Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2008; 4(2):71-80. doi:10.7150/ijbs.4.71 This issue Cite
Research Paper
Shedding Light on the Role of Vitreoscilla Hemoglobin on Cellular Catabolic Regulation by Proteomic Analysis
Chartchalerm Isarankura-Na-Ayudhya1, Patcharee Panpumthong1, 2, Teerawit Tangkosakul1, Somchai Boonpangrak1, Virapong Prachayasittikul1 ![]()
1. Department of Clinical Microbiology, Faculty of Medical Technology, Mahidol University, Bangkok 10700, Thailand,
2. Department of Medical Technology, Faculty of Allied Health Science, Thammasat University, Pathum Thani 12120, Thailand
Received 2008-1-7; Accepted 2008-3-2; Published 2008-3-3
Abstract
Heterologous expression of Vitreoscilla hemoglobin (VHb) has been reported to improve cell growth, protein synthesis, metabolite productivity and nitric oxide detoxification. Although it has been proposed that such phenomenon is attributed to the enhancement of respiration and energy metabolism by facilitating oxygen delivery, the mechanism of VHb action remains to be elucidated. In the present study, changes of protein expression profile in Escherichia coli as a consequence of VHb production was investigated by two-dimensional gel electrophoresis (2-DE) in conjunction with peptide mass fingerprinting. Total protein extracts derived from cells expressing native green fluorescent protein (GFPuv) and chimeric VHbGFPuv grown in Luria-Bertani broth were prepared by sonic disintegration. One hundred microgram of proteins was individually electrophoresed in IEF-agarose rod gels followed by gradient SDS-PAGE gels. Protein spots were excised from the gels, digested to peptide fragments by trypsin, and analyzed using matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry. Results revealed that expression of VHbGFPuv caused an entire disappearance of tryptophanase as well as down-regulated proteins involved in various metabolic pathways, e.g. glycerol kinase, isocitrate dehydrogenase, aldehyde dehydrogenase, and D-glucose-D-galactose binding protein. Phenotypic assay of cellular indole production confirmed the differentially expressed tryptophanase enzymes in which cells expressing chimeric VHbGFP demonstrated a complete indole-negative reaction. Supplementation of δ-aminolevulinic acid (ALA) to the culture medium enhanced expression of glyceraldehyde-3-phosphate dehydrogenase and glycerol kinase. Our findings herein shed light on the functional roles of VHb on cellular carbon and nitrogen consumptions as well as regulation of other metabolic pathway intermediates, possibly by autoregulation of the catabolite repressor regulons.
Keywords: Vitreoscilla hemoglobin (VHb), Two-dimensional gel electrophoresis (2-DE), Proteomic, Catabolic regulation, Peptide Mass Fingerprinting (PMF), Mass spectrometry
1. Introduction
Vitreoscilla hemoglobin (VHb) is an oxygen binding protein produced by the obligate aerobic bacterium Vitreoscilla stercoraria. It is comprised of two identical subunits of relative molecular mass of 15.7 kDa and two protoheme IX per molecule. Expression of VHb in various organisms (e.g. bacteria, yeasts, fungi, and plant cells), particularly under hypoxic conditions, is known to improve growth, enhance protein secretion, increase metabolic productivity and stress resistance, mediate ATP synthesis and detoxify the deleterious effects of nitric oxide. Much attention has currently been dedicated to the use of VHb for various cell-based biotechnological processes including metabolic engineering, production of valuable metabolites, and fermentation (for recent review please see [1]).
Because of the diverse functions of VHb in various organisms, many investigations have been conducted to elucidate their cellular mechanisms and biological reactivities. The physiological function of VHb is postulated to act as terminal oxidases as to facilitate efficient delivery of oxygen under microaerobic conditions. Expression of VHb in Escherichia coli triggers increased ATP production, improved growth rate and final cell density, and enhanced foreign protein production. A plausible explanation is that the presence of VHb within the respiratory membrane promotes the oxygen flux to one or two terminal oxidases: aerobic terminal oxidase (Cyo) and microaerobic terminal oxidase (Cyd) [2]. Such effect is expected to cause an increase in proton-pumping efficiency and concomitantly lead to a remarkable generation of ATP [3]. An increased production of translational components (the active 70S ribosomes and tRNA levels) can be detected using asymmetrical flow field-flow fractionation (AFFFF), suggesting another important role of VHb on the protein synthesis machinery [4, 5]. Recently, it has been established that the prosthetic heme group of VHb possesses peroxidase-like activity like that of mammalian hemoglobins [6, 7]. These findings support the hypothesis that VHb not only acts as an oxygen carrier but possesses other important functional roles. However, the underlying mechanism of VHb on cellular catabolic regulation is not yet entirely understood.
In the present study, two-dimensional gel electrophoresis (2-DE) combined with peptide mass fingerprinting was used to investigate changes of protein expression profile in E. coli cells with VHb expression. Experimentation was initiated by fusing VHb with green fluorescent protein (GFP). GFPuv was selected as a reporter molecule to confirm vgb gene expression, which is under the control of lac promoter, because of the following reasons: i) its autofluorescence property is 18 times brighter than wt GFP, which can easily be detected by standard long wave UV, ii) this variant provides a high translational efficiency and high protein solubility when expressed in E. coli, iii) it is a small monomeric protein with molecular size of approximately 30 kDa, which makes protein fusion manageable, and iv) the location of fusion protein (VHbGFP) on the 2D gels can easily be discriminated from other high abundant proteins. Using 2-DE, protein expression profiles of cells harboring chimeric VHbGFP can potentially be scrutinized with cells expressing native GFP and cells bearing the control plasmid. Spots of proteins are then identified by MALDI-TOF mass spectrometry. In some circumstances, supplementation of δ-aminolevulinic acid (ALA; a precursor in the heme biosynthetic pathway formed in three steps enzymatic reaction of the five-carbon skeleton of glutamate; C5 pathway) to the culture medium has been performed for comparison. Plausible explanations on the novel role of VHb on cellular catabolic regulation have been proposed.
2. Materials and methods
Bacterial strains and plasmid
Escherichia coli (E. coli) strain TG1 (lac-pro), Sup E, thi l, hsd D5/F' tra D36, pro A+ B+, lacI, lacZ, M15; (ung+, dut+) was used as host for transformation and gene expression. Plasmids pGFPuv (Clontech Laboratories, USA) and pVHb [6] were used for construction of chimeric genes. Cells harboring pUC19 were used as control.
Enzymes, chemicals and reagent kits
High Fidelity Taq DNA polymerase, restriction endonucleases and T4 DNA ligase were purchased from Roche (Mannheim, Germany). Molecular weight marker (λ/EcoRI + HindIII) was obtained from New England Biolabs, USA. Purification of plasmid DNA was performed with NucleoSpin Plasmid kit (Macherey-Nagel, Germany). Purified DNA was extracted from agarose via NucleoSpin Extract II kit (Macherey-Nagel, Germany). The oligonucleotides were synthesized by the Bioservice Unit, Thailand. All chemicals were of analytical grade and commercially available.
Construction of chimeric gene encoding Vitreoscilla hemoglobin-green fluorescent protein
DNA fragment (438 bp) encoding the Vitreoscilla hemoglobin was obtained by PCR amplification using plasmid pVHb as template and the two primers (sense: 5′-ATAACTCTGCAGCATGTTAGACCAGCAAACCAT -3′, antisense: 5′-ATTAATGGTACCAATTCAACCGCTTGAGCGTACA -3′). Since the primers contained 5′ overhang of PstI site in the sense and KpnI site in the antisense, therefore, the PCR products were digested with these enzymes and subsequently inserted into the pGFPuv. These resulted in an in-frame fusion of the VHb encoding gene at the 5′-end of the gfpuv gene. Cloning procedures were performed according to the standard protocol as previously described [8]. The newly constructed plasmid, designated as pVHbGFP, was verified for correct insertion by restriction endonucleases digestion and further confirmed by DNA sequencing.
Analysis of excitation and emission spectra of chimeric protein
The chimeric VHbGFP was partially purified by 50% ammonium sulfate precipitation followed by DEAE ion exchange column chromatography. Fractions possessing green fluorescence were collected and subjected to fluorescence spectra scanning using Perkin-Elmer spectrofluorometer FP6300 at ambient temperature. To obtain the fluorescence emission spectra of VHb and GFP, excitation wavelengths were fixed at 313 and 400 nm, respectively. The excitation spectra were further scanned upon setting the emission wavelengths at 630 and 509 nm.
Preparation of protein samples for proteomic analysis
Cells carrying pUC19, pGFPuv and pVHbGFP were grown at 37ºC for overnight in 5 ml Luria-Bertani (LB) broth (10 g/L tryptone, 5 g/L NaCl and 5 g/L yeast extract, pH 7.2) supplemented with 100 μg/ml Ampicillin. Cells were subcultured and further incubated at 37ºC for 6 hours to mid-exponential phase. Cells were then inoculated in 50 ml LB/Amp and incubated at 37ºC for 16 hours. In some circumstances, cells were incubated in the medium supplemented with 75 μM δ-aminolevulinic acid (ALA) for comparison. Cells were collected by centrifugation at 9,000 rpm for 10 min at 4ºC and washed for 3 times using 40 mM Tris, pH 8.0. Pellet was then resuspended in 1 ml of Tris buffer and mixed with 250 μl of lysis solution (7 M urea, 2 M thiourea, 4% CHAPS; freshly prepared by supplementation with 10 mg/ml dithiothreitol (DTT) and 10 μl/ml protease inhibitor cocktail). Cells were disrupted on ice by sonic disintegration at power 7 for 30 sec using Branson sonifier (model 450) equipped with a microtip. Whole cell lysates were collected by centrifugation at 15,000 rpm for 60 min at 4ºC. Protein concentrations were quantified by Bradford's method [9] using bovine serum albumin as a standard. The protein solution was mixed with 1 M acrylamide (at 1:10 of total volume) and kept at room temperature for 10 min.
Two-dimensional electrophoresis (2-DE)
The 2DE was carried out using IEF disc gel (Atto Corporation, model AE-6541, Japan) and mPAGE (Atto Corporation, model AE-6531, Japan) according to manufacturer's recommendations with minor modifications as follows. One hundred micrograms of samples were carefully applied on top of the precasting IEF agarose rod gels (Atto Corporation; pH ranges 3-10 and 5-8). Approximately 150 μl of upper electrode buffer (0.2 M NaOH) was filled into the column. Separation of proteins was carried out at 300 volts for 3.5 hours. Proteins in agarose gels were subsequently fixed in 1D gel solidified solution (2.5 g% Trichloroacetic acid) for 3 min. The gels were rinsed several times with distilled water and kept for overnight at 4ºC. The gels were soaked in SDS equilibration buffer (50 mM Tris-Cl, pH 8.8 containing 6 M urea, 30% (v/v) glycerol, 2% SDS and bromphenol blue) for 10 min at room temperature. The gels were thoroughly rinsed with SDS electrophoresis buffer and immediately placed onto the precasting gradient (5-20%) SDS-PAGE gel (Atto Corporation). Standard protein markers were spotted onto 3 mm2 Whatman filter paper and laid down in close contact with the SDS-PAGE gels. The rod gels were mounted onto the SDS-PAGE gels by applying one hundred microliter of pre-warmed agarose gel (1% of low melting agarose gel). Initially, separation of protein was conducted at 10 mA for 10 min and continued at 40 mA for 90 min. The gels were further stained with colloidal Coomassie blue for 2-3 hours. Excess dye was removed by rinsing several times with deionized distilled water. Gels were visualized with the Canoscan LiDE20 scanner (Canon, USA).
Mass spectrometry and peptide mass fingerprinting (PMF) analysis
Protein spots were manually excised from the gels. Gel pieces from each spot were transferred to a polypropylene 96 well microtitre plate and then soaked in 50% methanol and 5% acetic acid for overnight. Tryptic digestion using sequencing grade of modified trypsin (Promega, UK) and protein extraction were performed on a Spot Handling Workstation (GE Health Care, USA) using the preset protocols from the manufacture.
Protein identification was mainly based on peptide fingerprint map obtained from the MALDI-TOF mass spectrometer (Model ReflexIV, Bruker Daltonics, Germany). Briefly, the extracted peptides were mixed with solution of 10 mg/ml α-cyano-4-hydroxycinnamic acid (LaserBio Labs, France) in 66% acetonitrile and 0.1% trifluoroacetic acid (TFA) and then spotted onto a 96-well target plate. The mass spectra were acquired in the positive ion reflector delayed extraction mode using approximately 200 laser shots. Peak lists were generated using the XMASS software (Bruker Daltonics). The BioTool 2.0 software (Bruker Daltonics) integrated with the MASCOT 2.2 search engine (MatrixScience, http://www.matrixscience.com/) was used for the identification of protein spots by querying the trypsin-digested peptide fragment data. The reference database used for the identification of target proteins was NCBInr with 5470121 sequences and 1894087724 residues. The searching criteria exploited on complete carbamidomethylation of cysteine and partial methionine oxidation. One missed cleavage per peptide was allowed and an initial mass tolerance of ±1 Da was used in all searches. Search result scores which is greater than 80 was considered to be of significant difference (p<0.05). The accuracy of the experimental to theoretical pI and molecular weight of proteins were also taken into consideration.
Indole assay
Cells were inoculated into the tryptophan broth (10 g/L peptone from meat, 1 g/L DL-tryptophan, 5 g/L sodium chloride, pH 7.2) and grown at 37ºC for 24 hours. Kovac's Indole reagent was then added to the broth and the presence of indole ring (pink color) on the surface of broth was observed. For comparison, cells were cultured in LB broth [10, 11] and assay of indole production was performed in a similar manner. Results were taken from two independent experiments.
3. Results
Genetic construction of chimeric Vitreoscilla hemoglobin-green fluorescent protein
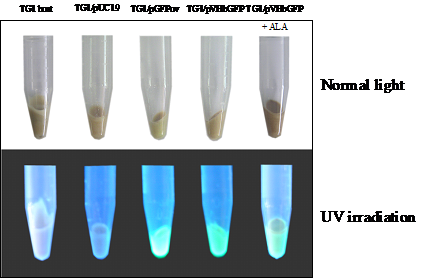
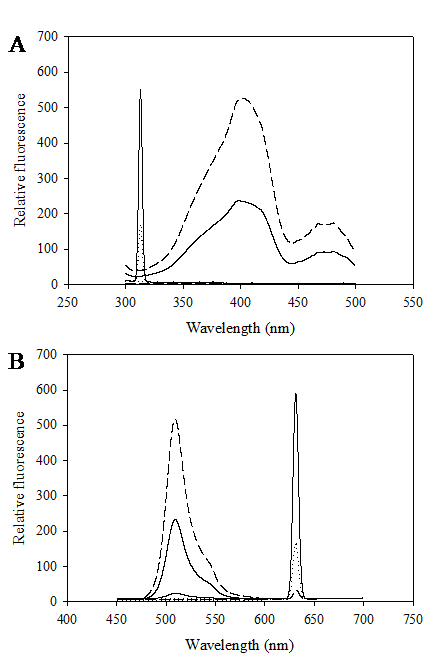
Construction of the chimeric Vitreoscilla hemoglobin and green fluorescent protein, designated as VHbGFP, was carried out successfully. VHbGFP was found to possess dual characteristics in which supplementation of ALA (heme precursor) to the culture medium gave rise to red pigment and strong greenish fluorescence under normal light exposure and UV irradiation, respectively (Fig. 1). Results from excitation and emission maxima scanning confirmed successful construction of the protein fusion. As depicted in Fig. 2, the chimeric VHbGFP exhibited two excitation maximum peaks at 313 and 400 nm, which represented the combined characteristics of VHb and GFPuv, respectively. Likewise, the emission maxima were found to be at 509 and 630 nm, respectively. This indicated that the red pigments (originated from the 'heme' structure of VHb [12]) did not interfere with the fluorescence excitation and emission spectra of GFP. However, it should be noted that the chimeric VHbGFP displayed a relative fluorescence emission (at 630 nm) of approximately 5 times higher than that of the native VHb. However, a marked decrease in the green fluorescence emission (at 509 nm) of up to 20-30 times was detected as compared to that of the native GFP.
Total amount of Escherichia coli TG1 host and engineered cells harboring different kinds of plasmid grown in 50 ml LB/Amp broth in the presence and absence of 75 μM δ-aminolevulinic (ALA) at 37°C for 16 hours observed under normal light and UV irradiation.
Excitation (A) and fluorescence emission (B) spectra of chimeric VHbGFP (solid line), native VHb (dot line) and native GFPuv (dash line).
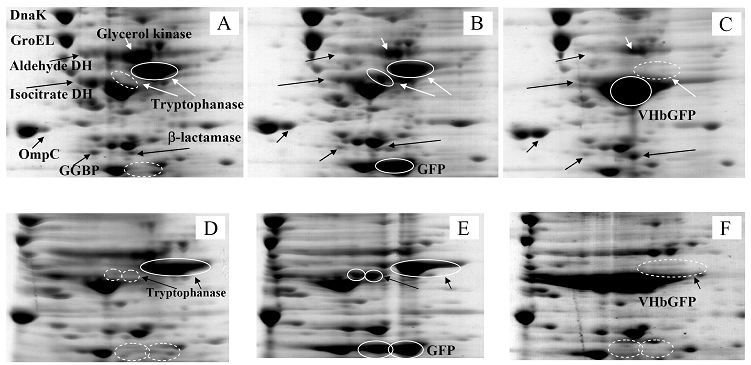
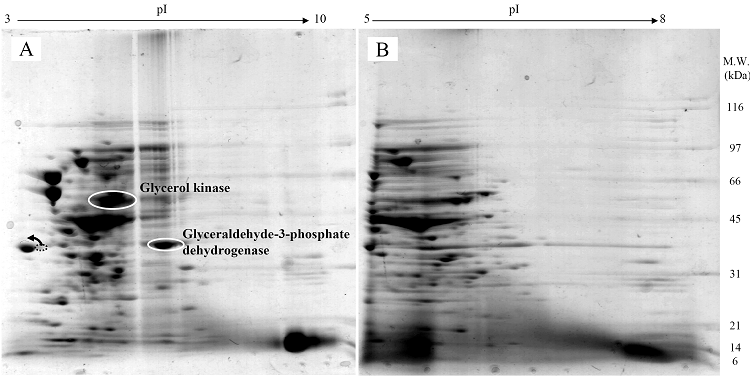
Proteomic analysis of whole cell lysate of E. coli expressing chimeric VHbGFP
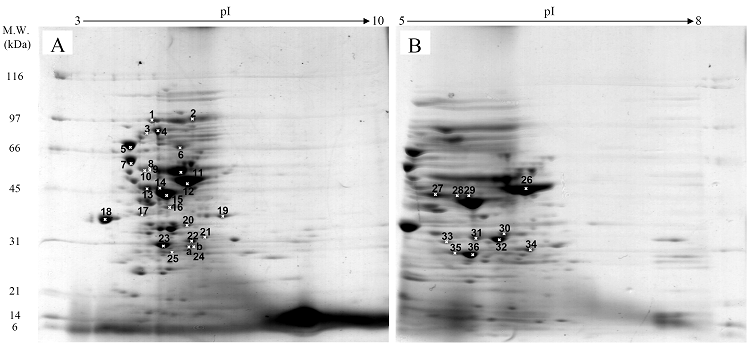
Fig. 3 demonstrated two complete maps of protein expression profiles of control cells harboring pUC19 at different ranges of pH (3-10 and 5-8). Protein spots were then identified by mass spectrometry and the results are summarized in Table 1. In some locations, several spots of protein were picked up in order to differentiate the co-migration of proteins. Spots number 5 and 7 identified as molecular chaperones DnaK and GroEL, respectively were usually located as markers in several proteomic studies [13, 14]. High abundant proteins were analyzed to be glycerol kinase (no. 11), tryptophanase (no. 12 and 26), translational elongation factor Tu (no. 15), outer membrane porin protein C (no. 18) and beta-lactamase (no. 23 and 36). Expression of GFPuv in E. coli resulted in minor changes of protein profiles (Figs. 4 A and C). GFPuv with a size of about 30 kDa and a pI of 5.8 was represented as a high abundant protein (no. 50, 62 and 63). The presence of GFPuv was found to mediate the down-regulation of glycerol kinase (no. 40) and isocitrate dehydrogenase (no. 44). Importantly, the presence of GFPuv somehow rendered the tryptophanase to be of two isoforms (no. 42, 43, 52-54). This is in contrast to those observed in the case of VHbGFP (Figs. 4 B and D) where the major band of tryptophanase was found to be completely disappeared from the gel. Changes of protein profiles as a consequence of VHbGFP are illustrated in detail in Fig. 5. It seemed that the presence of GFP and VHbGFP up-regulated the main cellular chaperones, e.g. DnaK and GroEL. On the contrary, glycerol kinase, isocitrate dehydrogenase, aldehyde dehydrogenase and D-galactose-D-glucose binding protein (GGBP) were remarkably down-regulated. Disappearance of tryptophanase was found only upon expression of VHb protein. As expected, VHbGFP was found to be located at approximately 45 kDa (no. 70a and 70b) since the molecular weights of GFP and VHb monomer were of 30 kDa and 15 kDa in size, respectively. Experimental results indicated that this protein was the major protein present in the gel. However, identification of protein in this area revealed that the VHbGFP co-migrated with elongation factor (no. 71, 72 and 85) and showed none of the tryptophanase isoform. This confirmed the notion that tryptophanase was the major target responsible for expression of bacterial hemoglobin.
Effect of δ-aminolevulinic acid (ALA) on protein expression profiles
The next question to address was whether supplementation of ALA rendered changes of protein expression profile. Results revealed that none of the tryptophanase was observed while glycerol kinase and glyceraldehyde-3-phosphate dehydrogenase were remarkably up-regulated (Fig. 6). Transformation of outer membrane porin protein C isoform from higher to lower pI values was also taken into account (as indicated by arrow).
Phenotypic changes of E. coli expressing chimeric VHbGFP
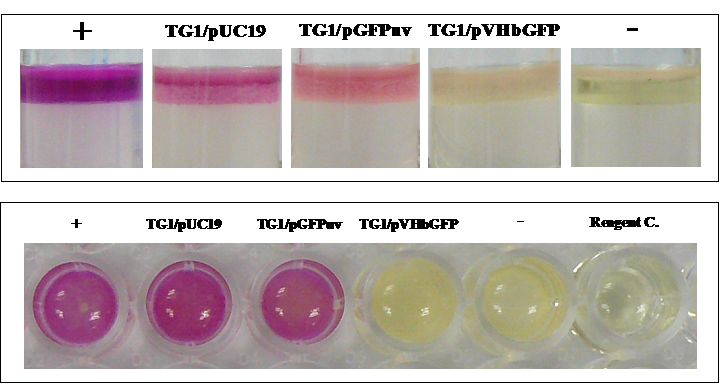
To further confirm the differentially expressed tryptophanase, an assay of indole (a major product of tryptophan degradation by tryptophanase) production was performed on the different cell types. As illustrated in Fig. 7, our results clearly demonstrated that only cells expressing chimeric VHbGFP showed strong indole-negative reaction comparable to that of Klebsiella pneumoniae. Results from both tryptophan broth (Top panel) and LB broth (Bottom panel) were in good agreement.
Proteins of Escherichia coli TG1 host and engineered cells expressing GFPuv and chimeric VHbGFP identified by Mass spectrometry and peptide mass fingerprinting (PMF) analysis.
| Spot No. | Accession No. | Description | Calculated pI value | Nominal mass (Mr) | Protein score | Sequence Coverage (%) |
|---|---|---|---|---|---|---|
| 1, 37 | gi|75236921 | Aconitase B | 5.21 | 93996 | 170, 145 | 55, 64 |
| 2, 38 | gi|15830005 | 2-oxoglutarate dehydrogenase decarboxylase component | 6.04 | 105566 | 81, 92 | 43, 59 |
| 3 | gi|75197115 | Polyribonucleotide nucleotidyltransferase | 5.11 | 77110 | 90 | 62 |
| 4 | gi|15803853 | Elongation factor EF-2 | 5.24 | 77704 | 102 | 55 |
| 5 | gi|15799694 | Molecular chaperone DnaK | 4.83 | 69130 | 155 | 70 |
| 6 | gi|15830002 | Succinate dehydrogenase catalytic subunit | 5.85 | 65008 | 99 | 42 |
| 7 | gi|15834378 | Chaperonin GroEL | 4.85 | 57464 | 214 | 68 |
| 8 | gi|38704234 | Aspartate ammonia-lyase | 5.19 | 52950 | 82 | 44 |
| 9, 10 | gi|15801728 | Aldehyde dehydrogenase | 5.07 | 52377 | 147, 86 | 63, 53 |
| 11, 40, 65 | gi|442946 | Glycerol kinase | 5.36 | 56349 | Range 84-123 | 52 |
| 12, 26, 42, 43, 52, 53, 54 | gi|41936 | Tryptophanase | 5.88 | 53098 | Range 80-143 | Range 41-60 |
| 13, 27, 44, 66 | gi|33383669 | Isocitrate dehydrogenase | 5.33 | 43192 | Range 101-127 | Range 35-55 |
| 14, 28, 29, 45, 55, 56 | gi|16975437 | Enolase, chain A | 5.32 | 45552 | Range 80-127 | Range 54-64 |
| 15, 16, 46 | gi|124532037 | Translation elongation factor Tu | 5.01 | 40514 | Range 103-122 | 68 |
| 17 | gi|14277926 | Transaldolase B, chain A, | 5.18 | 35193 | 95 | 44 |
| 18, 75, 76 | gi|16130152 | Outer membrane porin protein C | 4.58 | 40343 | Range 141-176 | Range 69-77 |
| 19 | gi|15802193 | Glyceraldehyde-3-phosphate dehydrogenase | 6.61 | 35681 | 101 | 53 |
| 20, 47, 77 | gi|9507742 | Outer membrane protease precursor [Plasmid F] | 5.91 | 35477 | Range 98-129 | Range 61-72 |
| 21 | gi|15800433 | Succinyl-CoA synthetase alpha subunit | 6.32 | 30044 | 83 | 60 |
| 22, 34, 79, 92 | gi|75196144 | Fructose/tagatose bisphosphate aldolase | 5.87 | 31021 | Range 80-96 | Range 62-65 |
| 23, 35, 36, 51, 61, 80, 93 | gi|145306442 | Beta-lactamase | 5.35 | 31247 | Range 147-197 | Range 49-80 |
| 24a | gi|1000056 | Ribose-binding protein complexed with Beta-D-Ribose | 5.99 | 28457 | 101 | 70 |
| 24b | gi|26246721 | Phosphoglyceromutase | 6.27 | 29204 | 97 | 62 |
| 25 | gi|75209666 | Glutathione S-transferase | 5.22 | 24328 | 91 | 61 |
| 30 | gi|226907 | Malate dehydrogenase | 5.61 | 32417 | 93 | 55 |
| 31, 32, 89, 90 | gi|15800816 | Outer membrane protein 3a | 5.99 | 37292 | Range 80-88 | Range 42-45 |
| 33, 60 | gi|230520 | D-Galactose-D-Glucose Binding Protein (GGBP) | 5.25 | 33347 | 111, 189 | 62, 70 |
| 39, 64 | gi|15829780 | Heat shock protein 90 | 5.09 | 71378 | 220, 80 | 49, 42 |
| 41 | gi|15804332 | ATP synthase subunit B | 4.90 | 50351 | 86 | 44 |
| 48, 78, 91 | gi|2293126 | Beta-lactamase | 5.93 | 31730 | Range 138-180 | Range 68-75 |
| 49, 50, 62, 63, 67, 68, 69, 73, 81, 83, 84, 86 | gi|1490529 | GFPuv | 5.8 | 26893 | Range 99-121 | Range 53-60 |
| 57, 85 | gi|26249935 | Elongation factor Tu | 5.25 | 44993 | 112, 108 | 65, 68 |
| 58, 88 | gi|75237909 | Malate/lactate dehydrogenases | 5.61 | 32502 | 97, 150 | 68, 68 |
| 59 | gi|13399487 | L-Asparaginase | 5.66 | 34671 | 84 | 54 |
| 70a | gi|1490529 | GFPuv | 5.8 | 26893 | 85 | 65 |
| 70b | gi|114816 | Bacterial hemoglobin (Soluble cytochrome O) | 5.31 | 15821 | 81 | 50 |
| 71, 72 | gi|1942721 | Chain A, Elongation Factor Complex Ef-TuEF-Ts | 5.22 | 42321 | 106, 102 | 58, 65 |
| 74, 87 | gi|15833050 | Fructose-bisphosphate aldolase | 5.52 | 39351 | 107, 122 | 42, 48 |
| 82 | gi|26250698 | ATP-dependent protease ATP-binding subunit | 5.24 | 49664 | 92 | 41 |
Note: Protein scores greater than 80 are significant (p<0.05). Protein scores and sequences coverage of multiple protein spots were given as the range of values.
Protein expression profiles of control E. coli cells bearing pUC19 plasmid separated under pH ranges of 3-10 (A) or 5-8 (B) and stained with colloidal Coomassie blue. (Numbers of protein spot denoted as identified protein represented in Table 1)
Protein expression profiles of E. coli expressing native GFPuv (A, C) and chimeric VHbGFP (B, D) separated under pH ranges of 3-10 and 5-8, respectively and stained with colloidal Coomassie blue. (Numbers of protein spot denoted as identified protein represented in Table 1)
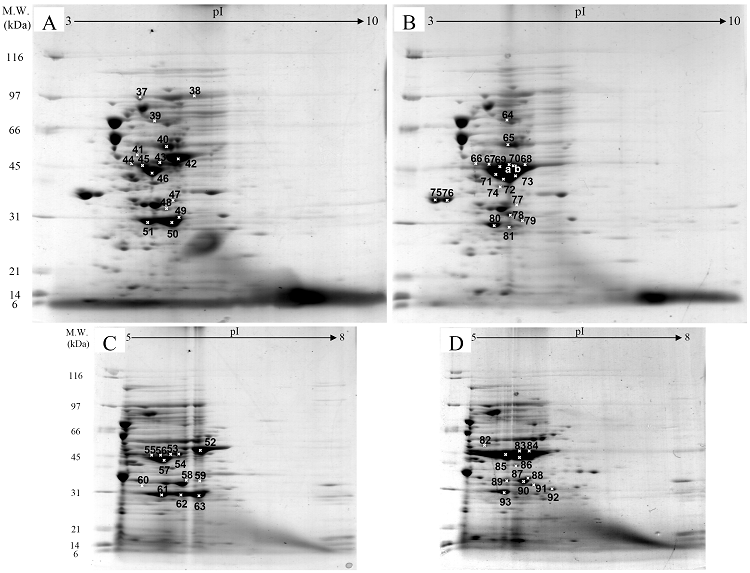
Zoomed 2D gels of protein profiles of crude proteins from control cells (A, D), cells expressing native GFPuv (B, E), and cells expressing chimeric VHbGFP (C, F) located at the same regions under pH ranges of 3-10 and 5-8, respectively.
Protein expression profiles of E. coli expressing chimeric VHbGFP grown in the presence of 75 μM δ-aminolevulinic (ALA) separated under pH ranges of 3-10 and 5-8, respectively and stained with colloidal Coomassie blue.
Biochemical assay of indole production in E. coli expressing chimeric VHbGFP cultured in tryptophan broth (Top panel) and LB broth (Bottom panel). Positive (+) and negative (-) controls were tested using Escherichia coli ATCC 25922 and Klebsiella pneumoniae ATCC 700603, respectively.
4. Discussion
Using two-dimensional gel electrophoresis in conjunction with peptide mass fingerprinting analysis, novel functional roles of Vitreoscilla hemoglobin on cellular catabolic regulation has been explored for the first time. Intracellular expression of VHb triggered a complete disappearance of tryptrophanase (Fig. 5 C), an enzyme involved in tryptophan, cysteine and serine catabolism. Confirmation of differentially expressed tryptophanase by indole assay revealed a pertinent indole-negative reaction by the VHb-expressing cells (Fig. 7). Our results coincided with the statement that expression of VHb perturbs the carbon flux of pentose phosphate metabolism, which may in turn affect key cellular enzymes and exert effects on the synthesis of aromatic amino acids [15]. More importantly, it has previously been reported that tryptophanase, aldehyde dehydrogenase and citrate synthase are drastically induced as a consequence of heme depletion [16]. In this study, parallel results revealed the complete loss of tryptophanase and down-regulation of aldehyde dehydrogenase (Fig. 5). Our presumptive evidence lends support to the notion that VHb potentially mediates alterations in intracellular carbon and nitrogen consumptions. This infers the relationship of ATP generation for metal ions transportation [17].
Detailed explanations for the aforementioned observation suggest that the tryptophanase and aldehyde dehydrogenase are under the control of catabolite repressor protein (Crp), a primary transcriptional regulator of carbon metabolism [18, 19]. It is known that the regulatory response of Crp is controlled by either stress signals for the utilization of alternative carbon sources or by cyclic-AMP under energy-deficient conditions [20]. Furthermore, there are also transcriptional regulatory connections between carbon and iron metabolism whereby Crp can modulate the transcription of fur gene (encoding the ferric uptake regulator; Fur) [21]. This autoregulation between Crp and Fur regulons is believed to be the underlying mechanism in maintaining intracellular Fur levels below a certain limit since iron is required for aerobic Krebs cycle and electron flow. Such evidences discern the relationship between iron regulation and metabolic status of cells. It is expected that VHb plays a role in the electron transport chain of oxidative phosphorylation during energy conservation in the same fashion as other heme-containing derivatives (e.g. the prosthetic group of cytochromes and tetrapyrroles). Therefore, it can be speculated that excessive intracellular heme production [22] due to the appearance of bacterial hemoglobin may in turn regulate Crp and other metabolic pathway intermediates.
Effects of VHb on regulation of metabolic pathway intermediates are summarized as follows. Changes of other metabolic-regulated proteins including glycerol kinase, isocitrate dehydrogenase, and D-galactose-D-glucose binding protein (GGBP) have been observed (Fig. 5). Glycerol kinase is a key enzyme involved in the regulation of glycerol uptake and lipolysis processes. Our findings revealed marked decrease of this enzyme subsequently leading to rapid accumulation of free glycerol. This coincides with the complete loss of tryptophanase and a decrease in aldehyde dehydrogenase since glycerol is established as a potent inhibitor and catabolic repressor [23, 24]. More supportive evidences have been reported on the inhibition and induction of glycerol kinase by fructose 1,6-biphosphate, L-alphaglycerol-3-phosphate and 2,4-dinitrophenol [25]. Reduction of isocitrate dehydrogenase may also be taken into consideration for retaining glycerol contents [24] and phosphorylating processes on the branch point between the glyoxylate pathway and the Krebs cycle [25]. Overexpression of intracellular VHb rapidly metabolizes huge amounts of carbon sources for energy production resulting in the consumption of GGBP [26].
Addition of ALA regains a remarkable amount of the glycerol kinase as well as the glyceraldehyde-3-phosphate dehydrogenase (an enzyme implicated in the glycolysis pathway) (Fig. 6). This controversial effect compared to the above mentioned remark may be attributable to the imbalance between the intermediates (involved in heme biosynthetic pathway) and overproduction of bacterial globin chains, subsequently leading to metabolic adaptation of the engineered cells. These observations coincide with the finding in Staphylococcus aureus that interruption of electron transport chain due to deficiency of heme causes an induction of proteins involved in the glycolytic pathway and other cluster of enzymes (e.g. glyceraldehyde-3-phosphate dehydrogenase, enolase, phosphoglycerate kinase, lactate dehydrogenase, alcohol dehydrogenase, and pyruvate formate lyase) [27]. It has been reported that supplementation of ALA in Pseudomonas cultures leads to an increased flux of heme pathway and excretion of high levels of porphyrins [28]. Further investigations, particularly on the equilibrium of heme contents and exogenous supplementation with iron salts and other catabolic activation/repression molecules are now taken into consideration as ongoing research in our laboratory.
Acknowledgements
T.T. is a postgraduate student supported by the collaborative Ph.D. fellowship from the Royal Thai Government under the supervision of V.P. This project was partially supported by an annual governmental grant under Mahidol University (2551-2555 B.E.). The authors would like to thank Mr. Surasak Jiemsup and Ms. Sirinuch Banyen for technical assistance.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Zhang L, Li Y, Wang Z. et al. Recent developments and future prospects of Vitreoscilla hemoglobin application in metabolic engineering. Biotechnol Adv. 2007;25:123-36
2. Tsai PS, Nageli M, Bailey JE. Intracellular expression of Vitreoscilla hemoglobin modifies microaerobic Escherichia coli metabolism through elevated concentration and specific activity of cytochrome o. Biotechnol Bioeng. 2002;79:558-67
3. Kallio PT, Kim DJ, Tsai PS. et al. Intracellular expression of Vitreoscilla hemoglobin alters Escherichia coli energy metabolism under oxygen-limited conditions. Eur J Biochem. 1994;219:201-8
4. Andersson CI, Arfvidsson C, Kallio PT. et al. Enhanced ribosome and tRNA contents in Escherichia coli expressing a truncated Vitreoscilla hemoglobin mutant analyzed by flow field-flow fractionation. Biotechnol Lett. 2003;25:1499-504
5. Nilsson M, Kallio PT, Bailey JE. et al. Expression of Vitreoscilla hemoglobin in Escherichia coli enhances ribosome and tRNA levels: a flow field-flow fractionation study. Biotechnol Prog. 1999;15:158-63
6. Suwanwong Y, Kvist M, Isarankura-Na-Ayudhya C. et al. Chimeric antibody-binding Vitreoscilla hemoglobin (VHb) mediates redox-catalysis reaction: new insight into the functional role of VHb. Int J Biol Sci. 2006;2:208-15
7. Kvist M, Ryabova ES, Nordlander E. et al. An investigation of the peroxidase activity of Vitreoscilla hemoglobin. J Biol Inorg Chem. 2007;12:324-34
8. Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning. A Laboratory Manual. New York, USA: Cold Spring Harbour Laboratory Press. 1989
9. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248-54
10. Lelong C, Aguiluz K, Luche S. et al. The Crl-RpoS regulon of Escherichia coli. Mol Cell Proteomics. 2007;6:648-59
11. Martino PD, Fursy R, Bret L. et al. Indole can act as an extracellular signal to regulate biofilm formation of Escherichia coli and other indole-producing bacteria. Can J Microbiol. 2003;49:443-9
12. Park KW, Webster DA, Stark BC. et al. Fusion protein system designed to provide color to aid in the expression and purification of proteins in Escherichia coli. Plasmid. 2003;50:169-75
13. Han MJ, Yoon SS, Lee SY. Proteome analysis of metabolically engineered Escherichia coli producing Poly(3-Hydroxybutyrate). J Bacteriol. 2001;183:301-8
14. Hatzimanikatis V, Choe LH, Lee KH. Proteomics: theoretical and experimental considerations. Biotechnol Prog. 1999;15:312-8
15. Tsai PS, Hatzimanikatis V, Bailey JE. Effect of Vitreoscilla hemoglobin dosage on microaerobic Escherichia coli carbon and energy metabolism. Biotechnol Bioeng. 1996;49:139-50
16. Rompf A, Schmid R, Jahn D. Changes in protein synthesis as a consequence of heme depletion in Escherichia coli. Curr Microbiol. 1998;37:226-30
17. Khleifat KM. Correlation between bacterial hemoglobin and carbon sources: their effect on copper uptake by transformed E. coli strain alpha DH5. Curr Microbiol. 2006;52:64-8
18. Gosset G, Zhang Z, Nayyar S. et al. Transcriptome analysis of Crp-dependent catabolite control of gene expression in Escherichia coli. J Bacteriol. 2004;186:3516-24
19. Isaacs HJr, Chao D, Yanofsky C. et al. Mechanism of catabolite repression of tryptophanase synthesis in Escherichia coli. Microbiology. 1994;140:2125-34
20. Saier MH, Ramseier TM, Reizer J. Regulation of carbon utilization. In: (ed.) Neidhardt FC. et al. Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd ed. Washington DC: American Society for Microbiology. 1996:1325-43
21. De Lorenzo V, Herrero M, Giovannini F. et al. Fur (ferric uptake regulation) protein and CAP (catabolite-activator protein) modulate transcription of fur gene in Escherichia coli. Eur J Biochem. 1998;173:537-46
22. Dikshit KL, Webster DA. Cloning, characterization and expression of the bacterial globin gene from Vitreoscilla in Escherichia coli. Gene. 1988;70:377-86
23. Botsford JL, Demoss RD. Catabolite repression of tryptophanase in Escherichia coli. J Bacteriol. 1971;105:303-12
24. Eppler T, Postma P, Schutz A. et al. Glycerol-3-phosphate-induced catabolite repression in Escherichia coli. J Bacteriol. 2002;184:3044-52
25. Han MJ, Lee SY. The Escherichia coli proteome: past, present, and future prospects. Microbiol Mol Biol Rev. 2006;70:362-439
26. Piszczek G, D'Auria S, Staiano M. et al. Conformational stability and domain coupling in D-glucose/D-galactose-binding protein from Escherichia coli. Biochem J. 2004;381:97-103
27. Kohler C, von Eiff C, Peters G. et al. Physiological characterization of a heme-deficient mutant of Staphylococcus aureus by a proteomic approach. J Bacteriol. 2003;185:6928-37
28. Harris WF, Burkhalter RS, Lin W. et al. Enhancement of bacterial porphyrin biosynthesis by exogenous aminolevulinic acid and isomer specificity of their products. Bioorg Chem. 1993;21:209-20
Author contact
![]() Correspondence to: Assoc. Prof. Dr.Virapong Prachayasittikul, Department of Clinical Microbiology, Faculty of Medical Technology, Mahidol University, 2 Prannok Rd., Bangkok-Noi, Bangkok 10700, Thailand. Tel: 662-418-0227; Fax: 662-412-4110; E-mail: mtvprac.th
Correspondence to: Assoc. Prof. Dr.Virapong Prachayasittikul, Department of Clinical Microbiology, Faculty of Medical Technology, Mahidol University, 2 Prannok Rd., Bangkok-Noi, Bangkok 10700, Thailand. Tel: 662-418-0227; Fax: 662-412-4110; E-mail: mtvprac.th