stained with 0.025% Trypan Blue in PBS for 3 min, after being treated by 1.0 and 10 μM 24p3 protein for 96 h. A minimum of 400 cells were counted for each treatment. The number of cells staining blue is expressed as a percentage. The data are expressed as mean ± SD, n=3. * p<0.01. (B) The percentage of cell death was detected by PI/annexinV-FITC double-staining was determined by flow cytometry. We calculated the percentages of dead cell in comparison to the untreated cells. Control cells are represented on the left plot, 0.5 μM 24p3 protein treated cells on the middle plot and 1.0 μM 24p3 protein treated cells on the right plot. Flow cytometry analysis of annexin V+ apoptotic cells. 2×106 L929 cells were incubated with 0.0, 0.5 , 1.0 μM 24p3 protein at 37 °C for 96h. At the indicated time, cells were stained simultaneously with annexin V-FITC and PI. The fluorescences of annexin V-FITC and PI were monitored by a flow cytometer. Dot plots are shown. For each sample, 10,000 cells were analyzed. The experiment shown is representative of three others performed.
Discussion
Studies by some laboratories have demonstrated that 24p3 protein is endocytosed after binding to cells [7, 15]. Recently, we also found that the 24p3 protein could bind to L929 cells and internalize into the cells [11]. Besides that, we could not observe the binding and internalizing of the 24p3 protein in A31 cell line (mouse embryo fibroblast), which is a negative control (data not shown). It indicated the cell type specificity of the 24p3 protein binding. A detailed understanding of the physiological role(s) of the protein is needed. We observed changes in 24p3 protein treated L929 cells. An increase in frequency of cells with blebs and lowered cell viability was observed with cells treated with both 1.0 and 10 μM 24p3 protein. The blebs may indicate the apoptotic body formation as the cell undergoing apoptosis. In fact, we found the appearance of apoptotic cells after further incubation with 24p3 protein. It has been suggested elsewhere that 24p3 protein may be involved in the cell death because of lower IL-3 levels that lead to T cell death [6, 16]. Our results reveal that 24p3 protein supplemented medium also induces L929 cell death directly. It has been suggested that 24p3 protein may be involved in an autocrine pathway [4, 6]. Richardson [17] has mentioned that 24p3 protein can be bound and internalized by its receptor, resulting in biological function. ROS can induce apoptotic or necrotic cell death dependent on the cell type and ROS concentration employed [18]. In this study, we showed that treatment of L929 cells with of 24p3 protein resulted in increase of the intracellular ROS and suggested the relationship between stress and L929 cell death. The reaction mechanism needs further study.
Although, we observed the necrotic cells at a 24p3 protein dosage of 1.0 μM in PI/annexinV double staining assay, it might result from the prolonged incubation of the cells in protein supplement medium. The data showed the late apoptotic cells after 96h protein treatment. 24p3 protein is likely to be a stress protein as EX-FABP [12]. Interestingly, we found that 24p3 protein blocked L929 cells at the S-phase and that they were unable to progress further in the cell cycle. This S-phase arrest is further supported by the 24p3 protein-induced BrdU incorporation. The incorporation of BrdU into DNA has been useful in various biological systems as a means of identifying S phase cells. It can bind with the replicating regions of chromosomes and is localized in the nuclei. Higher BrdU incorporation implies S phase arrest and that the cells are unable to enter the G2/M phase [19]. Besides that, one effect of S-phase arrest is accumulation of DNA damage or double-strand breaks, and this may lead to genomic instability [20]. Two approaches are used to detect and identify the apoptotic cells based on the DNA damage, viz., TUNEL assay and BrdUTP labeled assay. BrdUTP labeled assay was used as DNA damage assay in our study. DNA strand breaks were labeled with BrdUTP which was then detected by FITC-conjugated monoclonal antibody. The data allows one to relate apoptosis to cell cycle position [21, 22]. In our data, we found the DNA damage increased after 24p3 protein treatment via BrdU labeling assay. Thus, 24p3 protein inducing cell death may lead to S-phase arrest in L929 cells resulting in DNA damage. DNA damage evokes a complex of cellular responses, including delayed cell cycle progression and triggered cell death. Because of this, cell cycle arrest is complete before DNA repair and can activate the apoptotic cascade, leading to cell death [23, 24]. During cell apoptosis, the activation of endonuclease should initiate the DNA cleavage into small fragments and subsequently at the linkage of internucleosomal section (Fig. 5), is considered to be a hallmark of apoptosis [25, 26]. The observation of DNA damage, in our study, is the first to identify that 24p3 protein induces cell death via S-phase arrest and supports previous findings [6, 9, 16]. Current knowledge of cell death mechanisms, including apoptosis and necrosis [27], focuses on DNA damage under different mechanisms. Apoptosis is characterized by membrane blebing, DNA fragmentation, and nuclear condensation, all of which are influenced by the balance of pro-apoptotic and anti-apoptotic factors [28]. Based on our data, an increase in BrdUTP labeling, increase in the number of sub-G1 cells, the nuclear condensation and DNA fragments linkage with internucleosome appearing as the 24p3 protein treatment, indicated the cell apoptosis. As we did not find necrotic appearance, it suggests that 24p3 protein triggers the L929 undergoing an apoptotic pathway. It also corroborates the findings of other researchers [6, 9, 10]. Further studies will be needed in the future.
In summary, we demonstrated that 24p3 protein induces S-phase cell cycle arrest, followed by cell death in L929 cells. We suggest that 24p3 protein is a negative regulator of L929 cell growth. However, further investigation of the downstream events activated by 24p3 protein when it induces cell death, is required.
Acknowledgements
This work was partially supported by the grant NSC94-2311-B001-060 from the National Sciences Council, Taiwan.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Åkerstrom B, Flower DR, Salier J-P. Lipocalins: unity in diversity. Biochem Biophys Acta. 2000 ;1482:1-8
2. Flower DR. Beyond the superfamily: the lipocalin receptors. Biochem Biophys Acta. 2000 ;1482:327-336
3. Chu ST, Huang SL, Chen JM, Chen YH. Demonstration of a glycoprotein derived from the 243p gene in mouse uterine luminal fluid. Biochem J. 1996 ;316:545-550
4. Garay-Rojas E, Harper M, Hraba-Renevy S, Kress M. An apparent autocrine mechanism amplifies the dexamethasone- and retinoic acid-induced expression of mouse lipocalin-encoding gene 24p3. Gene. 1996 ;170:173-180
5. Chu ST, Lee YC, Nein KM, Chen YH. Expression immunolocalization and sperm-association of a protein derived from 24p3 gene in mouse epididymis. Mol Reprod Dev. 2000 ;57:26-36
6. Devireddy LR, Teodoro JG, Richard JA, Green MR. Induction of apoptosis by a secreted lipocalin that is transcriptionally regulated by IL-3 deprivation. Science. 2001 ;293:829-834
7. Yang J, Goetz D, Li JY, Wang W, Mori K, Setlik D, Du T, Erdjument-Bromage H, Tempst P, Strong R, Barasch J. Iron delivery pathway mediated by a lipocalin. Mol Cell. 2002 ;10:1045-1056
8. Liu Q, Nilsen-Hamilton M, Xiong SD. Synergistic regulation of the acute phase protein SIP24/24p3 by glucocorticoid and pro-inflammatory cytokines. Acta Phys Sinica. 2003 ;55:525-529
9. Tong Z, Wu X, Kehrer JP. Increased expression of the lipocalin 24p3 as an apoptotic mechanism for MK886. Biochem J. 2003 ;372:203-210
10. Bong JJ, Seol MB, Kim HH, Han O, Back K, Baik M. The 24p3 gene is induced during involution of the mammary gland and induces apoptosis of mammary epithelial cells. Mol Cells. 2004 ;17:29-34
11. Chou H-Y, Elangovan N, Lee Y-C, Lin H-H, Chu ST. Internaliztion and trafficking of mouse 24p3 protein in L929 cells. J Endocronol. 2006 ;191:1-9
12. Descalzi CF, Docin B, Zerege B, Cermelli S, Gentilt C, Cancedda R. Ex-FABP, extracellular fatty acid binding protein is a stress lipocalin expressed during chicken embryo development. Mol Cell Biochem. 2002 ;239:221-225
13. Hallowell B, Whiteman M. Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? British J. Pharm. 2004 ;14:231-225
14. Denecker G, Dooms H, van Loo G, Vercammen D, Grooten J, Fiers W, Declercq W, Vandenabeele P. Phosphatidylserine exposure during apoptosis proceeds release of cytochrome c and decrease in mitochondrial transmembrane potential. FEBS Lett. 2000 ;465:47-52
15. Elangovan N, Lee YC, Tzeng WF, Chu ST. Delivery of ferric ion to mouse spermatozoa is mediated by lipocalin internalization. Biochem Biophys Res Commun. 2004 ;319:1096-1104
16. Yousefi S, Simon H. Granulocyte apoptosis: death by a secreted lipocalin. Cell Death Different. 2002 ;9:595-597
17. Richardson DR. 24p3 and its receptor: dawn of a new iron age? Cell. 2005 ;123:1175-1177
18. Skulachev VP. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis. 2006 ;11:473-485
19. Lengronne A, Pasero P, Bensimon A, Schwob E. Monitoring S phase progression globally and locally using BrdU incorporation in TK+ yeast strains. Nuclei Acid Res. 2001 ;29:1433-1442
20. Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000 ;408:433-439
21. Li X, Melamed MR, Darzynkiewicz Z. Detection of apoptosis and DNA replication by differential labeling of DNA strand breaks with fluorochromes of different color. Exp Cell Res. 1996 ;222:28-37
22. Abend M, Frombeck S, Beuningen D Van. Indications for an active process underlying spontaneous and radiation-induced micronucleation in L929 cells. Int J Radiat Biol. 1999 ;75:1567-1578
23. O'Conner PM, Fan S. DNA damage check points:implications for cancer therapy. Prog Cell Cycle Res. 1996 ;2:165-173
24. Schwartz GK, Shah MA. Targeting the cell cycle:a new approach to cancer therapy. J Clin Oncol. 2005 ;23:9408-9421
25. Compton MM. A biochemical hallmark of apoptosis: internucleosomal degradation of the genome. Cancer Metastasis Rev. 1992 ;11:105-119
26. Walker PR, Weaver VM, Lach B, LeBlanc J, Marianna S. Endonuclease activities associated with high molecular weight and internucleosomal DNA fragmentation in apoptosis. Exp Cell Res. 1994 ;213:100-106
27. Darzynkiewicz Z, Juan G, Li X, Gorczyca W, Murakami T, Traganos F. Cytometry in cell necrobiology: analysis of apoptosis and accidental cell death (necrosis). Cytometry. 1997 ;27:1-20
28. Kim R, Emi M, Tanabe K, Murakami S, Uchida Y, Arihiro K. Regulation and interplay of apoptotic and anti-apoptotic cell death. J Pathol. 2006 ;208:319-326
Author contact
![]() Correspondence to: Dr. Sin-Tak Chu, Institute of Biological Chemistry, Academia Sinica, and P.O.BOX 23-106, Taipei, Taiwan. Tel: 886-2-23620261 Ext. 4531. Fax: 886-2-33664073. E-mail: stc316sinica.edu.tw.
Correspondence to: Dr. Sin-Tak Chu, Institute of Biological Chemistry, Academia Sinica, and P.O.BOX 23-106, Taipei, Taiwan. Tel: 886-2-23620261 Ext. 4531. Fax: 886-2-33664073. E-mail: stc316sinica.edu.tw.
Citation styles
APA
Li, P.T., Lee, Y.C., Elangovan, N., Chu, S.T. (2007). Mouse 24p3 Protein Has an Effect on L929 Cell Viability. International Journal of Biological Sciences, 3(2), 100-107. https://doi.org/10.7150/ijbs.3.100.
ACS
Li, P.T.; Lee, Y.C.; Elangovan, N.; Chu, S.T. Mouse 24p3 Protein Has an Effect on L929 Cell Viability. Int. J. Biol. Sci. 2007, 3 (2), 100-107. DOI: 10.7150/ijbs.3.100.
NLM
Li PT, Lee YC, Elangovan N, Chu ST. Mouse 24p3 Protein Has an Effect on L929 Cell Viability. Int J Biol Sci 2007; 3(2):100-107. doi:10.7150/ijbs.3.100. https://www.ijbs.com/v03p0100.htm
CSE
Li PT, Lee YC, Elangovan N, Chu ST. 2007. Mouse 24p3 Protein Has an Effect on L929 Cell Viability. Int J Biol Sci. 3(2):100-107.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.