Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2007; 3(7):402-407. doi:10.7150/ijbs.3.402 This issue Cite
Research Paper
DNA damage repair is unaffected by mimicked heterozygous levels of BRCA2 in HT-29 cells
Brian Tannenbaum, Tobechukwu Mofunanya, Alan R. Schoenfeld ![]()
Department of Biology, Adelphi University, Garden City, NY 11530, USA
Received 2007-6-11; Accepted 2007-9-28; Published 2007-9-30
Abstract
Functional loss of both alleles of the breast cancer susceptibility gene, BRCA2, facilitates tumorigenesis. However, the direct effects of BRCA2 heterozygosity remain unclear. Here, BRCA2 heterozygosity was mimicked in HT-29 colon cells by reducing levels of BRCA2 through stable RNA interference. No difference in RAD51 subcellular localization and focus formation was observed between control and mimicked heterozygous cell lines. DNA repair ability, as measured by colony survival following mitomycin C treatment and ultraviolet radiation exposure, was also unaffected by reduced levels of BRCA2. Interestingly, the growth rate of the mimicked BRCA2 heterozygous cell line was significantly lower than that of control cells. Increased expression of p53 in the mimicked heterozygous cells was observed, perhaps in response to BRCA2 deficiency. Levels of p27 were also found to be slightly increased in cells with reduced BRCA2, perhaps contributing to the slower growth rate. Overall, these results suggest that tumors are unlikely to arise directly from BRCA2 heterozygous cells without other genetic events such as loss of the wild-type BRCA2 allele and/or loss of p53 function or other cell cycle inhibitors.
Keywords: BRCA2, heterozygosity, RNAi, DNA damage repair
1. INTRODUCTION
The majority of inherited breast and ovarian cancers are caused by germline mutations of the BRCA1 and BRCA2 genes [1]. In addition, BRCA2 mutations have been associated with a number of other tumor types, including colon cancer [2, 3]. Loss of wild-type function of both BRCA2 alleles allows tumors to proliferate in affected individuals, classifying BRCA2 as a tumor suppressor gene [4]. The human BRCA2 gene encodes a nuclear protein of 3,418 amino acids [5], and is believed to play a pivotal role in DNA damage repair [6]. The BRCA2 protein has been shown to bind to RAD51, the mammalian homolog of the RecA recombinase [6-9], and thus is believed to be involved in the repair of DNA double-strand breaks [6]. In support of this notion, cells lacking a functional BRCA2 gene show hypersensitivity to DNA damaging agents such as mitomycin C (MMC) and sensitivity to chemicals such as methyl methane sulfonate [10]. In addition, cells with homozygous truncations in BRCA2 are genetically unstable [8, 9, 11] and are lacking in homology-directed DNA repair of chromosomal breaks [12, 13]. Moreover, BRCA2 regulates both the DNA binding ability of RAD51 and its intracellular location [14].
The effect of heterozygosity of the BRCA2 gene on human cells remains unclear. According to Knudson's two hit hypothesis for tumor suppressor genes [15], loss of both alleles must occur prior to tumor growth. However, it may be possible that tumor growth can be initiated in heterozygous cells prior to loss of the second, wild-type allele. In support of this hypothesis, heterozygosity for a BRCA2 mutation has been shown to cause sensitivity to DNA damage agents and reduced RAD51 focus formation after irradiation in the chicken B cell line DT40 [16]. Additionally, chromosomal rearrangements, increased rates of sister chromatid exchanges and double strand breaks have been observed in cells from heterozygous mutation carriers of BRCA2 [17, 18]. In this report, we have utilized HT-29 colon cancer cells and have mimicked the heterozygous state of BRCA2 in these cells through RNA interference. The characterization of the resulting cells with regard to key cellular BRCA2 functions is presented here.
2. MATERIALS AND METHODS
Retroviral vectors
For short hairpin RNA (shRNA) targeting of BRCA2, the pRETRO-SUPER plasmid [19] was utilized as previously described [20]. Pairs of oligonucleotides against BRCA2 base sequences 115-133 and 216-234 were separately annealed and ligated into BglII/HindIII sites in pRETRO-SUPER as described [20]. Targeting of luciferase using the pRETRO-SUPER shRNA plasmid was also as described [20]. All plasmids were confirmed by DNA sequencing and/or restriction digestion.
Cell culture and retroviral infection
HT-29 cells were grown in Dulbecco's Modification of Eagle's Medium (DMEM) containing Penicillin/Streptomycin (100 units/ml and 10μg/ml, respectively) and 10% Serum Supreme (Bio Whitaker). Retroviral supernatants were produced using 293T cells and cotransfection with the packaging plasmid pCL-Ampho [21], as previously described [20]. To achieve lower BRCA2 levels, retroviral supernatants directing expression of shRNA directed at both bases 115-133 and 216-234 of BRCA2 were mixed together 1:1, supplemented with 10 μg/ml polybrene, and incubated with HT-29 cells overnight (hereafter called BRCA2 RNAi cells). HT-29 cells were similarly infected with empty vector control retrovirus (hereafter called control RNAi cells). Forty-eight hours later, infected HT-29 cells were passaged into media containing 1µg/ml puromycin for selection. The cells were passaged and continued to undergo selection for approximately fourteen days.
Western blotting
For all western blots, cells in 100mm plates were rinsed with PBS and 500µl of lysis buffer [50 mM HEPES [pH 7.6], 250 mM NaCl, 0.1% NP-40, 5 mM EDTA, 1 mM PMSF, 2 µg each of aprotinin, bestatin, and leupeptin/ml] was added to plates and incubated for 30 minutes at 4°C. Cells were then collected using a plastic cell scraper and resuspended by pipetting. The lysates were clarified by microcentrifugation for 15 minutes at 4°C. The supernatants were collected and a Bradford assay (Bio-Rad) was conducted to determine protein concentration. 25µg of lysates for each cell line were separated on an SDS-PAGE gel. The gel was then transferred to a PVDF membrane and subjected to western blotting. Quantification of band intensities was performed using the gel analyzer function of Image J (version 1.34S).
Subcellular Fractionation
HT-29 cell lines grown to confluence in 100mm plates were trypsinized and collected with fresh media in 14ml tubes, washed with PBS, and recollected in 1.5ml eppendorf tubes. 500µl of homogenization buffer [250mM sucrose, 10mM Tris (pH 7.5), 10mM KCl, 1.5mM MgCl2, 1mM PMSF, 1µl/ml of aprotinin, 1µl/ml of bestatin, 1µl/ml of leupeptin, and 1mM Na2VO3 was added to each tube and mixed. Tubes were then placed on ice for twenty minutes and mixed every four to five minutes. Cells were collected at the bottom of the tubes and homogenized at the bottom using a plastic pestle. The eppendorf tubes were then spun at 4°C for ten minutes at 600 x g. The supernatant containing the cytosolic fraction was collected. 500µl of lysis buffer was added to the pellets (nuclei) in the original tube set. The tubes were then placed on ice for fifteen minutes, mixed every four to five minutes. The tubes were then spun down at 4°C for 15 minutes at 13,500 rpm in a refrigerated microcentrifuge. The supernatant containing the nuclear fraction was collected.
RAD51 focus assay
Two days prior to immunofluorescence microscopy, cells were passaged onto coverslips in a six-well plate. The following day, one set of cells was treated with 400ng/ml of MMC overnight. The next day, cells were fixed with 3.7% paraformaldehyde, permeablized with 0.5% Triton X-100, and blocked with 0.1% Goat Serum in PBS-Tween. Cells were then incubated with primary antibody (rabbit polyclonal anti-RAD51, Oncogene Sciences) and secondary antibody (Texas-Red conjugated anti-rabbit lgG) as directed by manufacturer, washed six times with PBS-Tween, stained with 4'-6-Diamidino-2-phenylindole (DAPI), and mounted on microscope slides. Cells were visualized on a Zeiss Axioskop fluorescence microscope.
Clonogenic survival assays
One day prior to treatment, cells were passaged into four 60mm culture plates and incubated until they reached fifty percent confluence. For MMC treatment, cells were incubated for one hour in media containing either 0 ng/ml, 200 ng/ml, 400 ng/ml or 800 ng/ml of MMC. Media was then removed and fresh media was added to each plate. The next day, the cells were resuspended and counted by hemacytometer. 1000 cells from each plate were added to new plates in triplicate. Fresh media was added to the plates every two to three days. After two weeks, the cells were fixed with 10% acetic acid/10% methanol for 10 minutes and stained with 1% crystal violet in methanol for 10 minutes. The cells were rinsed twice with water, dried, and the colonies on the plates were counted. Ultraviolet radiation DNA damage assay was performed as for MMC assay, except media from plates was removed and cells were struck with ultraviolet radiation from a germicidal lamp in the tissue culture hood at doses of 25 J/m2, 50 J/m2 and 100 J/m2.
Standard growth assay
Cells at approximately fifty percent confluence were trypsinized, resuspended in fresh media, and counted by hemacytometer. 30,000 cells from each plate were added to eighteen 60mm culture plates. Fresh media was added to the plates every three days. Cells were trypsinized and counted by hemocytometer (in triplicate for each cell line) at approximate 3 day intervals for up to 19 days. Cells approached confluency between days 12 and 15 of growth, approximately.
3. RESULTS
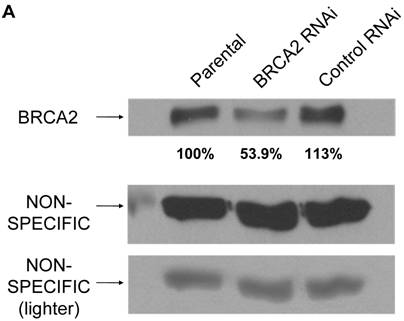
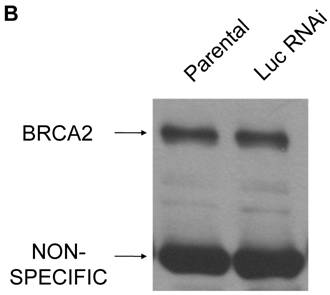
The heterozygous state of BRCA2 was mimicked in HT-29 colon cells via RNA interference in order to determine possible functional effects. BRCA2 RNAi cells were created via overnight incubation with 2 retroviruses containing shRNA BRCA2 targeting constructs (targeting BRCA2 bases 115-133 and 216-234) mixed together, as described in Materials and Methods. A control line of HT-29 cells was created with a control RNAi retrovirus, as also described. Following a 14 day selection, cells at fifty percent confluence were lysed and quantified for BRCA2 protein expression level by western blot to verify a reduction of BRCA2 protein levels to those likely to be seen with BRCA2 heterozygosity. In comparison to the parental and control RNAi cell lines, the BRCA2 RNAi cells expressed BRCA2 protein levels that were approximately half of the parental and control cells, verifying the mimic of BRCA2 heterozygosity (Figure 1a). The lower expression was not due to a non-specific effect of the shRNA since no reduction in BRCA2 was seen using a retroviral construct coding for a shRNA directed at the non-human target luciferase (Figure 1b).
BRCA2 heterozygosity mimicked through RNA interference in HT-29 colon cells. (A) 25µg of lysates from parental, BRCA2 RNAi, and control RNAi cells were loaded on a SDS-10% polyacrylamide gel and a BRCA2 western blot was performed (top panel). Quantification of BRCA2 band intensities as a percent of the parental band is indicated. A non-specific band was utilized as a loading control in the experiment (middle panel). A lighter exposure of the non-specific band is shown (lower panel). (B) Western blot was performed as in (A), except lysates from parental HT-29 cells were compared to HT-29 cells stably infected with a shRNA construct targeting luciferase (Luc RNAi).
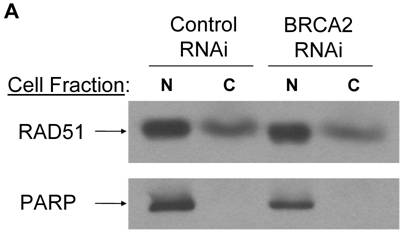
To determine whether the mimicked heterozygous state of BRCA2 altered the normal cellular activities of the BRCA2 protein, we examined aspects of the BRCA2 functional interaction with RAD51. Crude subcellular fractionation was conducted to determine whether reduced BRCA2 levels altered the cellular localization of RAD51. Cell lines were fractionated into nuclear and cytosolic fractions and analyzed by RAD51 Western blotting. There was no significant difference between the control RNAi and BRCA2 RNAi cell lines in either the nuclear or cytosolic levels of RAD51 (Figure 2a).
No effect of mimicked BRCA2 heterozygosity on RAD51 localization and foci formation. (A) Subcellular fractionation was conducted to separate nuclear fraction from cytosolic fraction. 25µg of each fraction was loaded on a SDS-10% polyacrylamide gel and a RAD51 western blot was performed. Poly (ADP-ribose) polymerase (PARP) was utilized as a control for the fractionation. N = nuclear fraction, C = cytosolic fraction. (B) Cell lines were grown on coverslips and treated with 400ng/ml of MMC overnight and RAD51 immunostaining was performed. RAD51 foci are seen as red sharp, punctuate staining in left panels; DAPI labeled nuclei are shown in blue in right panels. No RAD51 foci were seen with untreated cells (data not shown).
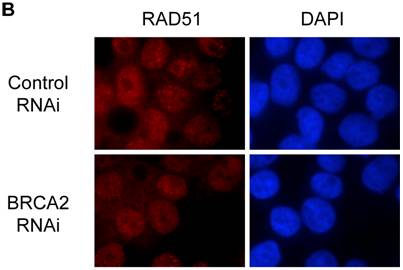
To further determine whether mimicked BRCA2 heterozygosity affected the BRCA2-RAD51 interaction, a RAD51 focus assay was conducted. BRCA2-null cells have been previously shown to be deficient in the formation of RAD51 foci [22]. However, no significant difference between the RAD51 foci of both the BRCA2 RNAi and control RNAi cells was observed (Figure 2a). Both cell lines had a similar abundance of RAD51 foci within the nuclei and there was no apparent difference between the sizes of the RAD51 foci seen in both cell lines (Figure 2b). Thus, reducing BRCA2 levels does not seem to affect this BRCA2 function.
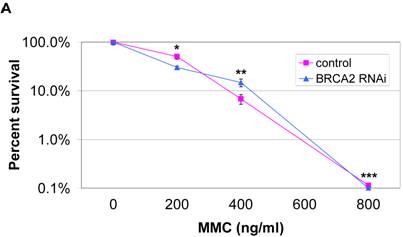
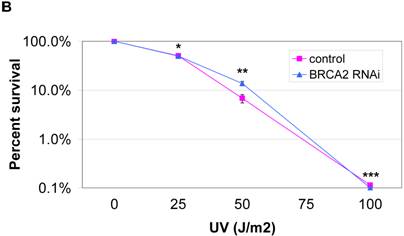
We next assayed whether the mimicked heterozygous state of BRCA2 has an effect on DNA damage repair. Since cells lacking the BRCA2 gene have shown hypersensitivity to MMC and UV, these treatments were utilized as damaging agents and DNA repair was assessed by clonogenic survival assays. No significant difference in clonogenic survival was observed with either the higher doses (400ng/ml and 800ng/ml) of MMC treatment (Figure 3a) or with UV treatment (Figure 3b). Thus, the mimicked heterozygous state of BRCA2 did not alter colony survival in response to either MMC and UV. However, in both DNA damage assays, there was a significantly greater number of control RNAi colonies than BRCA2 RNAi from the untreated cells at the end of the two-week assay, despite equal plating. Representative plates of colonies from the untreated cells of the UV survival assay are shown (Figure 3c). The amount of BRCA2 RNAi colonies was 50.9% (±13.2%) of the control colonies in the MMC survival assay (p < 0.002) and 46.1% (±7.8%) of the control colonies in the UV survival assay (p < 0.008).
No significant difference in survival after MMC DNA damage for BRCA2 RNAi and control RNAi HT-29 cells. (A) 1000 cells were plated in triplicate after treatment with 0 ng/ml, 200 ng/ml, 400 ng/ml, and 800 ng/ml of MMC in media and colonies were counted after two weeks of growth. No significant difference in DNA repair ability was observed at 400ng/ml and 800 ng/ml doses of MMC treatment (*p < 0.05, **p > 0.08, ***p > 0.40, Student's paired, two-tailed t-test). (B) 1000 cells were plated in triplicate after UV doses at 0 J/m2, 25 J/m2, 50 J/m2 and 100 J/m2 and colonies were counted after two weeks. No significant difference in DNA repair ability was observed as a result of UV radiation (*p > 0.05, **p > 0.50, ***p > 0.90, Student's paired, two-tailed t-test). (C) Colonies of untreated (0 J/m2 UV dose) control and BRCA2 RNAi cells from the UV colony survival assay in (B) are shown to demonstrate the intrinsic colony forming deficiency of BRCA2 RNAi cells.
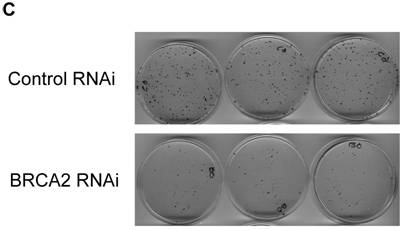
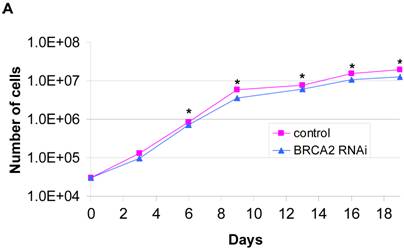
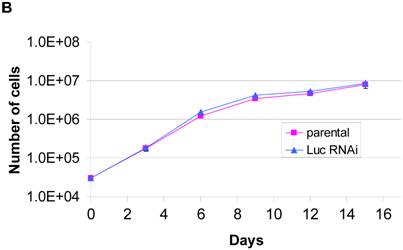
The augmented amount of colonies in the control RNAi line suggested that the growth of BRCA2 RNAi cells might be inhibited, a possible effect of the mimicked heterozygous state of BRCA2. To test this hypothesis, a standard growth assay was performed and differences between the cell lines were observed. A graphical representation of the standard growth of the BRCA2 RNAi and control RNAi cells is shown (Figure 4a). BRCA2 RNAi cells were observed to have a significantly decreased standard growth rate at all time points except the earliest (3 days), including time points prior to when the cells approached confluency, suggesting that the observed growth deficiency is intrinsic to the BRCA2 RNAi cells. In contrast, no difference in growth was seen between parental and luciferase RNAi cells (Figure 4b).
BRCA2 RNAi cells demonstrate inhibited growth. (A) A standard growth assay was performed on control and BRCA2 RNAi cells. 30,000 cells for each cell line were plated on 18 60-mm plates. Three plates of cells for each cell line were counted at each time point over the course of a nineteen-day period and graphed in a logarithmic scale. Time points demonstrating statistically significant differences between cell lines (p < 0.05, Student's paired, two-tailed t-test) are noted by asterisk. (B) A standard growth assay was performed as in (A) comparing parental HT-29 cells and cells stably infected with a shRNA construct targeting luciferase (Luc RNAi). (C) 25µg of cell lysates were loaded on a SDS-10% polyacrylamide gel and BRCA1, p53, p27 western blotting was performed. Alpha tubulin western blotting (lower middle panel) served as a control for equal loading of lysates.
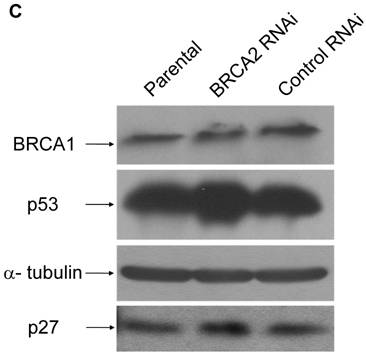
We then investigated the basis of the observed decreased growth rate. To see if a compensatory increase in BRCA1 might cause slower growth in BRCA2 RNAi cells, a BRCA1 Western blot was performed (Figure 4c, top panel). There was no observable difference of the BRCA1 levels among the parental, control RNAi, and BRCA2 RNAi cell lines. Because levels of p53 have been previously shown to increase due to loss of BRCA2 [23], a p53 Western blot was performed (Figure 4c, second panel). An observable increase in p53 was present in the BRCA2 RNAi cells as compared to the parental and control RNAi cell lines. However, since HT-29 cells express a mutant p53, it is very possible that the increased p53 levels in BRCA2 cells do not have any functional consequence. In support of this notion, the p53 target protein, p21, was not detectable in these cells (data not shown), suggesting that the slower growth of BRCA2 RNAi cells may occur through mechanisms other than p53-mediated growth arrest. Along these lines, a p27kip1 western was performed and levels of p27kip1 were found to be slightly increased in BRCA2 RNAi cells (Figure 4c, fourth panel).
4. DISCUSSION
Mimicked BRCA2 heterozygosity did not appear to alter the normal cellular functions of BRCA2 in HT-29 colon cells. No significant difference in either RAD51 localization or RAD51 focus formation was seen between the BRCA2 RNAi cell line and the control RNAi cell line, in contrast to BRCA2-deficient Capan-1 cells, which demonstrate decreased RAD51 focus formation [22]. Furthermore, lower BRCA2 levels did not seem to alter the DNA repair ability of HT-29 colon cells, with percent survival rates similar to control cells following MMC or UV treatment. Overall, these results indicate that it is unlikely that tumorigenesis occurs prior to loss of both BRCA2 alleles. The findings in this report are not in agreement with the reduced RAD51 focus formation after irradiation and sensitivity to DNA damage agents observed in a chicken B cell line, DT40, that was heterozygous for BRCA2 [16]. A possible explanation for these differences may be due to effects of the mutant allele in the DT40 cell line, which produces a truncated BRCA2 protein that is not present in the mimicked heterozygous cells.
The clearest effect of mimicked BRCA2 heterozygozity that was observed was a diminished rate of growth. There were no observable differences in the levels of BRCA1 protein in mimicked BRCA2 heterozygous cells, suggesting a lack of compensatory action of the BRCA1 and BRCA2 tumor suppressor genes. In contrast, a detectable increase in the levels of p53 was observed in the BRCA2 RNAi cell line. However, it is uncertain whether this p53 increase contributed to the observed reduced cell growth rate of BRCA2 RNAi cells since p53 is mutated in the HT-29 cell line. Furthermore, p21 levels were not observed to be increased concomitantly, suggesting that the p53 increase may have no functional significance with respect to the cell cycle. It is not certain whether non-transcriptional activities of p53 are also lost through this same mutation, and whether they may be partly responsible for the observed growth differences. Levels of p27kip1 were also slightly increased in BRCA2 RNAi cells, perhaps contributing to their growth deficiency. However, it is also possible that the observed increases in p53 and p27kip1 are merely cellular responses to other defects resulting from decreased BRCA2 levels (such as subtle defects in DNA repair that are not detectable by the assays employed here) that may have a more direct impact on cell division and cell growth rates.
The increased p53 that was observed in HT-29 cells as a result of mimicked BRCA2 heterozygosity is likely to also occur in cells with intact p53, where it may have more of a tumor suppressing role. Mouse embryonic fibroblasts that are homozygous for BRCA2 knockout alleles have been shown to have a defect in proliferation caused by the over-expression of p53 [23]. These results suggested that the loss of the p53 checkpoint may be vital for tumor progression generated by mutations in BRCA2. Loss of p53 has been suggested to be necessary for breast tumor formation following BRCA2 loss [24]. In this study, the increased levels of p53 in the BRCA2 RNAi cells may explain the infrequent incidences of colon cancer in BRCA2 carriers. It is probable that the same mechanisms may function in BRCA2 heterozygotes in vivo, with increased p53 levels preventing the proliferation of malignant tumors in the human colon and perhaps in other tissues as well. However, it is also possible that the effects of lower BRCA2 levels may differ depending on cell type. Investigations into the differential effects of BRCA2 levels on cell growth and p53 activation may provide insight into why BRCA2 mutations lead to a small subset of observed tumor types.
Acknowledgements
This work was supported by an Adelphi University Faculty Development Award (to A.R.S.).
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists.
References
1. Wooster R, Bignell G, Lancaster J. et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378(6559):789-792
2. Risch HA, McLaughlin JR, Cole DE. et al. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet. 2001;68(3):700-710
3. Berman DB, Costalas J, Schultz DC. et al. A common mutation in BRCA2 that predisposes to a variety of cancers is found in both Jewish Ashkenazi and non-Jewish individuals. Cancer Res. 1996;56(15):3409-3414
4. Collins N, McManus R, Wooster R. et al. Consistent loss of the wild type allele in breast cancers from a family linked to the BRCA2 gene on chromosome 13q12-13. Oncogene. 1995;10(8):1673-1675
5. Tavtigian SV, Simard J, Rommens J. et al. The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds. Nat Genet. 1996;12(3):333-337
6. Sharan SK, Morimatsu M, Albrecht U. et al. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997;386(6627):804-810
7. Chen PL, Chen CF, Chen Y. et al. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc Natl Acad Sci USA. 1998;95(9):5287-5292
8. Patel KJ, Yu VP, Lee H. et al. Involvement of Brca2 in DNA repair. Mol Cell. 1998;1(3):347-357
9. Yu VP, Koehler M, Steinlein C. et al. Gross chromosomal rearrangements and genetic exchange between nonhomologous chromosomes following BRCA2 inactivation. Genes Dev. 2000;14(11):1400-1406
10. Godthelp BC, van Buul PP, Jaspers NG. et al. Cellular characterization of cells from the Fanconi anemia complementation group, FA-D1/BRCA2. Mutat Res. 2006;601(1-2):191-201
11. Tutt A, Gabriel A, Bertwistle D. et al. Absence of Brca2 causes genome instability by chromosome breakage and loss associated with centrosome amplification. Curr Biol. 1999;9(19):1107-1110
12. Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7(2):263-272
13. Tutt A, Bertwistle D, Valentine J. et al. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. Embo J. 2001;20(17):4704-4716
14. Davies AA, Masson JY, McIlwraith MJ. et al. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell. 2001;7(2):273-282
15. Knudson AGJr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820-823
16. Warren M, Lord CJ, Masabanda J. et al. Phenotypic effects of heterozygosity for a BRCA2 mutation. Hum Mol Genet. 2003;12(20):2645-2656
17. Kim MK, Zitzmann S, Westermann F. et al. Increased rates of spontaneous sister chromatid exchange in lymphocytes of BRCA2+/- carriers of familial breast cancer clusters. Cancer Lett. 2004;210(1):85-94
18. Arnold K, Kim MK, Frerk K. et al. Lower level of BRCA2 protein in heterozygous mutation carriers is correlated with an increase in DNA double strand breaks and an impaired DSB repair. Cancer Lett. 2006;243(1):90-100
19. Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell. 2002;2(3):243-247
20. Schoenfeld AR, Apgar S, Dolios G. et al. BRCA2 is ubiquitinated in vivo and interacts with USP11, a deubiquitinating enzyme that exhibits prosurvival function in the cellular response to DNA damage. Mol Cell Biol. 2004;24(17):7444-7455
21. Naviaux RK, Costanzi E, Haas M. et al. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J Virol. 1996;70(8):5701-5705
22. Yuan SS, Lee SY, Chen G. et al. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 1999;59(15):3547-3551
23. Connor F, Bertwistle D, Mee PJ. et al. Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation. Nat Genet. 1997;17(4):423-430
24. Jonkers J, Meuwissen R, van der Gulden H. et al. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29(4):418-425
Author contact
![]() Correspondence to: Alan R. Schoenfeld, Adelphi University, Department of Biology, One South Avenue, P.O. Box 701, Garden City, NY 11530-0701. Phone: (516) 877-4211; Fax: (516) 877-4209; Email: schoenfeldedu
Correspondence to: Alan R. Schoenfeld, Adelphi University, Department of Biology, One South Avenue, P.O. Box 701, Garden City, NY 11530-0701. Phone: (516) 877-4211; Fax: (516) 877-4209; Email: schoenfeldedu