Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(5):335-341. doi:10.7150/ijbs.3.335 This issue Cite
Research Paper
The responses of HT22 cells to the blockade of mitochondrial complexes and potential protective effect of selenium supplementation
Jun Panee1, Wanyu Liu1, Kyoko Nakamura2, Marla J. Berry1 ![]()
1. Department of Cell & Molecular Biology, John A Burns Medical School, University of Hawaii, Honolulu HI 96813, USA
2. School of Medicine, Juntendo University, 2-1-1, Hongo, Bunkyo-ku, Tokyo 113-8421, Japan
Received 2007-6-5; Accepted 2007-7-11; Published 2007-7-13
Abstract
Mitochondria are the major reactive oxygen species (ROS) – generating sites in mammalian cells. Blockade of complexes in the electron transport chain (ETC) increases the leakage of single electrons to O2 and therefore increases ROS levels. Complexes I and III have been reported to be the major ROS-generating sites in mitochondria. In this study, using mouse hippocampal HT22 cells as in vitro model, we monitored the change of intracellular ROS level in response to the blockade of ETC at different complex, and measured changes of gene expression of antioxidant enzymes and phase II enzymes, also evaluated potential protective effect of selenium (Se) supplementation to the cells under this oxidative stress. In summary, our results showed that complex I was the major ROS-generating site in HT22 cells. Complex I blockade upregulated the mRNA levels of glutamylcysteine synthetase heavy and light chains, glutathione-S-transferases omega1 and alpha 2, hemoxygenase 1, thioredoxin reductase 1, and selenoprotein H. Unexpectedly, the expression of the enzymes that directly scavenge ROS decreased, including superoxide dismutases 1 and 2, glutathione peroxidase 1, and catalase. Se supplementation increased glutathione levels and glutathione peroxidase activity, indicating a potential protective role in oxidative stress caused by ETC blockade.
Keywords: electron transport chain, complex I, rotenone, ROS, antioxidant
1. Introduction
Over 90% of tissue respiration is due to oxygen consumption by mitochondria [1]. Superoxide is generated by one electron reduction of O2 by the electron transport chain (ETC), leading to formation of other reactive oxygen species (ROS). The percentage of O2 that is converted to superoxide in mitochondria has been reported ranging from 0.15 to 2% [2]. Production of ROS from the ETC is generally considered as the major continuous source of cellular oxidative stress, and as a major participant in pathophysiological processes, particularly aging and its associated degenerative diseases [3, 4].
The ETC in eukaryotic mitochondria consists of four complexes, transferring electrons from NADH to O2. When the ETC is blocked by an inhibitor, the reduction state of electron carriers increases on the substrate side of the inhibitor, while those on the oxygen side become more oxidized. Reduced complexes in the ETC have the ability to produce superoxide [1].
Mitochondrial complex I accepts electrons from NADH and passes them through flavin and iron-sulfur centers to ubiquinone [5]. Many structurally diverse hydrophobic compounds have been described to inhibit complex I by interfering with ubiquinone reduction, such as piericidin A (A type), rotenone (B type), and capsaicin (C type) [6]. Complex II uses succinate as substrate and provides electrons to ubiquinone. Malonate (MA) inhibits succinate dehydrogenase [7]. Complex III accepts electrons from ubiquinone and passes them on to cytochrome c [8]. It has two redox centers, known as Qo and Qi that can be inhibited by stigmatellin (ST) and Antimycin A (AA), respectively [8, 9]. Both complexes I and III have been reported to be the major ROS-generating sites in ETC [10-13].
Inhibition of the activity of complex I in the ETC, e.g. upon exposure to the pesticide rotenone (Rot), has been identified as one of the major pro-oxidative factors causing Parkinson's disease (PD) [14-16]. Animal studies showed that chronic exposure to Rot reproduces features of Parkinsonism in rats [17, 18], and selenium (Se), a trace element possessing antioxidant properties, prevented or slowed down neuronal injury in mouse PD models [19, 20].
In this study, we treated murine hippocampal HT22 cells with Rot, MA, AA/ST to block complexes I, II and III, respectively. We measured the changes of intracellular superoxide level induced by the treatments. We also monitored the change of gene expression of antioxidant and phase II enzymes in response to Rot treatment and investigated the potential protective effect of Se supplementation in this oxidative stress.
2. Materials and Methods
Cells
HT22 cells were maintained in Dulbecco's Modified Eagle Medium with 10% fetal bovine serum, 15 µg/ml gentamicin, 50 µg/ml ampicillin, and 4 mM L-glutamine supplement, in 5% CO2 at 37ºC, 50 % relative humidity.
Chemicals
Dihydroethidium (hydroethidine, HEt) was purchased from Molecular Probes (Carlsbad, CA). GSH/GSSG-412 TM assay kit was purchased from Oxis (Foster City, CA). RNeasy Mini Kit was purchased from Qiagen (Valencia, CA). SuperScript III First-Strand synthesis system and Platinum SYBR Green qPCR SuperMix-UDG kit were purchased from Invitrogen (Carlsbad, CA). All other chemicals were purchased from Sigma (St. Louis, MO).
Instruments
A Beckman Coulter Epics XL-MCL flow cytometer (Fullerton, CA) was used to measure the mean fluorescence intensity of the oxidized HEt to indicate the intracellular ROS level, a DU7500 Spectrophotometer (Beckman, Fullerton, CA) was used for total glutathione (GSH) and glutathione peroxidase (GPx) activity assays, and a LightCycler 2.0 real-time PCR machine (Roche, Indianapolis, IN) was used for the relative quantification on the mRNA levels.
Cell culture, treatment and harvesting
HT22 cells were seeded at a density of 4.5e4 cells /cm2 in multi-well cell culture plates (Costar, Bethesda, MA) 20 h before treatments. To monitor the superoxide-inducing effect of different chemicals, cells were incubated in culture media containing Rot, MA, AA or ST at desired concentrations for 30 min. The same amount of dimethyl sulfoxide (DMSO, solvent control) was applied in the culture media of the control cells.
To study the time-dependent change of intracellular superoxide level after withdrawal of the ETC inhibitors, the cells were allowed to recover in normal medium for 0.5 – 8 hours after treatment with ETC-inhibitors for 30 min and washed once with warm PBS. DMSO-treated cells were used as controls. Properly treated cells were then harvested and subjected to flow cytometric assay.
For Se supplementation, the cells were seeded at a density of 4.5e4 cells /cm2 in 100 mm cell culture dishes, and incubated with 100 nM sodium selenite for 24 h. Cells were then subjected to 1.25 μM Rot treatment (a concentration that causes decrease in total intracellular GSH level in HT22 cells) for 30 min, washed twice with cold PBS, collected and centrifuged at 5000 x g for 5 min. The pellet was sonicated in cold PBS. The cell lysate was centrifuged for 10 min at 10,000 x g, the supernatant was collected for GSH and GPx assays.
For gene expression quantification, the cells were seeded at a density of 4.5e4 cells /cm2 in 100 mm cell culture dishes and incubated with culture media containing 0.5 µM Rot for 16 h, with cells treated with equal amount of DMSO as control. The cells were also treated with 0.5 µM of Rot for 0, 0.5, 4, and 16 h to monitor the onset time in gene regulation. RNA was harvested from the cells for real-time PCR quantification of the expression of genes of interest.
Flow Cytometric assay
After desired treatments, the cells were incubated with Hank's Balanced Salt Solution containing 5 µM of HEt for 30 min, allowing the dye to enter into the cells and get oxidized by superoxide. Then the cell culture plates were placed on ice, cells were washed with ice-cold PBS once, trypsinized, and re-suspend in serum-free culture medium. Samples were kept on ice and protected from light before the flow cytometric assay. Five thousand cells were counted for each mean fluorescent intensity value.
Quantification of gene expression by real-time PCR
The expression levels of mRNAs encoding ROS scavenging enzymes, phase II enzymes and some selenoproteins were quantified using real-time PCR in the Rot-treated HT22 cells, using DMSO-treat cells as control. The enzymes directly scavenging ROS included glutathione peroxidase 1 (GPx1, also a selenoenzyme), catalase (Cat), superoxide dismutases 1 and 2 (SOD1, SOD2); phase II enzymes included glutamylcysteine synthetase heavy chain and light chain (GCS-HC, GCS-LC); glutathione S-transferase omega 1 and alpha 2 (GSTo1, GSTa2), and heme oxygenase-1 (HO-1); selenoproteins thioredoxin reductases 1 (TR1), and selenoprotein H (SelH) were also investigated.
Total RNA was harvested using RNeasy mini kit. cDNA were synthesized from the RNA extracts using SuperScript III First-Strand synthesis system (Invitrogen). The specific primers used for real-time PCR on the housekeeping genes (GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HPRT1, hypoxanthine phosphoribosyltransferase 1) and target genes are listed in Table 1. All PCR reactions were performed in 10 μl reaction volume using Platinum SYBR Green qPCR SuperMix-UDG kit (Invitrogen). The amplification and detection were carried out using a LightCycler 2.0 real-time PCR machine (Roche).
Primers used for real-time PCR for specific cDNA amplification.
| Gene | Sequence of primer |
|---|---|
| GAPDH forward | 5'-TGACATCAAGAAGGTGGTGAAGC-3' |
| GAPDH reverse | 5'-CCCTGTTGCTGTAGCCGTATTC-3' |
| HPRT1 forward | 5'-TCCTCCTCAGACCGCTTTT-3' |
| HPRT1 reverse | 5'-CCTGGTTCATCATCGCTAATC-3' |
| SelH forward | 5'-GGAAGAAAGCGTAAGGCGGG-3' |
| SelH reverse | 5'-GGTTTGGACGGGTTCACTTGC-3' |
| GPx1 forward | 5'-ACAGTCCACCGTGTATGCCTTC-3' |
| GPx1 reverse | 5'-CTCTTCATTCTTGCCATTCTCCTG-3' |
| TR1 forward | 5'-CCTATGTGGCCTTGGAATGTGC-3' |
| TR1 reverse | 5'-ATGGTCTCCTCGCTGTTTGTGG-3' |
| SOD1 forward | 5'-CAGGACCTCATTTTAATCCTCAC-3' |
| SOD1 reverse | 5'-CCCAGGTCTCCAACATGC-3' |
| SOD2 forward | 5'-CTGGACAAACCTGAGCCCTA-3' |
| SOD2 reverse | 5'-TGATAGCCTCCAGCAACTCTC-3' |
| Cat forward | 5'-CAGCGACCAGATGAAGCA-3' |
| Cat reverse | 5'-CTCCGGTGGTCAGGACAT-3' |
| GCS-HC forward | 5'-ATGATAGAACACGGGAGGAGAG-3' |
| GCS-HC reverse | 5'-TGATCCTAAAGCGATTGTTCTTC-3' |
| GCS-LC forward | 5'-TGACTCACAATGACCCGAAA-3' |
| GCS-LC reverse | 5'-GATGCTTTCTTGAAGAGCTTCCT-3' |
| GSTo1 forward | 5'-CAGCGATGTCGGGAGAAT-3' |
| GSTo1 reverse | 5'-GGCAGAACCTCATGCTGTAGA-3' |
| GSTa2 forward | 5'-TCTGACCCCTTTCCCTCTG-3' |
| GSTa2 reverse | 5'-GCTGCCAGGATGTAGGAACT-3' |
| HO-1 forward | 5'-GGTCAGGTGTCCAGAGAAGG-3' |
| HO-1 reverse | 5'-CTTCCAGGGCCGTGTAGATA-3' |
GSH measurement
The total intracellular GSH assay was carried out using the GSH/GSSG-412 TM (OXIS) assay kit following the manufacturer's instructions.
GPx activity assay
The assay was based on the classical principle [21] with optimization to the HT22 cell lysate. The peroxide used in this study was t-butyl hydroperoxide (0.32 mM), the concentration of GSH was 1.88 mM, and the pH of the assay was 7.6. The unit of GPx activity was defined as 1mU = 1 nmole NADPH oxidized per min.
Statistical analysis
GraphPad Prism 4 software was used for unpaired two-tailed t-test or two-way ANOVA analysis. P ≤ 0.05 was considered statistically significant. * indicates 0.01< P ≤ 0.05; ** indicates 0.001< P ≤ 0.01; *** indicates P ≤ 0.001.
3. Results
Rotenone treatment induced the highest intracellular superoxide level
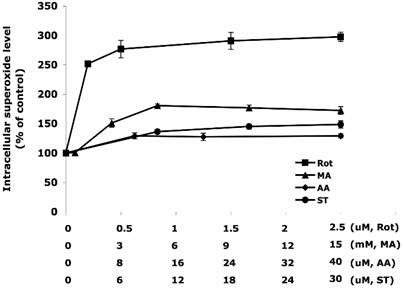
Different concentrations of ETC-inhibitors were used to treat HT22 cells. Specifically they were: Rot 0 – 2.5 µM, MA 0 – 15 mM, AA 0 - 40 µM, and ST 0 – 30 µM. The dose-response curves are summarized in Figure 1. Rot treatment most efficiently increased the intracellular superoxide generation to 300% of the control level, followed by the treatments of MA (180%), ST (150%), and AA (130%).
The increase of intracellular superoxide level induced by inhibitors of the electron transport chain. The concentration HT22 cells were treated for 30 min with rotenone (0-2.5 µM), malonate (0-15 mM), antimycin A (0-40 µM) and stigmatellin (0-30 µM). DMSO treated cells were used as control. The intracellular superoxide level was indicated by the fluorescent intensity of oxidized hydroethidine. Average values and SD are shown, N=3.
Change of superoxide overproduction after withdrawal of ETC-inhibitors
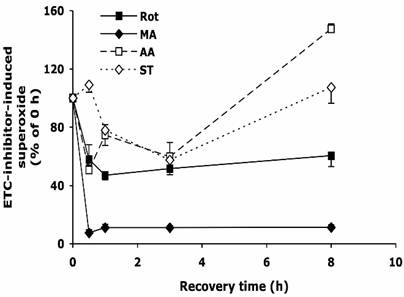
After 30 min treatment with Rot, MA, AA, or ST, the cells were allowed to recover in normal medium as described above. As shown in Figure 2, upon the withdrawal of Rot, within 30 min the Rot-induced superoxide decreased by ~40%, and the level was maintained for the next 7.5 hours. Removing MA from the medium reduced superoxide level by over 90% within 30 min. Surprisingly, when AA and ST were removed, the superoxide levels decreased to about 50% at 3 h, but increased to 110-150% at 8 h.
The change of intracellular superoxide level after withdrawal of the inhibitors of electron transport chain. HT22 cells were treated with rotenone (0.5 µM), malonate (5 mM), antimycin A (20 µM) and stigmatellin (20 µM) for 30 min, then washed and recovered in normal medium for up to 8 h. Average values and SD are shown, N=3.
The responses of oxidative stress-related genes to Rotenone treatment
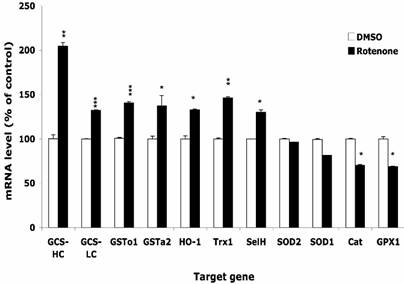
After treatment with 0.5 µM Rot or equal volume of DMSO for 16 h, the expression levels of selected genes were monitored by real-time PCR. Figure 3 shows that the expression level of enzymes that directly scavenge ROS, such as SOD1, SOD2, GPx1, and catalase, decreased by 10-30%. On the other hand, phase II enzymes including GCS-HC, GCS-LC, GSTo1, GSTa2 and HO-1, were up regulated by 30-100%. Two selenoproteins, selenoprotein H and thioredoxin reductase 1 were upregulated by 30-50%. Using either HPRT1 or GAPDH as housekeeping gene for normalization resulted in similar change. We also observed that the onset of the change of expression of different genes differed, e.g. the expression of GCS-HC increased 2 folds at 4 h, but the increase of HO-1 mRNA did not occur until 16 h.
Effects of rotenone treatment on gene expression in HT22 cells. The gene expression level of superoxide dismutase 1 and 2 (SOD1, SOD2), glutathione peroxide 1 (GPx1), catalase (Cat), glutamylcysteine synthetase heavy chain and light chain (GCS-HC, GCS-LC), glutathione s-transferase omega 1 and alpha 2 (GSTo1, GSTa2), heme oxygenase 1 (HO-1), thioredoxin reductase 1 (TR1), and selenoprotein H (SelH) in HT22 cells after treatment with 0.5 µM rotenone for 16 h. Average values and SD are shown.
The protective effects of Se supplementation on the antioxidant status
The HT22 cells were supplemented with 100 nM sodium selenite for 24 h before challenge with 1.25 μM Rot for 30 min, then the total cellular GSH level and GPx activity were analyzed. As shown in Figure 4A, without Se supplementation, the total cellular GSH level (normalized to a protein concentration of 1 mg/ml) decreased from 29.9 ± 0.9 µM to 24.4 ± 1.2 µM after the Rot treatment. Se supplementation significantly increased the baseline of GSH level in the cells to 44.0 ± 1.0 µM, and abolished the Rot-induced GSH decrease.
Effects of selenium supplementation and rotenone treatment on (A) total glutathione level and (B) glutathione peroxidase activity in HT22 cells. HT22 cells were incubated with medium containing 100 nM sodium selenite for 24 h, then treated by 1.25 µM rotenone for 30 min in normal medium. Average values and SD are shown, N=3. In Panel B, the statistical comparisons are between Se Supplementation and normal medium after DMSO or Rot treatment.
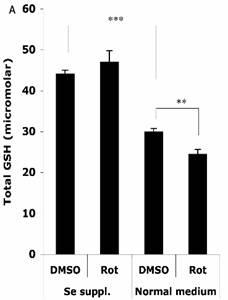
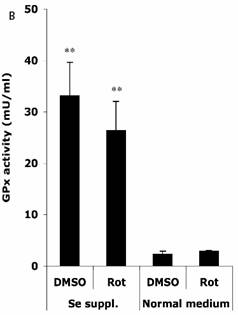
Figure 4B shows that in normal medium, similar activities of GPx (normalized to a protein concentration of 1 mg/ml) were observed in HT22 cells when treated with DMSO (2.3 ± 0.6 mU) and Rot (2.9 ± 0.2 mU). However, Se supplementation rendered significant increase in GPx activity under DMSO (33.1 ± 6.5 mU) and Rot (26.4 ± 5.7 mU) treatments.
Two-way ANOVA was also performed to analyze the influence of Se status and Rot treatment as independent factors and their combined effect. The result shows that for both GSH and GPx, the influence of Se status is significant (P<0.001), and the combination of Se status and Rot treatment also has significant influence on GSH level (P=0.0023).
4. Discussion
HT22 cell line is vulnerable to glutathione (GSH) depletion [22], and is a sensitive model for monitoring cellular responses to oxidative stress. It has been shown that the primary ROS-generating site in ETC can be different in different types of cells [10-13]. Our study indicates complex I is the major superoxide-producing site in HT22 cells since Rot treatment induced the highest level of intracellular ROS in comparison to the treatments with other ETC-inhibitors.
Rot treatment has been widely used on animals as in vivo models [18] and on neuronal cells as in vitro models to reveal the etiology of PD [23]. Rot treatment induces mitochondrial ROS prior to apoptotic events [24]. Our study shows that superoxide is highly inducible by Rot in HT22 cells, in accordance with previous studies on mitochondria isolated from rat brain and human parahippocampal gyrus [25], and on superoxide production by nonsynaptic brain submitochondrial particles from seven mammalian species [26]. The dose response of HT22 cells to Rot treatment is similar to those reported for isolated mitochondria and HL-60 cells in culture [24].
Approximately half of the Rot-induced superoxide was readily removable within 30 min after the withdrawal of Rot, but the rest remained for up to 8 h. This result indicates that Rot may induce superoxide through different mechanisms, e.g. reversible and high-dosage-demanding, versus irreversible and/or low-dosage-demanding. A neurotoxicity study showed that a short exposure to Rot at high concentrations reduced the viability of both dopaminergic and non-dopaminergic neurons, while chronic treatment with low-concentrations of Rot caused selective toxicity to dopaminergic neurons [27]. Considering the vulnerability of dopaminergic neurons in PD, the low-dosage-demanding Rot toxicity indicated in the present study may be more involved in the pathogenesis. This consideration also questions the neurological safety of the potential therapeutic strategy of administration of Rot before myocardial ischemia occurs to decrease damage to the myocardium [28, 29].
Complex II dysfunction has been related to neurodegenerative diseases such as Huntington's disease, as well as hypoxia-dependent generation of ROS in lungs [30]. In mice and rats, MA injection in the striatum produced a significant reduction in cytochrome oxidase activity in the tissue [31]. MA affected rat brain mitochondrial function and neuronal survival, mediated by ROS and mitochondrial permeability transition at a significantly lower level of respiratory inhibition in comparison to mitochondria isolated from liver and kidney [32]. The present study shows that superoxide is highly inducible by MA in neuronal HT22 cells, but the induced superoxide is readily reversible upon malonate withdrawal.
Stigmatellin blocks electron flow at the Qo site of complex III by binding to the heme b-566 domain of cytochrome b as well as to the iron-sulfur protein [8, 9]. Structural study of native complex III indicated that binding of stigmatellin to the reduced iron-sulfur protein at Qo center may be involved in the rate limiting reaction at this site [33]. Blockade of complex III by stigmatellin generates ROS through complex I by electron accumulation at this upstream complex [7]; at the same time, stigmatellin at higher concentrations also stimulates superoxide production by inhibiting complex I at the rotenone site [34]. Antimycin A inhibits reduction of ubiquinone and ubisemiquinone anion by cytochrome b heme-560 at Qi center [8], increasing the ROS generation from Qo center [7], which is released into the cytoplasm through an anion channel [30, 35, 36]. When mitochondria isolated from rat heart inhibited by antimycin A, a large amount of superoxide was produced [7, 37]. However, the present study showed only 20-30% of increase of superoxide generation in HT22 cells after antimycin A treatment at saturating concentration. The different responses might be due to tissue specificity, which alters the ratios and activities of complexes in the respiratory chain in mitochondrion [35]. The reason of the biphaseic response of the HT22 cells to the removal of inhibitors on complex III (Figure 2) is unknown.
Figure 3 indicates that the ability of direct scavenging Rot-induced ROS may be compromised in HT22 cells since the expression of the enzymes that dismutate superoxide (SOD1, SOD2) or reduce hydrogen peroxide (GPx1, catalase) was decreased following the Rot treatment. On the other hand, GCS-HC and GCS-LH, subunits of the rate-limiting enzyme in the GSH de novo synthesis pathway, was up-regulated by 30-100%. Other phase II enzymes such as GSTs, and HO-1 were also up-regulated, indicating the significance of involvement of phase II enzymes in combating the oxidative stress.
Se is thought to exert most of its biological effects through its incorporation into selenoproteins. Se status has been shown to modulate redox-regulated transcription factors such as AP-1 and NF-kappaB [38, 39]. It has been found to affect the pathogenesis of PD in animal models. For example, Se supplementation was reported to ameliorate brain tissue damage in 6-hydroxydopamine treated mice by sustaining the GSH level in the tissues and decreasing oxidative stress [19]. In this study we found in a cell culture model that Se supplementation increased the baseline level of total GSH concentration and prevented Rot-induced GSH depletion. We also found that the gene expression of 2 selenoproteins possessing important regulatory function [40] was upregulated by Rot treatment, indicating the possibility of existence of other pathways in Se-related protective effects.
The GPx activity assay in this study measured the total activity of GPxs that use BHPx and GSH as substrates. Out of the seven GPxs that have been identified to date, GPx7 and mouse GPx6 are non-selenoproteins, therefore they are not likely to be dramatically influenced by Se supplementation. GPx2 (gastrointestinal GPx) and GPx5 (sperm nuclei GPx) have tissue specific localization, while the expression of GPx3 (extracellular GPx) was found to be extremely low in mouse brain [41]. Our unpublished data showed that GPx4 (phospholipid hydroperoxide glutathione peroxidase) is highly expressed in HT22 cells, but its mRNA level was not significantly influenced by Se supplementation. On the other hand, the expression of GPx1 (cytosolic GPx) is highly responsive to Se status. Using BHPx as substrate, the activity obtained from GPx4 is only 10% of that contributed by GPx1 [42]. Therefore, the increased GPx activity after Se supplementation observed in this study is most likely attributable to GPx1, as potentially counteracts the downregulation of GPx1 expression by long-term Rot treatment.
5. Conclusion
This study indicates that complex I is the major ROS-generating site in HT22 cells. Rotenone treatment decreased the gene expression of enzymes directly scavenging ROS but increases that of phase II enzymes. Selenium supplementation increased glutathione levels and GPx1 activity in HT22 cells, may have a protective role in rotenone-induced oxidative stress.
Acknowledgements
This study was supported by Hawaii Community Foundation grant # 20050404 (Panee) and NIH grant RO1-NS40302 (Berry).
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707-716
2. Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527-605
3. Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondnial decay in aging. Proc.Nail. Acad.Sci. USA. 1994;91:10771-10778
4. Cortopassi C, Wang E . Modelling the effects of age-related mtDNA mutationaccumulation:complex I deficiency, superox-ide and cell death. Biochim. Biophys. Acta. 1995;1271:i7i-i76
5. Buchanan SK, Walker JE . Large-scale chromatographic purification of F1F0-ATPase and complex I from bovine heart mitochondria. Biochem. J. 1996;318:343-349
6. Okun JG, Lummen P, Brandt U. Three classes of inhibitors share a common binding domain in mitochondrial complex I (NADH:ubiquinone oxidoreductase). J. Biol. Chem. 1999;274:2625-2630
7. Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnef-sky EJ. Production of reactive oxygen spqcies by mitochondria, central role of complex III. J. Biol. Chem. 2003;278:36027-36031
8. Trumpower BL. The protonmotive Q cycle. J. Biol. Chem. 1990;265:11409-11412
9. von Jagow G, Ohnishi T. The chromone inhibitor stigmatel-lin--binding to the ubiquinol oxidation center at the C-side of the mitochondrial membrane. FEBS Lett. 1985;185:311-315
10. Cadenas E, Boveris A, Ragan CI, Stoppani AOM. Produc-tion of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c re-ductase from beef-heart mitochondria. Arch. Biochem. Biophys. 1977;180:248-257
11. Turrens JF, Boveris A . Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Bio-chem. J. 1980;191:421-427
12. Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985;237:408-414
13. Liu Y, Fiskum G, Schubert D. Generation of reactive oxy-gen species by the mitochondrial electron transport chain. J. Neurochem. 2002;80:780-787
14. Kweon GR, Marks JD, Krencik R, Leung EH, Schumacker PT, Hyland K, Kang UJ. Distinct mechanisms of neurodegen-eration induced by chronic complex I inhibition in dopaminergic and non-dopaminergic cells. J Biol Chem. 2004;279:51783-51792
15. Petrovitch H, Ross GW, Abbott RD, Sanderson WT, Sharp DS, Tanner CM, Masaki KH, Blanchette PL, Popper JS, Foley D, Launer L, White LR. Plantation work and risk of Parkinson disease in a population-based longitudinal study. Arch Neurol. 2002;59:1787-1792
16. Semchuk KM, Love EJ, Lee RG. Parkinson's disease and exposure to agricultural work and pesticide chemicals. Neurol-ogy. 1992;42:1328-1335
17. Betarbet R, Sherer TB, Mackenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic sysmetic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301-1306
18. Alam M, Schmidt WJ . Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav Brain Res. 2002;136:317-324
19. Zafar KS, Siddiqui A, Sayeed I, Ahmad M, Salim S, Islam F. Dose-dependent protective effect of selenium in rat model of Parkinson's disease: neurobehavioral and neurochemical evi-dences. J Neurochem. 2003;84:438-46
20. Kim HC, Jhoo WK, Choi DY, Im DH, Shin EJ, Suh JH, Floyd RA, Bing G. Protection of methamphetamine nigrostriatal toxicity by dietary selenium. Brain Res. 1999;851:76-86
21. Paglia D, Valentine W . Studies on the quantitative and qualitative characteristics of the erythrocyte glutathione per-oxidase. J Lab Clin Med. 1967;70:158-169
22. Davis JB, Maher P . Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Research. 1994;652:169-173
23. Sherer TB, betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanism of toxicity in rotenone models of Parkinson's dis-ease. J Neurosci. 2003;23:10756-10764
24. Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrail complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516-8525
25. Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS. Characterization of superoxide-producing sites in isolated brain mitochondria. J Biol Chem. 2003;279:4127-4135
26. Herrero A, Barja G . Localization of the site of oxygen radi-cal generation inside the complex I of heart and nonsynaptic brain mammalian mitochondria. J Bioenerg Biomembr. 2000;32:609-615
27. Sakka N, Sawada H, Izumi Y, Kume T, Katsuki H, Kaneko S, Shimohama S, Akaike A. Dopamine is involved in selec-tivity of dopaminergic neuronal death by rotenone. Neurore-port. 2003;14:2425-2428
28. Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL. Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem. 2004;279:47961-47967
29. Ockaili RA, Bhargava P, Kukreja RC. Chemical precondi-tioning with 3-nitropropionic acid in hearts: role of mitochon-drial K(ATP) channel. Am J Physiol Heart Circ Physiol. 2001;280:H2406-H2411
30. Weissmann N, Ebert N, Ahrens M, Ghofrani HA, Schermuly RT, Hanze J, Fink L, Rose F, Conzen J, Seeger W, Grim-minger F. Effects of mitochondrial inhibitors and uncouplers on hypoxic vasoconstriction in rabbit lungs. Am J Respir Cell Mol Biol. 2003;29:721-732
31. Lastres-Becker I, Bizat N, Boyer F, Hantraye P, Brouillet E, Fernandez-Ruiz J. Effects of cannabinoids in the rat model of huntington's disease generated by an intrastriatal injection of malonate. Neuroreport. 2003;14:813-816
32. Maciel EN, Kowaltowski AJ, Schwalm FD, Rodrigues JM, Souza DO, Vercesi AE, Wajner M, Castilho RF. Mito-chondrial permeability transition in neuronal damage promoted by Ca2+ and respiratory chain complex II inhibition. J Neuro-chem. 2004;90:1025-1035
33. Crofts AR, Shinkarev VP, Dikanov SA, Samoilova RI, Kolling D. Interactions of quinone with the iron-sulfur protein of the bc(1) complex: is the mechanism spring-loaded? Biochim Biophys Acta. 2002;1555:48-53
34. Degli Esposti M, Ghelli A, Crimi M, Estornell E, Fato R, Lenaz G. Complex I and complex III of mitochondria have common inhibitors acting as ubiquinone antagonists. Biochem Biophys Res Commun. 1993;190:1090-1096
35. St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. To-pology of superoxide production from dofferent sites in the mi-tochondrial electron transport chain. J. Biol. Chem. 2002;277:44784-44790
36. Han D, Williams E, Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J. 2001;353:411-416
37. Raha S, McEachern GE, Myint AT, Robinson BH. Super-oxide from mitochondrial complex III: the role of manganese superoxide dismutase. Free Radic Biol Med. 2000;29:170-180
38. Spyrou G, Bjornstedt M, Kumar S, Holmgren A. AP-1 DNA-binding activity is inhibited by selenite and selenodiglu-tathione. FEBS Lett. 1995;368:59-63
39. Kim IY, Stadtman TC . Inhibition of NF-kappaB DNA binding and nitric oxide induction in human T cells and lung adenocarcinoma cells by selenite treatment. Proc Natl Acad Sci U S A. 1997;94:12904-7
40. Rundlof AK, Arner ES . Regulation of the mammalian se-lenoprotein thioredoxin reductase 1 in relation to cellular phe-notype, growth, and signaling events. Antioxid Redox Signal. 2004;6:41-52
41. Maser RL, Magenheimer BS, Calvet JP. Mouse plasma glutathione peroxidase. cDNA sequence analysis and renal proximal tubular expression and secretion. J Biol Chem. 1994;269:27066-73
42. Maiorino M, Gregolin C, Ursini F. Phospholipid hydrop-eroxide glutathione peroxidase. Methods Enzymol. 1990;186:448-57
Author contact
![]() Correspondence to: Marla J. Berry, 651 Ilalo Street BSB 222, Honolulu HI 96813, USA. Telephone: +1-808-692 1506; Fax: +1-808-692 1970; Email: mberryedu
Correspondence to: Marla J. Berry, 651 Ilalo Street BSB 222, Honolulu HI 96813, USA. Telephone: +1-808-692 1506; Fax: +1-808-692 1970; Email: mberryedu