Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(4):205-211. doi:10.7150/ijbs.3.205 This issue Cite
Research Paper
Structural stability and endonuclease activity of a PI-SceI GFP-fusion protein
Alireza G. Senejani, J. Peter Gogarten ![]()
Department of Molecular and Cell Biology, University of Connecticut, Storrs, CT, 06269-3125 U.S.A.
Received 2006-11-16; Accepted 2007-2-15; Published 2007-2-16
Abstract
Homing endonucleases are site-specific and rare cutting endonucleases often encoded by intron or intein containing genes. They lead to the rapid spread of the genetic element that hosts them by a process termed 'homing'; and ultimately the allele containing the element will be fixed in the population.
PI-SceI, an endonuclease encoded as a protein insert or intein within the yeast V-ATPase catalytic subunit encoding gene (vma1), is among the best characterized homing endonucleases. The structures of the Sce VMA1 intein and of the intein bound to its target site are known. Extensive biochemical studies performed on the PI-SceI enzyme provide information useful to recognize critical amino acids involved in self-splicing and endonuclease functions of the protein. Here we describe an insertion of the Green Fluorescence Protein (GFP) into a loop which is located between the endonuclease and splicing domains of the Sce VMA1 intein. The GFP is functional and the additional GFP domain does not prevent intein excision and endonuclease activity. However, the endonuclease activity of the newly engineered protein was different from the wild-type protein in that it required the presence of Mn2+ and not Mg2+ metal cations for activity.
Keywords: Homing endonuclease, intein, GFP-fusion protein, PI-SceI, Sce VMA
1. Introduction
Homing endonucleases are found as domains in inteins, or encoded by open reading frames in genetically mobile introns [1]. Similar to restriction enzymes, homing endonucleases cleave double stranded DNA, but recognize a larger DNA sequence of 14-40 nucleotides. Based on the type of conserved sequence motifs, homing endonucleases are classified into four major families: LAGLIDADG, GIY-X-YIG, H-N-H, and His-Cys [2]. Homing endonucleases of the 'LAGLIDADG' type are the largest known family [3], and the ones most frequently found in inteins [4].
Homing endonucleases facilitate transfer of their host genetic elements into intronless, or inteinless sites via a process called 'homing'. The homing process begins when the homing endonuclease makes a site-specific, double-strand break in the intronless, or inteinless allele [5, 6]. During the repair of the cleaved gene, which relies on the host's repair machinery, the intron/intein is copied to the previously intron/intein free homolog [1, 7]. Homing endonucleases and the process of homing have been more intensively studied in self splicing introns [1, 2, 7], and the process is assumed to be similar for inteins.
PI-SceI homing endonuclease is encoded as the only active intein identified in Saccharomyces cerevisiae (Sce VMA1 intein). PI-SceI is the name used in the homing endonuclease field to denote the Sce VMA1 intein [1], which is also known as VMA1 Derived Endonuclease (VDE) [8]. The structure of the Sce VMA1 intein and of the intein bound to its target site are known, with the self-spicing and endonuclease catalytic domains clearly defined [9, 10]. In addition to these two major domains the Sce VMA1 intein contains a sub-domain, near the self-splicing domain, that is involved in DNA recognition [10]. The PI-SceI homing endonuclease recognizes a long DNA sequence of more than 30 nucleotides; however, unlike most other homing endonucleases, it acts as a monomer on its recognition sites [9, 10].
To study proteins in-vivo, green fluorescence protein (GFP) provides an excellent means for monitoring gene expression and protein localization in cells. GFP was first isolated from the jellyfish Aequorea in the early 1960's [11], however, identification of its sequence and its first use as a reporter gene didn't occur for another 30 years [12, 13]. Since then GFP has been used frequently throughout the biological sciences. Typically, the GFP marker is attached to the carboxyl or amino terminus of proteins. However, introducing the GFP inside another protein might not significantly distort GFP's structure. This is because, as shown in figure 1A, both termini of GFP appear at one end of the folded protein, and are rather flexible on the surface of the so-called β-can [14].
Cartoon representation of the PI-SceI GFP-fusion protein structure, SDS-PAGE of purified proteins, and illustration of PCR steps performed to create and amplify the PI-SceI-GFP encoding gene. Panel A shows the ribbon structure of GFP and PI-SceI intein bound to its target sequence [10, 14]. GFP was inserted into the loop containing G117 (the green ball-and-stick model indicated by arrows); this position does not interfere strongly with the intein's functions. Panel B illustrates the use of overlapping primers and the PCR based cloning technique as described in Material and Methods. After the three primary PCR products were combined, the primers VMA-1 and VMA-2 were used to amplify the entire recombinant gene. Panel C, Lane 1: Commassie blue stain of the purified PI-SceI protein, about 51kDa, from E. coli XL1B (DE3) transformed with pET-15b PI-SceI; lane 2: Commassie blue stain of the purified PI-SceI117GFP protein, about 78kDa, from E. coli XL1B (DE3) transformed with pET-28a PI-SceI 117GFP separated on 10% SDS-gel.
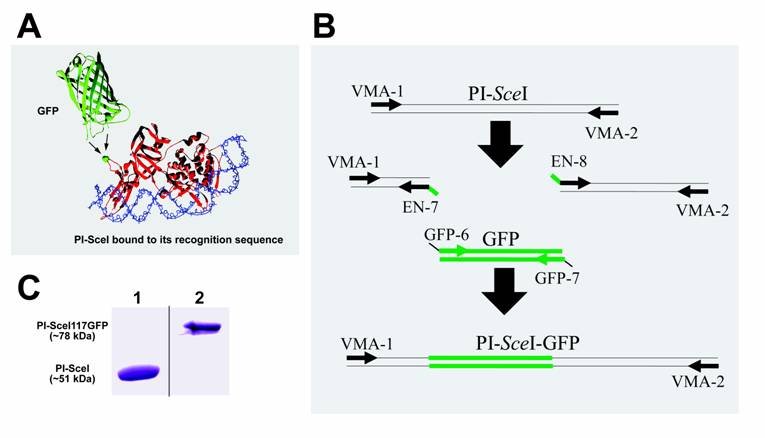
The PI-SceI homing endonuclease structure contains numerous loops which are distant from the active sites; suggesting that it might be possible to insert a functional GFP without disrupting either the self-splicing or the endonuclease activities. Here we describe insertion of a GFP domain within a loop of the sub-domain of the Sce VMA1 intein. The new recombinant protein was visualized using fluorescent microscopy indicating functional GFP; the tagged intein excises from the host protein; and the new GFP tagged homing endonuclease was functional. However, distinct from the wild type PI-SceI homing endonuclease, the new protein cleaves its target site only in presence of Mn2+.
2. Materials and Methods
PI-SceI cloning in pET expression plasmid
Plasmids containing the Saccharomyces cerevisiae V-ATPase catalytic subunit gene (vma1), with and without intein, [15] were kindly provided by Dr. Frederick S. Gimble (Purdue University). The gene encoding V-ATPase A-subunit intein was amplified by PCR using primers VMA-1 and VMA-2 (see Table 1). Following digestion with NdeI and BamHI, the coding sequence was cloned to the vector pET-15b (Novagen), behind the T7 promoter, for gene expression (pET-15b_PI-SceI). Six histidines are attached to the expressed protein product's N-terminus that can be used for purification of polyhistidine-tagged protein.
List of oligonuclotides used in this study.
| Oligo Name | Oligo Sequence | Use |
|---|---|---|
| VMA-1 | 5' CATATGGCTAGCTGCTTTGCCAAGGGTACC 3' | Sce VMA1 intein gene amplification and PI-SceI_117GFP construction |
| VMA-2 | 5' TTAGGATCCATTATGGACGACAACCTGG 3' | |
| Sc-0 | 5' GAAAAGAAACAATGGCTAGCGCAATTG 3' | vma1_PI-SceI117GFP construction |
| Sc-10 | 5' GGTGCTAGCCTGATGGAAACCATTTACG 3' | |
| Sc-11 | 5' CAGGATCCGAGAAATCACCAC 3' | |
| Sc-12 | 5' CTGGATCCGTAGATTCAGCAAATCTTTC 3' | |
| EN-7 | 5' CCTTTACTCATACCGTCGGGTGCTTTCTTTTG 3' | PI-SceI_117GFP construction |
| EN-8 | 5' CTATACAAAGGTAGAATTGTTGAGCTTGTC 3' | |
| GFP-6 | 5' CCCCCGACGGTATGAGTAAAGGAGAAGAAC 3' | |
| GFP-7 | 5' CAACAATTCTACCTTTGTATAGTTCATCCATG 3' |
PI-SceI_117GFP construction and cloning into a pET expression plasmid
Plasmids pVO190 containing the green fluorescent protein (GFP) mutant 1 gene [16] were kindly provided by Dr. Daniel J. Gage (University of Connecticut). This GFP exhibits a maximum excitation at 488nm and an emission maximum at 507nm [16]. To insert the GFP coding sequences in frame within the Sce VMA1 intein coding gene between the G117 and R118 encoding codons, a 363 bp fragment from the 5' end of the Sce VMA1 intein encoding gene was amplified using primers VMA-1 and EN-7 (Fig. 1B). The remaining 1020 bp fragment of the intein encoding gene was amplified using primers EN-8 and VMA-2. The GFP encoding gene was amplified using primers GFP-6 and GFP-7 (see Table 1; Fig. 1B). The three PCR products were first amplified separately. As shown in figure 1B, the entire PI-SceI 117GFP gene was amplified from the three shorter products using the beginning and end primers, VMA-1 and VMA-2. The primers EN-7 (PI-SceI reverse primer) and GFP-6 (GFP forward primers), and the primers EN-8 (PI-SceI forward primer) and GFP-7 (GFP reveres primers) have 22 nucleotides overlapping sequences (see Fig. 1B) facilitating recombinant PCR. The new PI-SceI 117GFP gene was initially cloned into the TA cloning vector to verify the identity and sequence of the cloned product. The complete coding sequences were then digested with NheI and BamHI and sub-cloned into the pET-28a (Novagene) expression vector. The new recombinant vector was named pET-28a_PI-SceI 117GFP.
vma1_PI-SceI117GFP construction and cloning into a pET expression plasmid
The complete vma1 gene, 3.2kb, was amplified by PCR using Sc-0 and Sc-12 primers. A truncated portion of the vma1 gene, containing the intein and surrounding sequence with a total length of 2.3kb, was amplified by PCR using Sc-10 and Sc-11 primers (see Table 1). After digestion with NheI and BamHI, the two sequences were cloned to the vector pET-28a (Novagene). To insert the GFP coding gene, 0.7kb, the two newly constructed recombinant pET-28a_vma1_PI-SceI 3.2kb and pET-28a_vma1_PI-SceI 2.3kb plasmids were digested with KpnI and SacII restriction enzymes. In parallel, the pET-28a_PI-SceI 117GFP plasmid was digested with the same restriction endonucleases. The internal part of the pET-28a_vma1 plasmids, 1.2kb in length, was replaced with the 1.9kb fragment, from pET-28a_PI-SceI 117GFP, which contains the GFP coding gene. The two recombinant plasmids were named pET-28a_vma1_PI-SceI 117GFP3.9kb and pET-28a_vma1_PI-SceI 117GFP3kb, respectively. Also, the intein free vma1 gene, which has the integration site, was amplified using primers Sc-0 and Sc-12 yielding a product of 1.8kb (see Table 1). The resulting PCR product is used as a target DNA to test the homing endonuclease activity.
Bacterial Strains and lysogenization of E. coli with DE3
The pET-15b and pET-28a expression systems are T7 promoter based. To create E. coli that contains an inducible bacteriophage T7 RNA polymerase gene [17] we transferred the T7 RNA polymerase gene into the genome of E. coli XL1-Blue MRF′ (Stratagene) using the λDE3 lysogenization kit (Novagen). The resulting E. coli strain, XL1B (DE3), was then used for the pET expression system.
Expression and purification of polyhistidine-tagged protein product
If not stated otherwise, transformed E. coli were grown overnight in a shaking incubator at 37oC in LB medium (10 g Tryptone, 5 g Yeast Extract, and 5 g NaCl in 1 L dH2O, pH 7.0) with tetracycline (15 μg/mL) supplemented with either 100 μg/mL ampicillin (for pET-15b), or 50 μg/mL Kanamycin (for pET-28a). One mL of the overnight culture was used to inoculate 10 mL of LB containing antibiotics and incubated at 37oC (225 rpm). Isopropyl thio-β-D-galactoside (IPTG) was added to a final concentration of 0.05 - 0.1 mM to induce the cultures. Expression was followed at 15oC (225 rpm) for 16 hours. Cells were harvested by centrifugation at 6000 rpm for 10 min, resuspended in 1 mL of sonication buffer (0.1 M Tris-HCl, pH 7.4, 1mM imidazole, 5% glycerol and 1 mM phenyl methyl sulfonyl fluoride), and disrupted by sonication (0.5 duty cycle; power output approximately 120) with micro-tip for 5 x 1 min at 4oC. The disrupted cells were centrifuged at 12,000 rpm for 15 minutes and the supernatant, which contains soluble proteins, was collected for further purification.
In the case of vma1_PI-SceI 117GFP3.9kb and vma1_PI-SceI 117GFP3kb expression, the transformed E. coli [XL1B (DE3)] were grown overnight in a culture wheel at 37oC in 10 ml LB with tetracycline (15 μg/mL) and kanamycin (50 μg/mL). The cultures were centrifuged at 5,000 rpm for 10 minutes and the pellets were re-suspended in minimal media supplemented with 0.01 - 0.05mM IPTG. The induced cultures were incubated in a shaker with 200rpm at 10oC for 72 hours. Cells were sonicated followed by centrifugation as described above.
Using a BD TALON™ Purification Kit (Clontech) the polyhistidine-tagged PI-SceI 117GFP protein was purified and 50 microliters of the samples from before and after the purification step were diluted by adding 6X sample buffer (10% SDS, 1.2mg/mL bromphenol blue, 0.6M DTT, 30% glycerol, 0.1M Tris/HCl pH 6.8) and heated to 95oC for 5 minutes to denature the protein. 10 - 50 microliters of this preparation were run in 10% denaturating Tris/Tricine SDS polyacrylamide gels electrophoresis system as described by Schagger and von Jagow [18].
In-vitro homing endonuclease activity assay
Using the purified protein, the in-vitro activity of the homing endonuclease was assayed with the target DNA re-suspended in PI-SceI reaction buffer [10 mM Tris-HCl, 100 mM dithiothreitol (pH 8.6 @ 25oC) plus 1-10 mM divalent ion (MgCl2, MnCl2, CoCl2, CaCl2, or ZnCl2) supplemented with 25 μg/ml BSA [15]].
Immunoblotting assay
For immunoblotting studies, protein extracts were denatured, separated on 10% SDS-polyacrylamide gels, and transferred to a nitrocellulose membrane. The resulting blots were blocked with 5% nonfat dry milk dissolved in TBST buffer (10mM Tris-HCl, 150mM NaCl, pH 7.5 + 0.05% tween-20). The blocked nitrocellulose filters were then incubated with primary antibody, rabbit anti-Sce VMA1 intein (New England Biolabs), diluted 1/2000 in blocking solution. After washing three times with TBST, the blots were incubated with secondary antibody conjugate (BIO-RAD anti-Rabbit IgG), diluted 1/3000 in blocking solution followed by washing three times with TBST. The blots were then developed by incubating with alkaline phosphate color substrate buffer (100mM Tris-HCl, 100mM NaCl, 50mM MgCl2, 0.33mg/ml NBT, and 0.17mg/ml BCIP).
3. Results
Expression and solubility of the wild type and GFP-fused PI-SceI protein
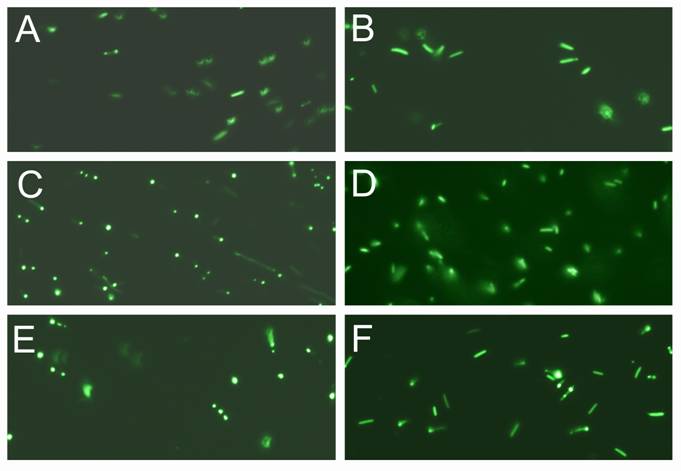
Expression and purification of the S. cerevisiae VMA1 intein was confirmed by denaturing SDS gel electrophoresis (Fig. 1C). Using overlapping primers and a PCR based cloning technique the complete GFP coding sequence was inserted in frame within the intein coding gene, between the G117 and R118 encoding codons (see experimental procedure). The new gene, encoding PI-SceI 117GFP, was cloned into the pET-28a expression vector and the histidine-tagged protein was purified (Fig. 1C). E. coli Xl1B (DE3) was used as a host for protein expression. The cells were grown overnight at 37oC followed by induction at 15oC. Using this low temperature expression condition the entire cytoplasm of the bacteria appeared green under the fluorescent microscope, indicating the GFP was soluble (Fig. 2). In contrast, when the induction was conducted at higher temperatures of 20 oC - 37 oC, the GFP-fused endonuclease was found in inclusion bodies. The aggregation was visible under the fluorescent microscope, where green clusters were found inside the bacteria cells (see Fig. 2). Results from SDS gel confirmed the observations made by microscopy (data not shown). The excitation maximum for the purified soluble fusion protein is at 488 nm (data not shown), as was reported for the isolated GFP domain used in our study [16]. The emission maximum for the purified fusion protein was at 510 nm compared to 507 nm for the isolated GFP domain [16].
Expression and visualization of the GFP inserted into PI-SceI and VMA1 PI-SceI proteins. A and B, E. coli XL1B (DE3) transformed with pET-28a PI-SceI 117GFP vector. C and D, E. coli XL1B (DE3) transformed with pET-28a vma1 PI-SceI 117GFP3.9kb vector. E and F, E. coli XL1B (DE3) transformed with pET-28a vma1 PI-SceI 117GFP3kb vector. A, C, and E expression was conducted in LB media at 10oC for 72 hours while B, D, and F expression was performed in minimal media at 10oC for 72 hours. Induction and cell grow in minimal media result in expression of soluble protein since the GFP is seen distributed in cytoplasm of the cells. In LB grown cells more expression was detected; however, the expressed proteins, formed inclusion body in parts of cells.
Homing endonuclease activity of wild type and GFP-fused PI-SceI proteins
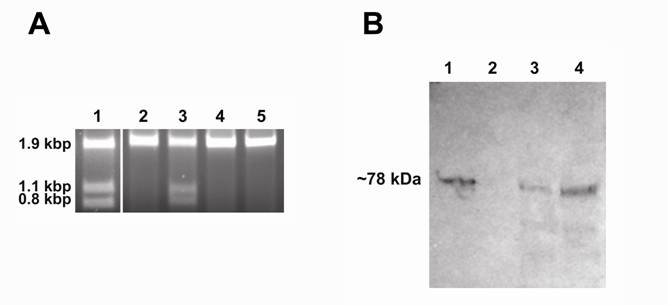
The wild type PI-SceI protein, purified from the pET expression system, was tested for homing endonuclease activity (Fig. 3A). Although the expressed protein carries six histidines at its N-terminal sequence followed by a thrombin cleavage site, the presence of these residues did not prevent normal function of the protein. Catalysis requires divalent metal ions. As previously reported the PI-SceI protein is active in reaction buffer in presence of Mg2+ or Mn2+ metal ions. However, the new GFP-fused PI-SceI enzyme did not show any sign of activity when tested in reaction buffer containing 1 - 10mM Mg2+. No activity was observed when pHs between 6 – 9 were tested using the reaction buffer containing 2.5mM Mg2+. Furthermore, we replaced Mg2+ with Mn2+, Co2+, Ca2+, or Zn2+ ions. Only in presence of Mn2+ divalent ions (Fig. 3A) did the GFP-fused PI-SceI cleave the target site. The greatest amount of the activity was observed only in the presence of 2.5mM Mn2+ metal ions; however, endonuclease activity was seen when tested in reaction buffer containing 1-10mM Mn2+.
Demonstration of endonuclease activity and immunoblot assay. Panel A: Digestion of the S. cerevisiae V-ATPase catalytic subunit gene (vma1) without intein. The reactions were performed for one hour at 37oC in PI-SceI reaction buffers in presence of 2.5 mM metal ions. The target site is present in a 1.9kb DNA sequence and after cleavage two fragments of 1.1 kb and 0.8 kb should be observed. Lane 1: Digestion with purified PI-SceI homing endonuclease using the reaction buffer in presence of 2.5mM MgCl2. Lane 2, 3, 4, and 5: Digestion with purified PI-SceI117GFP using the reaction buffer in presence of 2.5mM MgCl2, MnCl2, ZnCl2, or CaCl2 respectively. As shown, the GFP-fusion PI-SceI homing endonuclease only cleaves the target site in presence of MnCl2. Panel B: Immunoblot assay using commercial anti-Sce VMA1 intein antibodies. Lane 1: Purified PI-SceI117GFP protein (positive control). Lane 2: Protein extract from XL1B (DE3) without plasmid (negative control). Lane 3 and 4: Protein extracts from XL1B (DE3) with plasmids expressing PI-SceI 117GFP3.9kb and PI-SceI 117GFP3kb respectively. Expression was performed in minimal media as described in the experimental procedure.
Excision of the PI-SceI and GFP-fused PI-SceI proteins from their host proteins
Expression of the entire and truncated S. cerevisiae V-ATPase catalytic subunit gene (vma1) containing the Sce VMA1 intein was confirmed using SDS gel electrophoresis (data not shown). Since expression of these relatively large proteins at normal temperature caused misfolding and protein aggregation, the protein expression was conducted at low temperature of 12oC - 20 oC. Lower temperature facilitates proper protein folding for some portion of the expressed protein. Protein splicing was not efficient in E.coli for both, the entire and the truncated VMA1 proteins, about one third of the expressed proteins were detected as partially spliced proteins (data not shown).
After the insertion of GFP into the designated site, the sizes of the two new protein molecules were further increased to about 150kDa for vma1_PI-SceI 117GFP3.9kb and 118kDa for vma1_PI-SceI 117GFP3kb. The low temperature did not provide a sufficient environment for appropriate protein folding of these two massive multi-domains proteins. Therefore, as a result of expression, almost all of the expressed proteins were found as aggregated molecules in an inclusion body in the host. To overcome this aggregation problem, expression was performed in minimal media at low temperature. As shown in figure 2, under these conditions the proteins were soluble in the cytoplasm. Because of the low level of expression, the amount of proteins expressed was insufficient for purification. To test for in-vivo excision of the GFP-intein fused protein, immunoblot assay was performed using the antibody against S. cerevisiae vma1 intein. As shown in figure 3B, the GFP-intein fusion migrated with a size of about 78 kilo-Dalton (kDa) which corresponds to the excised protein without the extein attached. Under the chosen conditions autocatalytic self-excision was detected in E. coli for both the PI-SceI 117GFP3.9kb and the PI-SceI 117GFP3kb proteins.
4. Discussion
We describe an insertion of the GFP into the Sce VMA1 intein found in the S. cerevisiae V-ATPase catalytic subunit protein. The Sce VMA1 intein (also known as PI-SceI in the homing endonuclease field) is composed of two major domains, which are responsible for protein splicing and endonuclease activity, respectively. The self-splicing domain is structurally similar to the small intein found in Mycobacterium xenopi gyrase [6, 19]. Residues found at the N and C termini of the inteins are critical for splicing activity [4]. Therefore, addition of peptides to the N or C terminus likely would prevent the auto-splicing activity. The endonuclease activity exists as an additional distinct domain and a brief sub-domain associated with the splicing domain. The sub-domain was shown to be essential for homing endonuclease activity and is involved in DNA recognition [10]. Some of the residues found in the sub-domain region act as primary DNA attachment site to provide a structural deformation of the DNA. These changes facilitate the bending of the DNA into the major endonuclease domain and active sites. Mutation and photo cross-linking studies have demonstrated that interactions of the sub-domain with the DNA are critical: altering either some residues in the sub-domain or the DNA bases abolish binding [20, 21].
As demonstrated in figure 1, we have implanted the GFP protein into the sub-domain between Glycine 117 and Arginine 118. These residues, Glycine 117 and Arginine 118, are located fairly distant from both the DNA-protein interaction region, as well as from the protein splicing domain. GFP has a unique cylinder shape with both termini found at one end. Eleven beta-strands make up the beta-barrel of the can and an alpha-helix that runs through its center. By replacing the stop codon found at the end of the GFP encoding gene to encode for a Glycine we provided more flexibility to the carboxyl terminus of the GFP protein. The resulting protein, which consists of three distinct domains, is capable of performing all three functions: self excision, endonuclease activity and GFP fluorescence.
The majority of known inteins contain homing endonuclease and they are considered to be formed as a result of the splicing and homing endonuclease domains fusion [6]. Most natural proteins have more than one domain and gene fusion is known to be one of the key contributors to evolution of multi-domain proteins [22-24]. A naturally occurring example of interacting proteins being merged into a multi-domain protein are the Gyr A and Gyr B subunits of Escherichia coli DNA gyrase that , in yeast, have been fused into a single subunit protein, the topoisomerase II [25]. Multi-domain proteins are generally formed by covalent linkage between the N and C-terminus of domains; however, there are also examples of proteins created as a result of domain insertion [26].
Homing endonucleases, like restriction endonucleases, require divalent cations for cleavage to generate DNA fragments with a 5'-phosphate and a 3'-hydroxyl. The PI-SceI homing endonuclease is known to cleave the target site in presence of Mg2+ or Mn2+. Ca2+ alone, in the absence of Mg2+ or Mn2+, does not promote DNA cleavage [27]. In our study, the newly engineered GFP-fused PI-SceI protein did not show detectable endonuclease activity in presence of Mg2+, Co2+, Ca2+, or Zn2+ metal cations. However, the engineered protein cleaved the target site in presence of Mn2+ as cofactor. This altered ion specificity might be due to conformational changes in the metal binding pocket, or the substitution of Mn2+ for Mg2+ may cause structural modifications that relax the target site specificity, as shown previously for PI-SceI and some restriction endonucleases [28, 29].
Similar to inteins, many proteins cannot tolerate addition of GFP marker peptides to their termini. Fusion of the GFP to the C-terminus of mini-inteins, i.e., inteins without the endonuclease domain, has been reported previously [30]. However, if splicing and endonuclease activity of the intein are desired, the GFP needs to be inserted inside the protein. As demonstrated in this study, insertion of GFP into exposed loops on the outer structure, and away from protein's active site, can be accomplished without disrupting protein function. Fusing a functional GFP, or other markers, into a loop not interfering with protein function would greatly facilitate monitoring of protein expression. An intein construct that has endonuclease, excision and GFP functions may be useful to study the homing cycle of inteins in laboratory populations [31]. The ability to monitor the frequency with which a homing endonuclease containing allele occurs in such a population might allow using experimental evolution to select for homing endonucleases with novel properties. Furthermore, inserting the intein with GFP into a host protein can lead to a functional host protein, after splicing, without interference of the intein or GFP. If the endonuclease function of the large intein is not desired, the endonuclease domain can be replaced with a GFP domain; similarly GFP can be inserted into a mini intein. This can lead to expression of a relatively smaller intein-GFP protein and development of a system with more efficient expression of soluble proteins and splicing. Thus the expression of the host protein could be monitored without modifying it.
Acknowledgements
The authors thank Dr. James Knox for stimulating discussions on the GFP and PI-SceI protein structure; Drs. Daniel Gage and Frederick Gimble for providing plasmids encoding GFP and VMA1, respectively; Kristen Swithers and Holly Gardner for reading the manuscript, Drs. Harry Frank and Dariusz Niedzwiedzki for help with fluorescence spectroscopy, and Michael Wosczyna for his suggestions. The research was supported in part through the NASA Exobiology (NAG5-12367 and NNG05GN41G) and UCONN Research Foundation Programs.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Belfort M, Roberts RJ. Homing endonucleases: keeping the house in order. Nucleic Acids Res. 1997;25(17):3379-88
2. Guhan N, Muniyappa K. Structural and functional characteristics of homing endonucleases. Crit Rev Biochem Mol Biol. 2003;38(3):199-248
3. Stoddard BL. Homing endonuclease structure and function. Q Rev Biophys. 2005;38(1):49-95
4. Perler FB. InBase, the Intein Database. Nucleic Acids Res. 2000;28(1):344-5
5. Chevalier BS, Stoddard BL. Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001;29(18):3757-74
6. Gogarten JP, Senejani AG, Zhaxybayeva O, Olendzenski L, Hilario E. Inteins: structure, function, and evolution. Annu Rev Microbiol. 2002;56:263-87
7. Jurica MS, Stoddard BL. Homing endonucleases: structure, function and evolution. Cell Mol Life Sci. 1999;55(10):1304-26
8. Bremer MC, Gimble FS, Thorner J, Smith CL. VDE endonuclease cleaves Saccharomyces cerevisiae genomic DNA at a single site: physical mapping of the VMA1 gene. Nucleic Acids Res. 1992;20(20):5484
9. Duan X, Gimble FS, Quiocho FA. Crystal structure of PI-SceI, a homing endonuclease with protein splicing activity. Cell. 1997;89(4):555-64
10. Moure CM, Gimble FS, Quiocho FA. Crystal structure of the intein homing endonuclease PI-SceI bound to its recognition sequence. Nat Struct Biol. 2002;9(10):764-70
11. Shimomura O, Johnson FH, Saiga Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J Cell Comp Physiol. 1962;59:223-39
12. Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263(5148):802-5
13. Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ. Primary structure of the Aequorea victoria green-fluorescent protein. Gene. 1992;111(2):229-33
14. Yang F, Moss LG, Phillips GNJr. The molecular structure of green fluorescent protein. Nat Biotechnol. 1996;14(10):1246-51
15. Gimble FS, Thorner J. Purification and characterization of VDE, a site-specific endonuclease from the yeast Saccharomyces cerevisiae. J Biol Chem. 1993;268(29):21844-53
16. Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene. 1996;173:33-8
17. Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189(1):113-30
18. Schagger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166(2):368-79
19. Klabunde T, Sharma S, Telenti A, Jacobs WRJr, Sacchettini JC. Crystal structure of GyrA intein from Mycobacterium xenopi reveals structural basis of protein splicing [see comments]. Nat Struct Biol. 1998;5(1):31-6
20. Christ F, Steuer S, Thole H. et al. A model for the PI-SceIxDNA complex based on multiple base and phosphate backbone-specific photocross-links. J Mol Biol. 2000;300(4):867-75
21. He Z, Crist M, Yen H. et al. Amino acid residues in both the protein splicing and endonuclease domains of the PI-SceI intein mediate DNA binding. J Biol Chem. 1998;273(8):4607-15
22. Enright AJ, Iliopoulos I, Kyrpides NC, Ouzounis CA. Protein interaction maps for complete genomes based on gene fusion events. Nature. 1999;402(6757):86-90
23. Koonin EV, Wolf YI, Karev GP. The structure of the protein universe and genome evolution. Nature. 2002;420(6912):218-23
24. Pasek S, Risler JL, Brezellec P. Gene fusion/fission is a major contributor to evolution of multi-domain bacterial proteins. Bioinformatics. 2006;22(12):1418-23
25. Wang JC. DNA topoisomerases. Annu Rev Biochem. 1996;65:635-92
26. Aroul-Selvam R, Hubbard T, Sasidharan R. Domain insertions in protein structures. J Mol Biol. 2004;338(4):633-41
27. Noel AJ, Wende W, Pingoud A. DNA recognition by the homing endonuclease PI-SceI involves a divalent metal ion cofactor-induced conformational change. J Biol Chem. 2004;279(8):6794-804
28. Gimble FS, Thorner J. Homing of a DNA endonuclease gene by meiotic gene conversion in Saccharomyces cerevisiae. Nature. 1992;357(6376):301-6
29. Hsu M, Berg P. Altering the specificity of restriction endonuclease: effect of replacing Mg2+ with Mn2+. Biochemistry. 1978;17(1):131-38
30. Zhang A, Gonzalez SM, Cantor EJ, Chong S. Construction of a mini-intein fusion system to allow both direct monitoring of soluble protein expression and rapid purification of target proteins. Gene. 2001;275(2):241-52
31. Gogarten JP, Hilario E. Inteins, introns, and homing endonucleases: recent revelations about the life cycle of parasitic genetic elements. BMC Evol Biol. 2006;6:94
Author contact
![]() Correspondence to: J. Peter Gogarten, E-mail: gogartenedu, Phone: (860) 486-4061, Fax: (860) 486-4331
Correspondence to: J. Peter Gogarten, E-mail: gogartenedu, Phone: (860) 486-4061, Fax: (860) 486-4331