Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(5):318-327. doi:10.7150/ijbs.3.318 This issue Cite
Review
In search of a function for the most frequent naturally-occurring length polymorphism (MFNLP) of the HIV-1 LTR: Retaining functional coupling, of Nef and RBF-2, at RBEIII?
Mario Clemente Estable ![]()
Department of Chemistry and Biology, Ryerson University, 350 Victoria Street, Toronto, Ontario, Canada, M5B 2K3
Received 2007-5-15; Accepted 2007-6-7; Published 2007-6-11
Abstract
Although the prototypical HIV-1 LTR sequences were determined 22 years ago from the initial isolate, elucidating which transcription factors are critical to replication in vivo, has been difficult. One approach has been to examine HIV-1 LTRs that have gone through the gamut of in vivo mutation and selection, in search of absolutely conserved sequences. In this vein, RBEIII sequences are virtually 100% conserved in naturally occurring HIV-1 LTRs. This is because when they are mutated, the MFNLP recreates an RBEIII site. Here, I enumerate some retroviral mutation mechanisms, which could generate the MFNLP. I then review the literature corresponding to the MFNLP, highlighting the discovery in 1999, that RBEIII and MFNLP sequences, bind USF and TFII-I cooperatively, within the context of earlier and later work that suggests a role in HIV-1 activation, through T-cell receptor engagement and the MAPK cascade. One exception to the nearly absolute conservation of RBEIII, has been a group of long term non progressors (LTNP). These patients harbor deletions to the Nef gene. However, the Nef gene overlaps with the LTR, and the LTNP deletions abrogate RBEIII, in the absence of an MFNLP. I suggest that the MFNLP retains functional coupling between the MAPK-mediated effects of Nef and the HIV-1 LTR, through RBEIII. I propose that difficult-to-revert-mutations, to either Nef or RBEIII, result in the convergent LTNP Nef/LTR deletions recently observed. The potential exploitation of this highly conserved protein-binding site, for chimeric transcription factor repression (CTFR) of HIV-1, functionally striving to emulate the LTNP deletions, is further discussed.
Keywords: HIV-1 polymorphisms, MFNLP
1. HIV-1 polymorphisms
In the 24 years since the discovery of HIV-1 [1], with the impetus to cure AIDS, there have been an unprecedented number of publications examining retroviral polymorphisms. These have led to significant advances in understanding mechanisms generating them. In particular, when a provirus replicates along with host cellular DNA during mitosis, progeny are extremely homogenous. A case in point is clonal expansion of HTLV-1 [2], resulting in small variability over centuries [2-5]. This can be attributed to the high fidelity of mammalian cellular DNA replication, mediated by the 3'-5' exonuclease proofreading activity of DNA polymerase [6].
When a provirus replicates exogenously by infection, progeny can be extremely heterogeneous [4, 7]. Since exogenous retroviral replication involves transcription and reverse transcription, both could contribute towards generating polymorphisms. However, RNA polymerase appears to have transcript-assisted proofreading activity [8]. Therefore, for exogenously replicating retroviruses, such as HIV-1, polymorphisms are primarily generated during error prone reverse transcription (Fig. 1) [9, 10].
The MFNLP is generated during reverse transcription. (A) Synthesis of minus strong stop DNA, begins with t-RNA (clover-leaf structure) binding to the primer binding site (pbs), followed by RNA-templated polymerization of minus strong stop DNA (dashed arrow). RNase H degrades the template RNA after polymerization (arrows); (B) Template switching to the other RNA genome copy or to the same RNA, occurs via complementary sequences in the Repeat region (R). During the template switch, slippage can occur; (C) Minus strand extension could combine with slippage, templated and non-templated polymerization, resulting in duplication of the URE/RBEIII/partial E-box region (single white box, in RNA U3 region), generating a first copy of the MFNLP (two white boxes in U3 region), in the minus DNA. RNAse H continues to degrade the RNA genome, leaving just the polypurine tracts. (D) Initial synthesis of plus strand DNA begins from the central polypurine tract (cppt) and from the 3'-polypurine tract (3'-ppt), resulting in synthesis of a plus strand MFNLP (four white boxes); (E) Annealing of the free pbs, further extension and rearrangements, are followed by nuclear import and integration of the proviral HIV-1 sequences. (F) The final integrated proviral sequences contain a 5'-LTR and a 3'-LTR with U3, R, U5 sequences. In this model, both the 5' and 3'-LTR would contain the MFNLP. Note the Nef ORF in the 3'-LTR and the Nef/LTR sequences in the 5'-LTR. Integrity of the Nef TGA stop codon (ACTGCTGA) is critical for RBF-2 binding to the MFNLP.
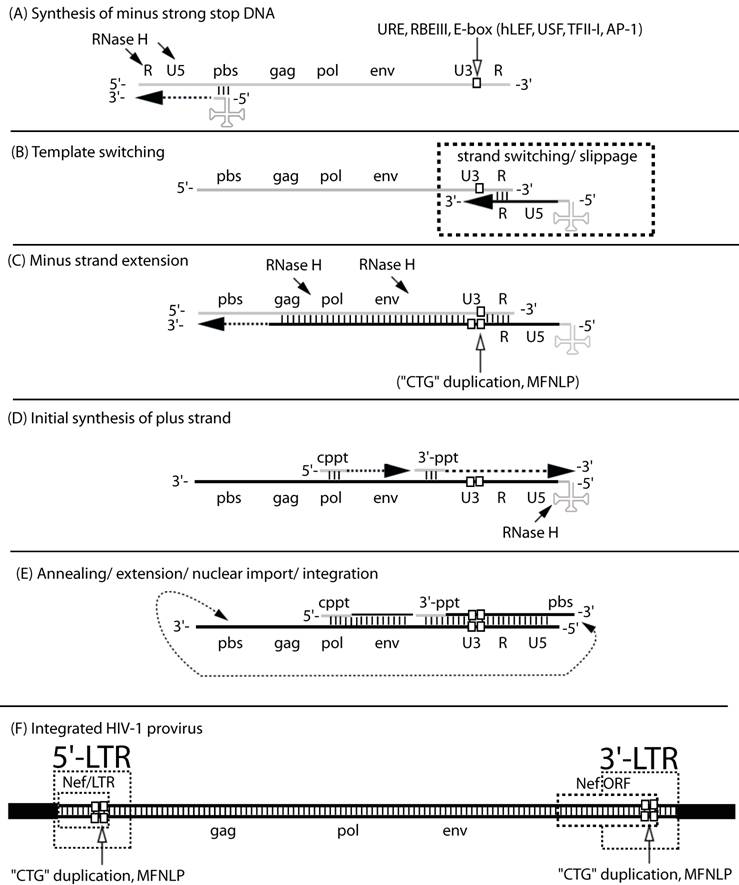
In particular the mis-incorporations produced by RNA-templated DNA synthesis and DNA-templated DNA synthesis (Fig. 1), have been proven to be exacerbated by dNTP imbalances both in vitro and in vivo [11-14]. Proof of the existence of these dNTP imbalances in a proportion of retroviral permissive cells [13] has provided a mechanistic explanation for observed retroviral G to A hypermutations in vivo [11, 13-17]. These mis-incorporations result in polymorphisms, since reverse transcriptase lacks 3'-to-5' exonuclease proof-reading activity [18], provided that the mutations result in replication competent virus that is naturally selected in vivo. More recently, RNA editing, through APOBEC enzymes, that deaminate cytosine to uracil, have also been proposed as a mechanism for retroviral G to A hypermutation [19]. In fact, the newest role for the HIV-1 Vif protein, is to counter the effect of APOBEC [20, 21]. Interestingly, Vif may also be exploiting the mutation potential afforded by APOBEC, in generating more HIV-1 diversity [21], rather than simply providing immunity from the innate host APOBEC defense mechanism.
Importantly, both templated and non-templated miss-incorporations, as well as “slippage”, can occur during the jumps of reverse transcription (Fig. 1) [22]. Therefore, in addition to point mutations, in vivo sampled retroviruses also frequently harbor deletions and insertions. Here, among the most probable mechanistic explanations, in addition to, or in combination with the above mentioned non-templated additions, is forced copy-choice strand switching during reverse transcription followed by mis-alignment and continued polymerization on the new template [23-28]. In essence this is a forced emulation of the reverse transcription "jumps" depicted in figure 1 [29]. This mechanism reconciles the fact that retroviruses can synthesize cDNA with only one RNA genomic copy [30] whereas viral particles carry 2 copies. The second would then serve the failsafe function of providing new template to "jump to" when reverse transcriptase encounters a nick. In addition, forced copy choice strand-switching provides a probable mechanistic explanation for phylogenetic revelations of inter-subtype recombinant HIV isolates [15, 31]. The implication is that chimeric exogenous retroviruses, containing transcribed genomic RNA from two different proviruses, are produced from doubly or multiply infected cells. In fact, recombination has now been shown to be the primary driving force for generating HIV-1 variants [32, 33], despite cellular retroviral superinfection resistance mechanisms [34].
The above retroviral mutation mechanisms highlight how retroviruses can generate sequence diversity. These mechanisms are shared by the larger family of elements capable of retro-transposition [7, 35]. However, HIV-1 undergoes such rapid exogenous viral replication and turnover [36-38], that natural selection generates enormously heterogeneous populations termed quasispecies [16, 39], or swarms, which have either temporally adapted to cell-specific microenvironments, or exist because their specific polymorphisms are neutral.
2. The MFNLP
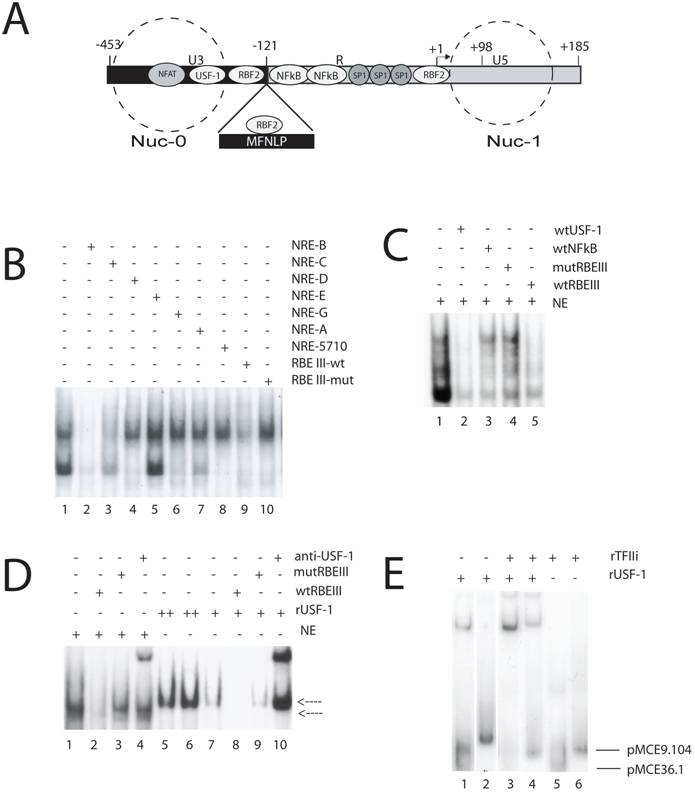
The proviral HIV-1 LTR sequences first investigated, were from the initial isolate [40] and rapidly became the prototypical LTR. These sequences orchestrate the cellular RNA polymerase II transcription machinery, in conjunction with viral proteins, and include a TATA-box, 3 SP1 sites, 2 NFkB enhancer elements, an RBEIII site and an upstream USF site (Fig. 2A) (reviewed in [41]). Importantly, the extensive HIV-1 polymorphism detected in vivo, extends to the LTR ([15, 42] and references therein).
MFNLPs and RBEIII, bind USF-1 and TFIIi. (A) Schematic representation of the HIV-1 LTR. Some transcription factors landmarks (NFAT, USF-1, RBF2, NFkB, SP-1) known to interact with target sequences on the HIV-1 LTR are indicated in their relative positions. The -121 position where the MFNLPs occur is indicated, with the MFNLP above, bound by RBF2 at the MFNLP RBEIII site. The -453 and +185 sites indicate the extremes of the LTR. The +1 site indicates the start of transcription and the +98 site, the end of Tar sequences. The dashed circles represent the approximate positions of two nucleosomes (Nuc-0 and Nuc-1). (B) RBEIII sequences specifically compete with a USF EMSA. The dsNREB oligosed as targets for the respective Hela nuclear extracts. Inclusion of competitor DNA sequences is indicated by a plus sign. (C) A USF-1 site competes specifically with RBF2 for an RBEIII site. A p32-labelled wt RBEIII ds oligo was used as target in EMSA with nuclear extracts from HeLa-s cells (NE, lanes 1-5). The target site that the respective competitor ds oligo sequences encompass, are designated with a + sign above each lane, and are listed on the right top of the panel. (D) rUSF-1 specifically binds to RBEIII, but results in a higher mobility EMSA than RBF2. A p32-labelled wt RBEIII ds oligo was used as target in EMSA with nuclear extracts from HeLa-s cells (NE, lanes 1-4) or with rUSF-1 (lanes 5-10). The target site that the respective competitor ds oligo sequences encompass, are designated with a + sign above each lane, and are listed on the right top of the panel. Lower arrow: NE-RBEIII (RBF2) complex. Upper arrow: rUSF-1-RBEIII complex. (D) rTFIIi and rUSF-1 bind cooperatively to MFNLP and wt RBEIII sites from 2 patients. A p32-labelled ds oligonucleotide target, encompassing the RBEIII site of either clone pMCE36.1 (lanes 1, 3, 5) or pMCE9.104 (lanes 2, 4, 6) was used as target in EMSA with either rUSF-1, TFIIi, or both, as indicated with a + sign above each lane, and listed at the right top of the panel. Lower arrow: Free pMCE36.1 ds oligonucleotide. Upper arrow: Free pMCE9.104 ds oligonucleotide. (B to E) The NRE, RBEIII, USF, NFκB sequences and EMSA conditions have been previously described [55, 64, 65, 67, 69].
In particular, a length polymorphism immediately 5' of the enhancer region, has been detected, investigated, or referred to by a growing list of publications [42-60]. This polymorphism was initially detected as a 24 base pair duplication by Golub, Li and Volsky, looking for LTR variants; but no regulatory cis-acting motifs were described within it, despite a correlation with increased transcription [43]. Blumberg and co-authors, described a similar duplication, in adult and pediatric patients, while looking for variant nef sequences, but again did not find any motifs that might explain functional significance [44]. Koken and co-authors, were the first to refer to this duplication as the “CTG”-motif, and noted that it encompassed a repeat of the sequence 5'-ACTGCTGA-3', and further found that a single “CTG”-motif virus, could out-compete an isogenic virus containing the duplication, in co-culture experiments, in 42 days [45]. These authors also noted similarities between the duplicated region and the recently-described TCF-1α (hLEF) binding site, and pointed out that HIVANT 70, as well as SIVCPZ, have the duplicated “CTG”-motif ([45] and references therein). These authors also pointed out that Nakanishi, Masamune and Kobayashi, had described the “CTG”-motif region as a novel cis-acting, cell-specific element [61]. In fact, in their paper, Nakanishi and co-authors designated this region (-157 to -121) the Upstream Regulatory Element (URE) and showed that it bound a transcription factor (designated URE-binding factors C1 and C2, by EMSA-specific analysis with nuclear extracts) distinct from AP-1 [61]. Furthermore, Koken and co-authors [45] noted that Zeichner, Kim and Alwine, had published data suggesting positive regulation in the “CTG”-motif region [62]. Soon afterwards, Kim, Gonzales, Zeichner and Alwine, published another paper, showing a more profound effect of mutations in the URE-region (“CTG”-motif), in the context of chromatin and viral replication, but attributed the effect to mutation of the TCF-1α (hLEF) binding-site [63]. In another paper by Koken and co-authors, they extended their initial finding, first showing that “CTG”-motif deleted, and “CTG”-motif duplicated isogenic viruses, are out-competed in cell culture assays by the monomeric isomeric virus [46]. These authors also showed that a Circa 64kD protein, binds with specificity to the 5'-ACTGCTGA-3' sequence, duplicated in the “CTG”-motif duplications and obtained similar EMSA results to those of Nakanishi and co-authors, but further reported no correlation between the “CTG”-motif duplication and disease stage or viral phenotype [46, 61]. So, by 1994, it was already clear that a cis-element, named URE, enhanced viral replication, contained a putative binding site for TCF-1α (hLEF), could specifically bind proteins from nuclear extracts, and was encompassed in a frequent Nef/LTR duplication (“CTG”-motif duplication). Furthermore it was clear that whatever the in vivo function of the “CTG”-motif duplication, in tissue culture assays with isogenic viruses, no dramatic effects were observed.
Further reports on this region of the HIV-1 LTR, included a paper from Michael and co-authors, that detected the TCF-1α duplication in uncultured samples from patients, and suggested it may mediate a repressive effect [47], and a paper by Ait-Khaled, also detecting the TCF-1α duplication, in post-mortem samples [48]. However, in 1996, in a cross-sectional analysis of nearly 500 HIV-1 LTRs, we published that the “CTG”-motif or URE region duplications, could best be aligned as a single larger polymorphism, which we named the most frequent naturally-occurring length polymorphism (MFNLP) of the HIV-1 LTR [42]. Furthermore, we proposed that the MFNLP encompassed a binding site for an unpublished nuclear factor named RBF-2, instead of TCF-1α [42]. This transcription factor was defined in a subsequent publication [64], by a TGA-specificity for the 5'-ACTGCTGA-3' sequence, in crude nuclear extracts, and correlated with a region of the HIV-1 LTR required for optimal responsiveness to Ras signaling through the MAPK pathway, in tissue culture transient transfections. This sequence was named the Ras- responsive binding element III (RBEIII) [64]. Strikingly, a subsequent publication by Linqi Zhang and co-authors, reported a TCF-1α duplication in a patient that progressed towards AIDS, despite the lack of NFκB binding [49]. Interestingly, these authors suggested a compensatory role for the TCF-1α duplication, in the face of NFκB binding-site abrogation, yet they found very little effect of the duplication in tissue culture [49]. Soon after, Kirchhoff and co-authors, reported on the presence of the MFNLP sequence in clinical samples, but did not point out that they duplicated RBEIII sequences, responsive to Ras [50]. Similarly, Zhang and co-authors, reported on more clinical samples with TCF-1α duplications, for which they found no phenotype, but failed to point out that the MFNLPs duplicated RBEIII [51]. Quinones-Mateu, reported similar detection of sequence duplications, which we called MFNLPs [53]. In 1998, we further expanded our knowledge about the RBEIII and MFNLP-binding proteins, by showing: (i) that hLEF (TCF-1α) can footprint on the wild type site but not on all MFNLPs or RBEIII sites; (ii) that all MFNLPs bind RBF-2; and (iii) that a small repressive effect was mediated through the MFNLP in transient transfections [65]. In 1999 Naghavi and co-authors also reported TCF-1α duplications in clinical samples [55, 66]. At this time, we had already attempted to purify the components of RBF-2 by chromatography, including an oligo-affinity step [67]. Purified RBF-2 included proteins of approximately 120 kD and 45kD, however our repeated attempts to obtain peptide sequences from large scale purifications were inconclusive [67].
Importantly, in the lab of Robert Roeder, Naghavi and I began doing some experiments to confirm her sequencing data that predicted various HIV-1 subtypes would not bind to the NRE USF site, since the sequence was not highly conserved, as I had shown for samples from the long-standing Vancouver Lymphadenopathy Study (VLAS) cohort [42]. While studying the role of USF-1 in HIV-1 LTR-directed transcription and viral replication, we used a double stranded RBEIII site and a mutant RBEIII site, as a non-specific competitor in an EMSA with labeled double stranded E-box targets, encompassing the upstream USF-1 site in the prototypical LAV HIV-1 LTR sequence, and nuclear extracts (NRE B USF site, Fig 2B, lane1). Surprisingly, we found that at 100X molar excess, RBEIII competed with the band shift, but the RBEIII mutant did not (Fig 2B, lane 9 versus lane 10). Although we at first assumed we had made a mistake, we immediately tested our new hypothesis that RBF2 contains USF-1, by competing a standard nuclear extract RBF2-RBEIII EMSA, with the E-box unlabelled target (Fig. 2C, lane 2). The hallmark of the RBF2 EMSA (Fig 2C, lane 1) is competition with 100X wild type RBEIII (Fig. 2C, lane 5) in the face of no competition with 100X mutant RBEIII (Fig. 2C, lane 4). The specific RBF2 bands were further tested with the non-specific 100X wtNFκB sequences from the prototype LAV HIV-1 LTR (Fig. 2C, lane 3). This important result indicated that our initial observation was true, and suggested that RBF2 contains USF-1. We therefore compared the EMSA mobility of rUSF-1 versus nuclear extracts, with an RBEIII target (Fig. 2D). The standard RBF-2 EMSA (Fig. 2D, lanes 1-4, lower arrow) appears to have a specific band that is lower in mobility than the specific band forming with rUSF-1 (Fig. 2D, lanes 5-10). Importantly, although the mobility is slightly different, both bands are specific, since the nuclear extract band can be competed with 100X molar excess of wtRBEIII (lane 2) but not with 100X mut RBEIII (lane 3) and the rUSF-1 band can be competed with 100X wtRBEIII (lane 8) but not 100X mutRBEIII (lane 9). Additionally, antibodies to rUSF-1 appear to supershift both RBF-2 and rUSF-1 (lanes 4 and 10), consistent with USF-1 being in RBF-2. Since our focus is on the MFNLP and MFNLPs encompass RBEIII sequences, the above results strongly suggested that MFNLPs would also bind USF-1. However, the canonical E-box CANNTG site, recognized by USF-1, is not present in RBEIII sites. Indeed Target Detection Analysis (TDA), with random targets, using USF-1, does not select an RBEIII site [68]. However, we noted that in the purified RBF-2 preparations there was a circa 120 kD component [67]. It was then Robert Roeder who immediately suggested to me that RBF-2 was comprised of USF and TFII-I. Because TFIIi and USF-1 bind the initiator/E-box region of the HIV-1 LTR, and RBF-2 had been shown to bind an RBEI site (in addition to the RBEIII site) that overlaps with the initiator sequences (Fig.2A) [64], the hypothesis that RBF-2 was comprised of USF and TFII-I seemed plausible. In fact, I found this was indeed the case, for at least 2 naturally occurring RBEIII HIV-1 LTR sequences (from clones pMCE36.1 and pMCE9.104, described elsewhere [65]), one of which is encompassed in an MFNLP (pMCE9.104) (Fig. 2D), in addition to the prototype HIV-1 LTR LAV sequences (above). Importantly, since binding of rUSF-1 alone (Fig. 2D, lanes 1 and 2) and binding of rTFIIi alone (lanes 5 and 6) is significantly weaker than when USF-1 is combined with TFIIi (lanes 3 and 4), it is likely that cooperative binding was occurring. Indeed, this important result suggested that USF-1 and TFIIi, when integrated at an RBEIII site, exert a stronger effect than individually, thus potentially explaining why their individual binding to an RBEIII site has not been previously observed.
Subsequent to our discovery, we asked the laboratory of Ivan Sadowski to confirm our results. Martin Hirst detected both USF and TFII-I in RBF-2 purifications [67], confirming our results. All the above initial findings were excluded from our publication about the NRE USF subtype sites [69], in order to better develop them into a clearer picture of the function of USF/TFII-I at the RBEIII sites, in collaboration with the lab of Ivan Sadowski (see below) [70].
Importantly, Jeeninga and co-authors, confirmed in 2000, that RBEIII sites are absolutely conserved, in LTR subtypes A through G [66]. A publication in 2000 by Gomez-Roman and co-authors, detected MFNLP sequences, duplicating RBEIII, in a long term non progressor (LTNP), but no deletions to RBEIII sites were found [56]. These LTNP are clinically distinct groups with lower viral replication and a slower or non-progressive form of HIV-1 infection (see below).
In 2000, Chen and co-authors found that an MFNLP from one of their patient samples, created a new AP-1 binding site, and identified c-Fos and JunB as binding proteins for their MFNLP [57]. Birch and co-authors described in 2001, LTNP from the Sydney Blood Bank Cohort (SBBC), that are accumulating progressively more LTR/Nef deletions, and some of these persons harbor RBEIII duplications in the form of MFNLPs [58]. Similarly, another LTNP study appears to have LTR/Nef sequences progressively accumulating greater deletions over time [60]. However it has not been at all clear if the LTR or the Nef deletions are responsible for the LTNP phenotype.
In 2002, Hiebenthal-Millow and Kirchhoff, found that in primary T cells, the MFNLP appears not to confer a major effect upon viral replication in culture [59]. A noteworthy observation about this paper is that these authors actually found enhanced transcriptional activity occurred when both the RBEIII site and MFNLPs were deleted, and that the MFNLP slightly reduced transcriptional activity.
Perhaps the most important paper about the role of the RBEIII site and the MFNLP is that from Chen and co-authors [70]. This paper expands on our initial discovery that RBF-2 is composed of TFIIi and USF, based on the TGA specificity of the EMSA used by Bell and Sadowski [64]. RBF-2 appears to be a heterodimer of USF-1/USF-2; and TFII-I appears to load USF-1/ USF-2 onto the RBEIII site. Importantly, this paper shows that RBEIII mutant LTRs are unresponsive to cross-linking of the T cell receptor or stimulation with PMA and ionomycin. Thus it appears that the phenotype of the RBEIII site and hence the compensatory MFNLP role, is to provide a target for Ras/MAPK activation of the HIV-1 LTR, in the context of chromatin [70, 71].
3. Nef, LTNP and RBF-2
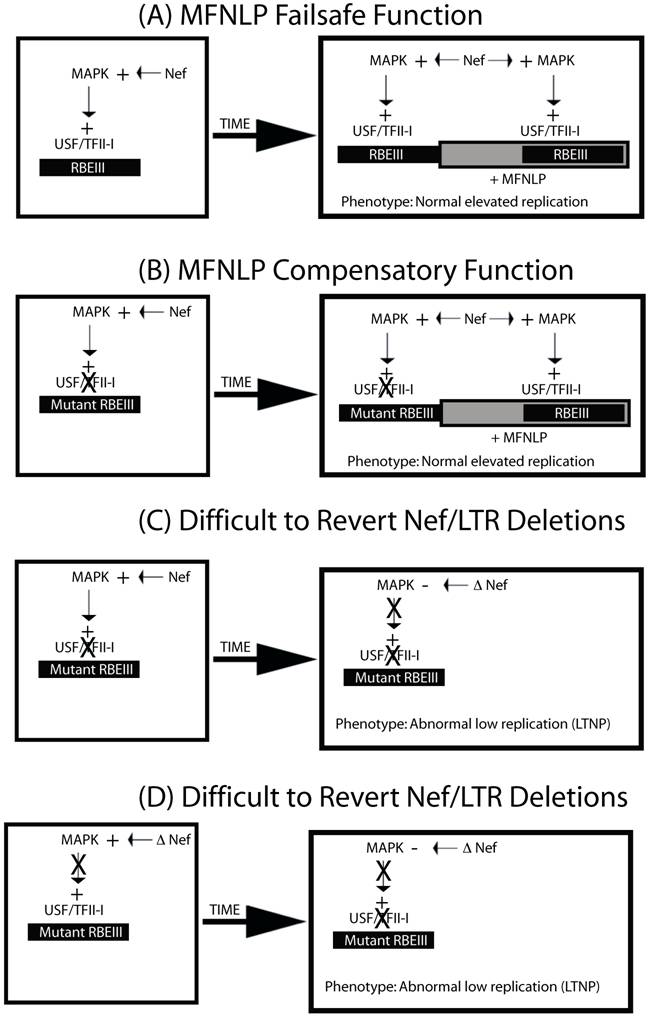
Importantly, the most recent SBBC LTNP study, indicates that there is a convergence in LTR/Nef deletions for these LTNPs [72]. These deletions appear to leave the sequences downstream of the NFκB sites intact, yet appear to be abrogating RBEIII sites, since they are deleted, without MFNLPs. Importantly, there are publications indicating that Nef can activate the MAPK pathway [73] and increase T cell activation [74]. Therefore Nef could be actually acting through the Nef/LTR cis-acting RBEIII site, which is abrogated in the SSBC LTNPs [72]. I therefore propose the model in figure 3. LTRs with normal RBEIII sites and a normal Nef sequence, could contribute to high viral replication and progress to AIDS (Fig 3A). The presence of MFNLPs in the context of normal RBEIII sites would simply provide a failsafe function. However, LTRs with mutated RBEIII site would require an MFNLP, as a compensatory mechanism (Fig 3 B). If however, LTRs receive difficult-to-revert Nef deletions, either after an RBEIII mutation (Fig. 3C) or prior to an RBEIII mutation (Fig. 3D), then the selective pressure to maintain the RBEIII site and the MFNLP disappears. The result of this model, would explain the convergent deletions observed in the LTR/Nef SSBC LTNP study, as well as being consistent with the role for RBF-2, RBEIII and MFNLPs.
MFNLPs compensate for RBEIII mutations, by maintaining functional coupling, of Nef and RBF-2. In this schematic representation four states are considered. In A & B, normal progressive disease with high viral titer would develop. In C & D, difficult-to-revert-mutations to Nef or the RBEIII site, would result in LTNP, and deletions of both RBEIII LTR sequences and Nef sequences. This is because, in our model, Nef can activate the MAPK pathway, and exert its effects through RBEIII.
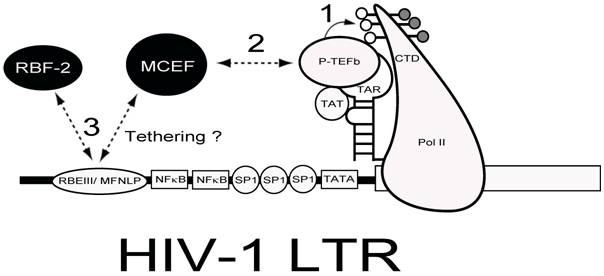
Whether my model about LTNP is true or not, the conserved nature of the RBEIII sites in vivo, makes it an important potential target for delivery of chimeric transcription factor repressors, which could emulate the LTNP phenotype, by blocking activation through this highly conserved site [75]. This is illustrated in Figure 4, where a chimeric transcription factor encompassing a repressor (such as MCEF[75, 76]) and a DNA binding partner (RBF-2), might have the potential to both block tat-transactivation and interfere with the function of RBEIII.
Chimeric Transcription Factor Repression (CTFR) of HIV-1. Delivery of a repressor with the ability to interfere with Tat-transactivation and bind to the RBEIII/ MFNLP is hypothesized. The HIV-1 LTR is drawn with some salient transcription factors indicated. The RNA stem-loop structure termed TAR, can form a ternary complex with HIV-1 TA and P-TEFb, to hyperphosphorylate the carboxy terminal domain (CTD) of RNA Polymerase II (PolII). Because MCEF interacts with P-TEFb and represses HIV-1 Tat-transactivation, if tethered to the RBEIII/ MFNLP site, MCEF could interfere with the binding of RBF-2 (3) and tat transactivation (1 and 2).
Competing interests
I have no competing interests.
Acknowledgements
I thank Mojgan Naghavi for providing her data (Fig. 2D), Robert Roeder for use of his lab and reagents between 1998 and 2001, Ivan Sadowski for the use of his lab and reagents between 1993 and 1998, and Martin Hirst for his role in confirming our RBF-2 preparations in the Sadowski lab contained USF and TFII-I. Maksymillian Niedzielski helped with making the figures and Robert Hopewell helped with the bibliography. The writing of this review was supported in part by grants from Canfar, CFI, OIT, NSERC and CIHR.
References
1. Barre-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vezinet-Brun F, Rouzioux C, Rozenbaum W, Montagnier L. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science. 1983;220:868-871
2. Wattel E, Vartanian JP, Pannetier C, Wain-Hobson S. Clonal expansion of human T-cell leukemia virus type I-infected cells in asymptomatic and symptomatic carriers without malignancy. J Virol. 1995;69:2863-2868
3. Gessain A, Gallo RC, Franchini G. Low degree of human T-cell leukemia/lymphoma virus type I genetic drift in vivo as a means of monitoring viral transmission and movement of ancient human populations. J Virol. 1992;66:2288-2295
4. Wain-Hobson S. Running the gamut of retroviral variation. Trends Microbiol. 1996;4:135-141
5. Komurian F, Pelloquin F, de The G. In vivo genomic variability of human T-cell leukemia virus type I depends more upon geography than upon pathologies. J Virol. 1991;65:3770-3778
6. Kunkel TA. DNA replication fidelity. J Biol Chem. 1992;267:18251-18254
7. Preston BD, Dougherty JP. Mechanisms of retroviral mutation. Trends Microbiol. 1996;4:16-21
8. Zenkin N, Yuzenkova Y, Severinov K. Transcript-assisted transcriptional proofreading. Science. 2006;313:518-520
9. Freed EO. HIV-1 replication. Somat Cell Mol Genet. 2001;26:13-33
10. Rausch JW, Le Grice SF. 'Binding, bending and bonding': polypurine tract-primed initiation of plus-strand DNA synthesis in human immunodeficiency virus. Int J Biochem Cell Biol. 2004;36:1752-1766
11. Cheynier R, Gratton S, Vartanian JP, Meyerhans A, Wain-Hobson S. G --> A hypermutation does not result from polymerase chain reaction. AIDS Res Hum Retroviruses. 1997;13:985-986
12. Meyerhans A, Vartanian JP, Hultgren C, Plikat U, Karlsson A, Wang L, Eriksson S, Wain-Hobson S. Restriction and enhancement of human immunodeficiency virus type 1 replication by modulation of intracellular deoxynucleoside triphosphate pools. J Virol. 1994;68:535-540
13. Vartanian JP, Plikat U, Henry M, Mahieux R, Guillemot L, Meyerhans A, Wain-Hobson S. HIV genetic variation is directed and restricted by DNA precursor availability. J Mol Biol. 1997;270:139-151
14. Vartanian JP, Meyerhans A, Sala M, Wain-Hobson S. G-->A hypermutation of the human immunodeficiency virus type 1 genome: evidence for dCTP pool imbalance during reverse transcription. Proc Natl Acad Sci U S A. 1994;91:3092-3096
15. Delassus S, Cheynier R, Wain-Hobson S. Evolution of human immunodeficiency virus type 1 nef and long terminal repeat sequences over 4 years in vivo and in vitro. J Virol. 1991;65:225-231
16. Goodenow M, Huet T, Saurin W, Kwok S, Sninsky J, Wain-Hobson S. HIV-1 isolates are rapidly evolving quasispecies: evidence for viral mixtures and preferred nucleotide substitutions. J Acquir Immune Defic Syndr. 1989;2:344-352
17. Vartanian JP, Meyerhans A, Asjo B, Wain-Hobson S. Selection, recombination, and G----A hypermutation of human immunodeficiency virus type 1 genomes. J Virol. 1991;65:1779-1788
18. Skalka A, Goff S. Reverse Transcriptase. New York: Cold Spring Harbor Press. 1993
19. Vartanian JP, Sommer P, Wain-Hobson S. Death and the retrovirus. Trends Mol Med. 2003;9:409-413
20. Suspene R, Rusniok C, Vartanian JP, Wain-Hobson S. Twin gradients in APOBEC3 edited HIV-1 DNA reflect the dynamics of lentiviral replication. Nucleic Acids Res. 2006;34:4677-4684
21. Simon V, Zennou V, Murray D, Huang Y, Ho DD, Bieniasz PD. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog. 2005;1:e6
22. Patel PH, Preston BD. Marked infidelity of human immunodeficiency virus type 1 reverse transcriptase at RNA and DNA template ends. Proc Natl Acad Sci U S A. 1994;91:549-553
23. Allain B, Lapadat-Tapolsky M, Berlioz C, Darlix JL. Transactivation of the minus-strand DNA transfer by nucleocapsid protein during reverse transcription of the retroviral genome. Embo J. 1994;13:973-981
24. Darlix JL, Vincent A, Gabus C, de Rocquigny H, Roques B. Trans-activation of the 5' to 3' viral DNA strand transfer by nucleocapsid protein during reverse transcription of HIV1 RNA. C R Acad Sci III. 1993;316:763-771
25. Guo J, Henderson LE, Bess J, Kane B, Levin JG. Human immunodeficiency virus type 1 nucleocapsid protein promotes efficient strand transfer and specific viral DNA synthesis by inhibiting TAR-dependent self-priming from minus-strand strong-stop DNA. J Virol. 1997;71:5178-5188
26. Klaver B, Berkhout B. Comparison of 5' and 3' long terminal repeat promoter function in human immunodeficiency virus. J Virol. 1994;68:3830-3840
27. Ramsey CA, Panganiban AT. Replication of the retroviral terminal repeat sequence during in vivo reverse transcription. J Virol. 1993;67:4114-4121
28. Temin HM. Retrovirus variation and reverse transcription: abnormal strand transfers result in retrovirus genetic variation. Proc Natl Acad Sci U S A. 1993;90:6900-6903
29. Panganiban AT, Fiore D. Ordered interstrand and intrastrand DNA transfer during reverse transcription. Science. 1988;241:1064-1069
30. Jones JS, Allan RW, Temin HM. One retroviral RNA is sufficient for synthesis of viral DNA. J Virol. 1994;68:207-216
31. Gao F, Robertson DL, Morrison SG, Hui H, Craig S, Decker J, Fultz PN, Girard M, Shaw GM, Hahn BH, Sharp PM. The heterosexual human immunodeficiency virus type 1 epidemic in Thailand is caused by an intersubtype (A/E) recombinant of African origin. J Virol. 1996;70:7013-7029
32. Jung A, Maier R, Vartanian JP, Bocharov G, Jung V, Fischer U, Meese E, Wain-Hobson S, Meyerhans A. Multiply infected spleen cells in HIV patients. Nature. 2002;418:144
33. Meyerhans A, Jung A, Maier R, Vartanian JP, Bocharov G, Wain-Hobson S. The non-clonal and transitory nature of HIV in vivo. Swiss Med Wkly. 2003;133:451-454
34. Nethe M, Berkhout B, van der Kuyl AC. Retroviral superinfection resistance. Retrovirology. 2005;2:52
35. Preston BD. Error-prone retrotransposition: rime of the ancient mutators. Proc Natl Acad Sci U S A. 1996;93:7427-7431
36. Coffin JM. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483-489
37. Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123-126
38. Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, Lifson JD, Bonhoeffer S, Nowak MA, Hahn BH. et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117-122
39. Biebricher CK, Eigen M. What is a quasispecies? Curr Top Microbiol Immunol. 2006;299:1-31
40. Wain-Hobson S, Sonigo P, Danos O, Cole S, Alizon M. Nucleotide sequence of the AIDS virus, LAV. Cell. 1985;40:9-17
41. Pereira LA, Bentley K, Peeters A, Churchill MJ, Deacon NJ. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res. 2000;28:663-668
42. Estable MC, Bell B, Merzouki A, Montaner JS, O'Shaughnessy MV, Sadowski IJ. Human immunodeficiency virus type 1 long terminal repeat variants from 42 patients representing all stages of infection display a wide range of sequence polymorphism and transcription activity. J Virol. 1996;70:4053-4062
43. Golub E, Li G, Volsky D. Differences in the basal activity of the long terminal repeat determine different replicative capacities of two closely related human immunodeficiency virus type 1 isolates. J Virol. 1991;64:3654-3660
44. Blumberg BM, Epstein LG, Saito Y, Chen D, Sharer LR, Anand R. Human immunodeficiency virus type 1 nef quasispecies in pathological tissue. J Virol. 1992;66:5256-5264
45. Koken SE, van Wamel JL, Goudsmit J, Berkhout B, Geelen JL. Natural variants of the HIV-1 long terminal repeat: analysis of promoters with duplicated DNA regulatory motifs. Virology. 1992;191:968-972
46. Koken SE, van Wamel JL, Geelen JL, Berkhout B. Functional Analysis of the ACTGCTGA Sequence Motif in the Human Immunodeficiency Virus Type-1 Long Terminal Repeat Promoter. J Biomed Sci. 1994;1:83-92
47. Michael NL, D'Arcy L, Ehrenberg PK, Redfield RR. Naturally occurring genotypes of the human immunodeficiency virus type 1 long terminal repeat display a wide range of basal and Tat-induced transcriptional activities. J Virol. 1994;68:3163-3174
48. Ait-Khaled M, McLaughlin JE, Johnson MA, Emery VC. Distinct HIV-1 long terminal repeat quasispecies present in nervous tissues compared to that in lung, blood and lymphoid tissues of an AIDS patient. Aids. 1995;9:675-683
49. Zhang L, Huang Y, Yuan H, Chen BK, Ip J, Ho DD. Identification of a replication-competent pathogenic human immunodeficiency virus type 1 with a duplication in the TCF-1alpha region but lacking NF-kappaB binding sites. J Virol. 1997;71:1651-1656
50. Kirchhoff F, Greenough TC, Hamacher M, Sullivan JL, Desrosiers RC. Activity of human immunodeficiency virus type 1 promoter/TAR regions and tat1 genes derived from individuals with different rates of disease progression. Virology. 1997;232:319-331
51. Zhang L, Huang Y, Yuan H, Chen BK, Ip J, Ho DD. Genotypic and phenotypic characterization of long terminal repeat sequences from long-term survivors of human immunodeficiency virus type 1 infection. J Virol. 1997;71:5608-5613
52. Salvi R, Garbuglia AR, Di Caro A, Pulciani S, Montella F, Benedetto A. Grossly defective nef gene sequences in a human immunodeficiency virus type 1-seropositive long-term nonprogressor. J Virol. 1998;72:3646-3657
53. Quinones-Mateu ME, Mas A, Lain de Lera T, Soriano V, Alcami J, Lederman MM, Domingo E. LTR and tat variability of HIV-1 isolates from patients with divergent rates of disease progression. Virus Res. 1998;57:11-20
54. Estable MC, Merzouki A, Arella M, Sadowski IJ. Distinct clustering of HIV type 1 sequences derived from injection versus noninjection drug users in Vancouver, Canada. AIDS Res Hum Retroviruses. 1998;14:917-919
55. Naghavi MH, Schwartz S, Sonnerborg A, Vahlne A. Long terminal repeat promoter/enhancer activity of different subtypes of HIV type 1. AIDS Res Hum Retroviruses. 1999;15:1293-1303
56. Gomez-Roman VR, Vazquez JA. et al. nef/long terminal repeat quasispecies from HIV type 1-infected Mexican patients with different progression patterns and their pathogenesis in hu-PBL-SCID mice. AIDS Res Hum Retroviruses. 2000;16:441-452
57. Chen P, Flory E, Avots A, Jordan BW, Kirchhoff F, Ludwig S, Rapp UR. Transactivation of naturally occurring HIV-1 long terminal repeats by the JNK signaling pathway. The most frequent naturally occurring length polymorphism sequence introduces a novel binding site for AP-1 factors. J Biol Chem. 2000;275:20382-20390
58. Birch MR, Learmont JC, Dyer WB, Deacon NJ, Zaunders JJ, Saksena N, Cunningham AL, Mills J, Sullivan JS. An examination of signs of disease progression in survivors of the Sydney Blood Bank Cohort (SBBC). J Clin Virol. 2001;22:263-270
59. Hiebenthal-Millow K, Kirchhoff F. The most frequent naturally occurring length polymorphism in the HIV-1 LTR has little effect on proviral transcription and viral replication. Virology. 2002;292:169-175
60. Kondo M, Shima T, Nishizawa M, Sudo K, Iwamuro S, Okabe T, Takebe Y, Imai M. Identification of attenuated variants of HIV-1 circulating recombinant form 01_AE that are associated with slow disease progression due to gross genetic alterations in the nef/long terminal repeat sequences. J Infect Dis. 2005;192:56-61
61. Nakanishi Y, Masamune Y, Kobayashi N. A novel cis-acting element that controls transcription of human immunodeficiency virus type 1 DNA, depending on cell type. J Virol. 1991;65:6334-6338
62. Zeichner SL, Kim JY, Alwine JC. Linker-scanning mutational analysis of the transcriptional activity of the human immunodeficiency virus type 1 long terminal repeat. J Virol. 1991;65:2436-2444
63. Kim JY, Gonzalez-Scarano F, Zeichner SL, Alwine JC. Replication of type 1 human immunodeficiency viruses containing linker substitution mutations in the -201 to -130 region of the long terminal repeat. J Virol. 1993;67:1658-1662
64. Bell B, Sadowski I. Ras-responsiveness of the HIV-1 LTR requires RBF-1 and RBF-2 binding sites. Oncogene. 1996;13:2687-2697
65. Estable MC, Bell B, Hirst M, Sadowski I. Naturally occurring human immunodeficiency virus type 1 long terminal repeats have a frequently observed duplication that binds RBF-2 and represses transcription. J Virol. 1998;72:6465-6474
66. Jeeninga RE, Hoogenkamp M, Armand-Ugon M, de Baar M, Verhoef K, Berkhout B. Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J Virol. 2000;74:3740-3751
67. Estable MC, Hirst M, Bell B, O'Shaughnessy MV, Sadowski I. Purification of RBF-2, a transcription factor with specificity for the most conserved cis-element of naturally occurring HIV-1 LTRs. J Biomed Sci. 1999;6:320-332
68. Bendall AJ, Molloy PL. Base preferences for DNA binding by the bHLH-Zip protein USF: effects of MgCl2 on specificity and comparison with binding of Myc family members. Nucleic Acids Res. 1994;22:2801-2810
69. Naghavi MH, Estable MC, Schwartz S, Roeder RG, Vahlne A. Upstream stimulating factor affects human immunodeficiency virus type 1 (HIV-1) long terminal repeat-directed transcription in a cell-specific manner, independently of the HIV-1 subtype and the core-negative regulatory element. J Gen Virol. 2001;82:547-559
70. Chen J, Malcolm T, Estable MC, Roeder RG, Sadowski I. TFII-I regulates induction of chromosomally integrated human immunodeficiency virus type 1 long terminal repeat in cooperation with USF. J Virol. 2005;79:4396-4406
71. Sadowski I, Mitchell DA. TFII-I and USF (RBF-2) regulate Ras/MAPK-responsive HIV-1 transcription in T cells. Eur J Cancer. 2005;41:2528-2536
72. Churchill MJ, Rhodes DI, Learmont JC, Sullivan JS, Wesselingh SL, Cooke IR, Deacon NJ, Gorry PR. Longitudinal analysis of human immunodeficiency virus type 1 nef/long terminal repeat sequences in a cohort of long-term survivors infected from a single source. J Virol. 2006;80:1047-1052
73. Schrager JA, Der Minassian V, Marsh JW. HIV Nef increases T cell ERK MAP kinase activity. J Biol Chem. 2002;277:6137-6142
74. Schrager JA, Marsh JW. HIV-1 Nef increases T cell activation in a stimulus-dependent manner. Proc Natl Acad Sci U S A. 1999;96:8167-8172
75. Niedzielski MF, Hopewell R, Ismail Z, Estable MC. MCEF is localized to the nucleus by protein sequences encoded within three distinct exons, where it represses HIV-1 Tat-transactivation of LTR-directed transcription. Int J Biol Sci. 2007;3:225-236
76. Estable MC, Naghavi MH, Kato H, Xiao H, Qin J, Vahlne A, Roeder RG. MCEF, the newest member of the AF4 family of transcription factors involved in leukemia, is a positive transcription elongation factor-b-associated protein. J Biomed Sci. 2002;9:234-245
Author contact
![]() Correspondence to: Dr. Mario Clemente Estable, mestableca
Correspondence to: Dr. Mario Clemente Estable, mestableca