ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2008; 4(3):133-142. doi:10.7150/ijbs.4.133 This issue Cite
Research Paper
Identification and characterization of cancer initiating cells from BRCA1 related mammary tumors using markers for normal mammary stem cells
Athanassios Vassilopoulos1, Rui-Hong Wang1, Constantinos Petrovas2, David Ambrozak2, Richard Koup2, Chu-Xia Deng1 ![]()
1. Genetics of Development and Disease Branch, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland, MD 20892, USA.
2. Immunology Laboratory, Vaccine Research Center, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health, Bethesda, Maryland, MD 20892, USA.
Abstract
It is hypothesized that cancer stem cells arise either from normal stem cells or from progenitor cells that have gained the ability to self-renew. Here we determine whether mammary cancer stem cells can be isolated by using antibodies that have been used for the isolation of normal mammary stem cells. We show that BRCA1 mutant cancer cell lines contained a subpopulation of CD24+CD29+ or CD24+CD49f+ cells that exhibited increased proliferation and colony forming ability in vitro, and enhanced tumor-forming ability in vivo. The purified CD24+CD29+ cells could differentiate and reconstitute the heterogeneity found in parental cells when plated as a monolayer. Under low-attachment conditions, we detected “tumorspheres” only in the presence of double positive cells, which maintained their ability to self-renew. Furthermore, CD24+CD29+ cells could form tubular structures reminiscent of the mammary ductal tree when grown in three-dimensional cultures, implying that these cancer cells maintain some of the characteristics of the normal stem cells. Nevertheless, they could still drive tumor formation since as low as 500 double positive cells immediately after sorting from BRCA1 mutant primary tumors were able to form tumors with the same heterogeneity found in the original tumors. These data provide evidence that breast cancer stem cells originate from normal stem cells and advance our understanding of BRCA1-associated tumorigenesis with possible implications for future cancer treatment.
Keywords: breast cancer, BRCA1, stem cells, CD24, CD29
1. Introduction
Breast cancer occurs at a high frequency and is the second leading cause of death from cancer in women [1, 2, 3, 4, 5]. One of the major problems in treating breast cancer is the heterogeneity of tumors that prevents a complete response of cancer cells to the treatment [6]. This situation is more evident especially in basal and breast cancer associated gene-1 (BRCA1)-related breast cancers. The apparent paradox is that despite the excellent initial response to chemotherapy [7], cancer is more likely to recur early [8]. Clinical treatment regimens operate under the assumption that all cancer cells have equal malignant potential, thus, suffer from their lack of specificity for only tumorigenic cells. Relatively successful cancer treatments shrink the bulk of tumor cells but often fail to eliminate the cancer stem cells [9]. The necessity for targeting cancer stem cells highlights the importance of defining markers for identification of the cancer initiating subpopulation of cells in order to better characterize and analyze these cells.
There are two possible ways in which cancer stem cells can be generated. They can arise either from normal stem cells or from progenitor cells that have gained the ability for self-renewal. In contrast to the progress in the hematopoietic system, where the entire cellular hierarchy is known [10, 11], our knowledge on mammary tissue is limited. The massive expansion of the mammary gland during puberty and pregnancy, together with the regenerative capacity during successive reproductive cycles implicate the presence of stem-like cells in the mammary gland [12]. In the adult human breast it was shown that staining for keratins K14 and K19 may reveal both the stem cell ductal zone and the stem cell hierarchy [13]. In mice, the existence of normal mammary stem cells has been postulated in the past by experiments showing that after serial transplantation of retrovirally tagged epithelial fragments, a single progenitor cell may repopulate an entire mammary gland [14]. More recently, discrete populations of mouse mammary cells have been isolated on the basis of cell-surface markers and a subpopulation (Lin-CD29hiCD24+ or Lin-CD49fhiCD24med) was identified that is highly enriched for mammary stem cells [15, 16]. On the other hand, a population of Lin-CD44+CD24-/Low cells has been identified as cancer stem cells in human breast cancers [17]. Although there has been much progress in the field, many questions still remain. For example, can we use markers for normal mammary stem cells in mice to isolate mammary cancer stem cells? Is it possible that markers for identifying human breast cancer stem cells can be used for the isolation of either mammary cancer stem cells or normal breast stem cells? What are the phenotypes of these cells by means of the particular used markers for cancer stem cells, normal mammary stem cells, and the progenitor cells?
These undefined issues are multiplied when considering the role of BRCA1 in mammary stem cells. Germline mutations in BRCA1 have been detected in approximately half of familial breast cancer cases and most cases of combined familial breast/ovarian cancers [1-3, 5, 18]. Numerous previous studies in mouse models uncovered an essential role for BRCA1 in multiple biological processes, including DNA damage repair, cell cycle checkpoint regulation, transcriptional activation and repression, mammary gland development and mammary tumor formation [19]. Interestingly, many characteristics of the BRCA1-related cancers fit with the stem cell origin of these cancers. First, data suggests that breast stem cells are estrogen receptor negative [20] and breast cancers related to BRCA1 are more likely to be estrogen receptor negative than other types of breast cancer [21-23]. Indeed, it was recently shown that BRCA1 plays a critical role in the differentiation of ER-negative stem/progenitor cells to ER-positive luminal cells [23]. Second, immortal cells, such as stem cells, need few mutational “hits” to become cancer, and this might explain the early age of onset of BRCA1-related cancers [24]. Third, women with BRCA1 mutations who breastfed for a cumulative total of more than one year have a statistically significant reduced risk of breast cancer, which is in accordance with the breast cancer stem cell phenotype [25].
Taking into consideration that in the hematopoietic system normal stem cells share the same cell-surface markers with leukemic stem cells [26, 27], we wanted to test whether CD24 and CD29, which have been used before for the isolation of normal mammary stem cells [15, 16], can be used as markers for the isolation of enriched subpopulations of cancer initiating cells. Moreover, our goal was to examine whether BRCA1 related mammary tumors contain CD24+CD29+ cells, which retain their capacity for self-renewal and tumor formation. Focusing on the isolation and characterization of breast cancer stem cells may be important in gaining a better understanding of cancer formation and the distinct nature of BRCA1-associated breast cancer.
2. Materials and Methods
Mice and cell culture
Mice carrying Cre-LoxP mediated mammary specific deletion of the full length form of BRCA1 and a heterozygous null mutation of p53 (Brca1Co/Co;p53+/-;MMTV-Cre) were generated as described earlier [28, 29] and maintained in a pathogen free facility. Cell lines were generated from tumors of either the third (W780) or fourth (W0069) mammary gland as reported previously [30]. Subclones were generated by a limiting dilution method. Briefly, W0069 cells were diluted (1 cell in 200 μl) and plated in 96-well plates. Colonies were grown from single cells and expanded by plating them sequentially in 24-well, 12-well, 6-well plates and finally in 10 cm dishes after trypsinization. All cells were grown in DMEM medium supplemented with 10% fetal bovine serum at 37oC in a 5% CO2 incubator. Cells at 80-85% confluence were trypsinized, washed with PBS and plated for each experiment.
Single-cell preparation and Lin depletion
Mammary tumors from BRCA1 conditional knockout mice were minced, washed in PBS and digested in DMEM containing 300 U/ml collagenase (Sigma) and 100 U/ml hyaluronidase (Sigma) for 1 hour at 37oC. The resultant organoid suspension was sequentially resuspended in 0.25% trypsin-EDTA for 1–2 min, 5 mg/ml dispase (Roche Diagnostics) and 0.1 mg/ml DNase (Worthington) for 5 min, and 0.64% NH4Cl for 3 min before filtration through a 40-mm mesh to obtain single-cell suspension. Enrichment of Lin- epithelial cells was achieved through selective depletion of hematopoietic and endothelial cells using specific antibodies. Briefly, the single cell suspension was blocked in rat γ-globulin (Jackson Laboratories) and anti-CD16/CD32 Fcγ III/II receptor antibody (BD Pharmingen) for 10 min. Biotinylated antibodies against endothelial cells (anti-CD31, BD Pharmingen) and hematopoietic cells (anti-CD45 and anti-TER119, BD Pharmingen) were added for 25 min at 4oC. Subsequently, cells were incubated with anti-Biotin Microbeads and depleted by using MACS columns according to the manufacturer (Miltenyi Biotec Inc.).
In case of cell lines in culture, cells were plated in 10 cm dishes, trypsinized and filtered through a 40-mm mesh or passed through 26G needles to obtain single-cell suspension followed by blocking as described above.
FACS analysis
Cells were stained at a concentration of 1x106 cells per 100 μl of buffer (PBS pH 7.2, 0.5% BSA, 2 mM EDTA). Antibodies against CD24 (anti CD24-PE, BD Pharmingen), CD29 (anti CD29-FITC, Chemicon) and CD49f (anti CD49f-FITC, BD Pharmingen) were used. Antibody incubations were performed at 4oC for 25 min. Fluorescence-activated cell sorting (FACS) analysis was done using FACSCalibur flow cytometer (Becton Dickinson). Live single cells were gated for analysis. Cell sorting was carried out using FACSAria Cell Sorter (Becton Dickinson).
Cell proliferation assays
Cell proliferation was measured either by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] or growth curve assay. For the MTT assay, 1–2x104 cells were plated per one well of a 24-well plate and medium was changed every 24 h. After 24 h, 48 h and 72 h, medium was removed and 0.3 ml of 0.1% MTT in PBS was added in each well. After incubation for 30 min in a 37°C CO2 incubator, MTT solution was removed and 0.8 ml of 2-propanol was added. After shaking for 30 min, OD560 was measured using a plate reader. Plating for each time point was done in triplicate. For the growth curve assay, 5x104 cells were plated per one well of a 6-well plate and medium was changed every 24 h. Plate was incubated at 37oC in 5% CO2 and every 24 h cells were detached by trypsinization and counted with a Z1 Coulter counter. Plating for each time point was done in triplicate.
In vitro colony assays
For colony assays, cells were plated into wells of either 6-well plates or 24-well plates. For soft agar assay, a bottom layer of 0.5% agar in enriched medium (containing FBS, glutamine and antibiotics) was poured first, after solidifying this was followed by a layer containing a lower amount of agar (0.3%) and containing a specified number of cells (500 cells/well). The plates were placed in the incubator and after two-three weeks, colonies were counted by naked eyes. For three-dimensional assays, cells were resuspended in matrigel by following the thick gel method according to the manufacturer. Cells were allowed to set before covering with culture medium. Pictures of the colonies were taken after two weeks by using an Olympus IX81 inverted microscope.
Histology and immunohistochemical staining
For histology, tissues were fixed in 10% formalin, blocked in paraffin, sectioned, stained with hematoxylin and eosin, and examined by light microscopy. Detection of mitotic cells was performed using a ZYMED Histostain staining kit (ZYMED, San Francisco, CA) according to the manufacturer's instructions by using a primary antibody against Ki67 (Novocastra, Newcastle upon Tyne, UK).
Tumorsphere culturing in vitro
For generation of tumorspheres, single cells were plated on ultra-low attachment plates (Corning) in DMEM medium containing 20 ng/mL basic fibroblast growth factor (Sigma), 20 ng/mL EGF (Sigma), 4 μg/mL heparin (Sigma) and B-27 supplement (1:50 dilution, Invitrogen), and cultured at 37oC, 5% CO2. For limiting dilution experiments cells were plated in low attachment 96-well plates (Corning). For testing regeneration capacity of these tumorspheres, single-cell generated tumorspheres were collected by centrifugation (800 g) and then were mechanically and enzymatically dissociated in 0.25% Trypsin-EDTA for 10 min at 37oC, followed by passing through 26G needles.
Tumor injection
Cells after trypsinization or cells immediately after sorting were resuspended in PBS, which then were mixed at 1:1 ratio with Matrigel (BD Biosciences) on ice. The cell mixture was then injected either subcutaneously bilaterally in the flanks and on the back or into the fourth mammary fat pad of female immunocompromised mice. At least five mice were used for each group. Mice were followed every other day for tumor formation. Tumor size was measured with a caliper when visible nodules were present. Tumor volume was calculated by using the formula V=2/3πrxryrz (r is radius and x, y, z refer to each axis, and π=3.14).
3. Results
BRCA1 mutant cancer cell lines and primary tumors from BRCA1 mutant mice contain a subpopulation of CD24+CD29+ cells
We first tested whether CD24 and CD29, markers for normal mammary stem cells [15, 16], could be used to isolate CD24+CD29+ double positive populations from BRCA1 mutant tumor cells. We first performed flow cytometry analysis of two tumor cell lines derived from mammary tumors of Brca1Ko/Co;p53+/-;WAP-Cre mice [30]. We identified a subpopulation of cells in both cancer cell lines that expressed cell surface markers CD24 and CD29 (Fig. 1A). The profile was similar when cells were stained with CD49f instead of CD29 (Fig. 1C). CD49f stains for α6-integrin, which forms heterodimers with β1-integrin (CD29) and it has also been shown before to enrich for mammary stem cells [16]. We have also isolated a discrete subpopulation of CD24+CD29+ cells from BRCA1 mutant primary mammary tumors after using antibodies against well-characterized endothelial (CD31) and hematopoietic (CD45 and TER119) antigens to deplete these lineage positive cells followed by fluorescence-activated cell sorting (FACS) (Fig. 1D). The percentage of double positive cells varied among different BRCA1 cancer cell lines and primary tumors (12.8±2.5% in W0069, 16.7±5.3% in W780, and from 15-25% in the primary tumors).
Both BRCA1 cancer cell lines and BRCA1 primary tumors contain a distinct subpopulation of CD24+CD29+ cells. (A, B) representative flow cytometry analysis of stained (A) and unstained (B) W0069 cells with CD24-PE and CD29-FITC. (C) representative flow cytometry analysis of W0069 cells stained with CD24-PE and CD49f-FITC. (D) representative flow cytometry analysis of CD31-, CD45-, Ter119- (Lin-) mammary epithelial cells purified from BRCA1 mammary tumors using anti CD24-PE and anti CD29-FITC. (E, F) representative flow cytometry analysis of two different subclones of W0069 cell line after staining with antibodies against CD24 and CD29 [(subclone 228 (E), and subclone 229 (F)]. Numbers represent the percentage of each cell population. G) morphology of different cell lines under microscope.
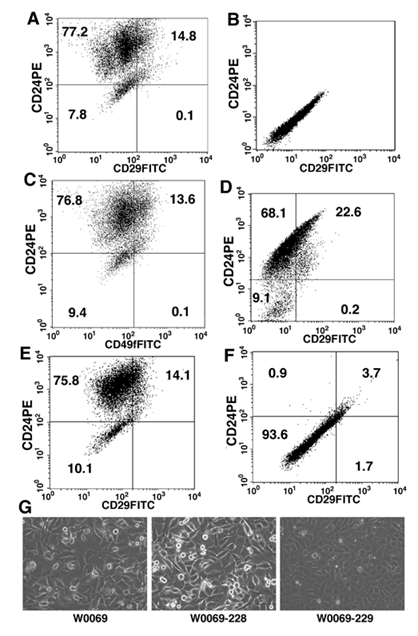
Because the CD24+CD29+ cells are only present as a small population in these cell lines and primary cancers, we next investigated whether this population of cells can be enriched in the subclones derived from these cells. Therefore, we derived subclones from one of the cell lines, W0069. Interestingly, we found that all clones examined exhibited heterogeneity containing from a high percentage of CD24+CD29+ cells at about 15% (W0069-228, Fig. 1E) to very low or no CD24+CD29+ (W0069-229, Fig. 1F) cells as revealed by FACS analysis. While the profile of the W0069-228 clone was similar to that of W0069, the W0069-229 clone showed a homogeneous profile by flow cytometry where we could not detect discrete cell populations of double positive cells. Notably, there was a difference in cellular morphology between the different subclones as well. The morphology of the W0069-228 subclone was similar to the parental cells characterized by a heterogeneous population of cells with different shapes as compared with the homogeneous population of W0069-229 (Fig. 1G). Thus, the heterogeneity found by FACS analysis was reflected in the morphology of these cell lines as observed by microscopy. Since these clones were derived from individual single cells, the failure to obtain a high purity of CD24+CD29+ cells suggests that these cells might differentiate and repopulate the heterogeneity, while the CD24-CD29- cells, such as W0069-229 might have a decreased ability to undergo differentiation, and therefore maintain their homogeneity (see next for further evidence).
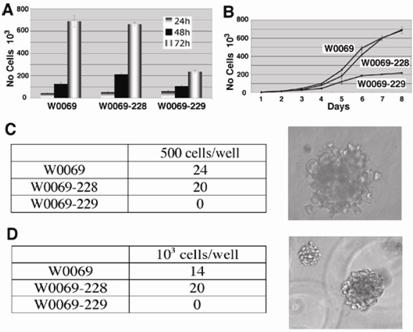
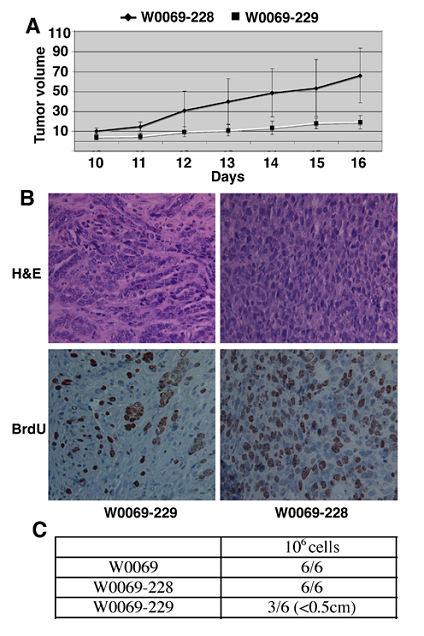
The differences in the number of CD24+CD29+ cells in subclones led us to test whether there are any different phenotypes between these clones. Specifically, we were interested in characteristics related to cancer stem cell phenotype such as the ability to proliferate or grow under low attachment conditions either in liquid culture or in matrigel. The proliferation capacity correlated with the amount of double positive cells since both W0069 and W0069-228 cells proliferated much faster than W0069-229 cells (Fig. 2A, B). Furthermore, both W0069 and W0069-228 could form colonies in soft agar and matrigel, respectively, while no colonies were formed when the same number of W0069-229 cells was tested under identical experimental conditions (Fig. 2C, D). To determine the tumor-initiating capacity of the cell lines, we injected subclones containing different percentages of double positive cells to nude mice subcutaneously and monitored tumor formation. W0069-228 cells showed enhanced tumorigenicity compared to W0069-229 cells as indicated by measuring the tumor volume at different time points after injection (Fig. 3A). This was further confirmed by staining tumors with the proliferation marker Ki67, since it was shown that in W0069-228 tumors the majority of cells exhibited positive staining compared to the lower amount of Ki67 positive cells found in W0069-229 tumors (61±4.9% vs. 34±1.1%, respectively), suggesting that indeed W0069-228 cells are more tumorigenic than W0069-229 cells (Fig. 3B). The same results were obtained when the different cell lines were injected directly to the mammary fat pad of nude mice. After 106 cells of either W0069 or W0069-228 cells were injected, all mice formed tumors (6/6) in both cases after one month. On the other hand, when W0069-229 cells were injected, only half of the mice (3/6) formed tumors. In addition, tumors generated from W0069 and W0069-228 cells were much larger (>1cm) than tumors formed from the same number of W0069-229 cells (<0.5cm) when the tumors were measured one month after injection (Fig. 3C). Based on previous studies it is believed that tumors are composed of a heterogeneous population of cells, with differences in their phenotypic characteristics and proliferative potential, including cancer cells with indefinite proliferative potential and those with limited or no proliferative potential [11, 31]. Thus, our data indicates that the presence of CD24+CD29+ cells may drive tumorigenicity of these cell lines, implying that this subpopulation shares the characteristics of cancer initiating cells.
CD24+CD29+ cells affect characteristics of cancer cell lines including proliferation and anchorage-independent growth. (A) W0069, W0069-228, and W0069-229 cells were grown for 72h and cell proliferation was assessed by MTT assay at 24h, 48h and 72h, respectively. (B) 5x104 cells for each cell line were grown in 6 well plates. Every 24h cells were collected and the number of cells was measured by using Z1 Coulter counter up to 8 days. (C, D) cells were grown either in soft agar (C) or in matrigel (D), respectively. Numbers represent the number of colonies in one out of three independent experiments. Characteristic pictures of colonies in both soft agar and matrigel are shown on the right.
Enhanced tumorigenicity in the presence of CD24+CD29+ cells. (A) 106 cells were injected subcutaneously and tumor size was measured with a caliper. Tumor volume is expressed in mm3 by using the formula mentioned in the Materials and Methods. (B) Paraffin-embedded sections of tumors were stained with hematoylin-eosin (H&E) or with an antibody recognizing the Ki67 antigen. (C) 106 cells were injected into the fourth mammary fat pad and mice were monitored for tumor formation for 30 days after injection. Denominators in the table represent the number of injections, and numerators represent the number of resultant tumors.
Double positive (CD24+CD29+) cells exhibit differentiation potential
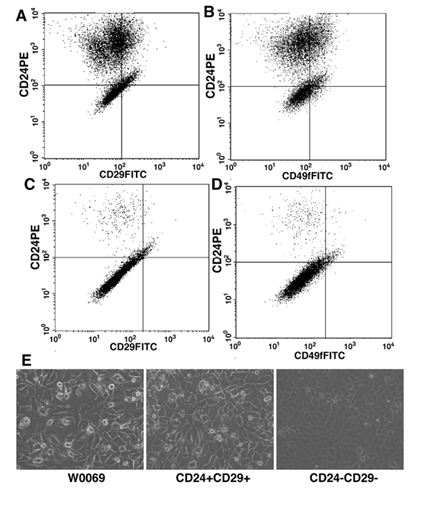
To provide further evidence to our hypothesis, we sorted out CD24 and CD29 double positive or negative cells and compared their ability for growth, differentiation, and tumorigenesis. After 2-3 passages in conventional tissue culture dishes, the sorted CD24+CD29+ cells were able to differentiate and reconstitute the heterogeneity found in the parental cell line, as revealed by FACS using markers for either CD24/CD29 (Fig. 4A) or CD24/CD49f (Fig. 4B). In contrast, the sorted CD24-CD29- cells could not differentiate and reconstitute the heterogeneity found in the W0069 cell line (Fig 4C, D). Notably, the heterogeneity of the double positive cells was again reflected in the morphology of the cells in culture (Fig. 4E), which was similar to the parental cell line. On the other hand, double negative cells in culture were homogeneous and looked very similar to the W0069-229 subclone. Thus, expansion of double positive cells in a monolayer leads to reconstitution of all different subpopulations found in parental cells, whereas cells depleted of double positive cells were unable to repopulate the CD24+CD29+ fraction.
CD24+CD29+ cells maintain their ability to differentiate. CD24+CD29+ cells were sorted out from the W0069 cell line and they were cultured as monolayers. After 3 passages, flow cytometry analysis was performed using anti CD24-PE and anti CD29-FITC (A) or anti CD24-PE and anti 49f-FITC (B). Same strategy was followed in the case of CD24-CD29- sorted out cells (C, D). Representative data after flow cytometry are shown. (E) morphology of double positive or double negative cell in culture under microscope.
As mentioned earlier, one of the hallmarks of cancer stem cells is their ability to grow and form colonies under anchorage-independent conditions. Thus, in order to check whether double positive cells are enriched for cancer initiating cells, sorted CD24+CD29+ cells were grown in matrigel. As shown in figure 5A, and 5B, double positive cells exhibited enhanced colony-forming ability, whereas no colonies were formed when 500, 200, or 100 double negative cells were grown under the same experimental conditions. It was shown that epithelial cells grown in three-dimensional cultures recapitulate numerous features of breast epithelium in vivo, including the formation of acini-like spheroids, apicobasal polarization of cells, basal deposition of basement membrane components (collagen IV and laminin V), and in some cases, the production of milk proteins [32, 33]. Since the cancer cell lines used were from mammary epithelial cancers, we wanted to investigate whether these cells may differentiate when cultured in three-dimensional cultures. Indeed we found that CD24+CD29+ cells exhibited an expanded differentiative repertoire compared to the double negative cells. While no differentiated structures were found in colonies formed by double negative cells, double positive cells could differentiate into complex tubular and branched structures reminiscent of the mammary ductal tree (Fig. 5C). Previous studies reported that normal mammary stem cells have the ability to recapitulate the architecture of the mammary gland [15, 34]. Since this subpopulation of cancer cells maintains the ability to differentiate in three-dimensional culture systems, it is possible that CD24+CD29+ cells share some of the characteristics of normal mammary stem cells.
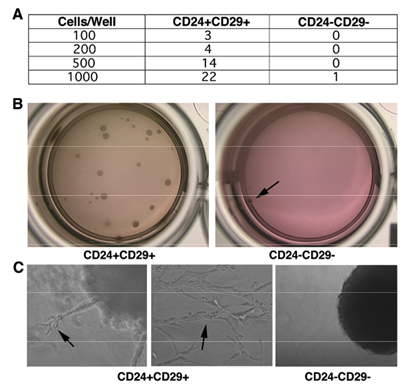
Double positive cells grow in three-dimensional cultures and form well differentiated structures. (A) Immediately after sorting, CD24+CD29+ and CD24-CD29- cells were grown in matrigel with varying number of cells per well. Numbers in table represent the number of colonies in one out of three independent experiments. (B) Pictures showing colonies formed when 1000 cells were inoculated in matrigel. Note, only one colony (arrow) was formed from 1000 double negative cells. (C) Characteristic colonies are shown in higher magnification. Arrows show differentiated structures as well as complex tubular structures formed from double positive cells grown in three dimensions.
Double positive cells exhibit self-renewal capacity and contribute to tumor initiation and growth
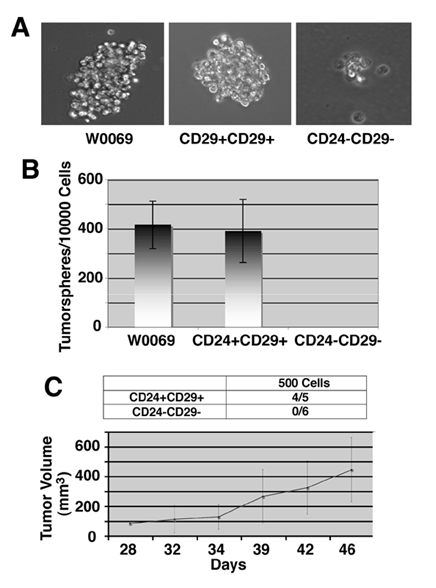
Most stem cell enrichment protocols rely on immunosorting according to the cell surface expression of some proteins. Alternatively, it has been shown that mammospheres, which are spherical colonies formed by epithelial cells when cultured on non-adherent surfaces are enriched in stem/progenitor cells [35]. Since cancer initiating cells resemble stem cells in their ability to grow as “tumorspheres” in culture, we wanted to check whether CD24+CD29+ cells maintain the potential to grow under conditions in which they cannot attach to a solid substratum. Whereas many cells underwent anoikis as expected, both parental and double positive cells were able to form “tumorspheres” after 3-4 days in culture (Fig. 6A). After enzymatic dissociation of these spheres, we could still get secondary “tumorspheres”, indicating that they are enriched for cancer initiating cells with self-renewal capacity. In contrast, the majority of double negative cells grown in suspension were non-viable floating cells with some cells growing as aggregates, which eventually deteriorated. To further confirm that these tumorspheres represented the progeny of an individual cell, we plated cells in limiting dilution with one cell, on the average, in each well of 96-well plates. Of note, we found that both parental and double positive cells had almost the same number of cells capable of giving rise to “tumorspheres” (419±96 vs. 391±128 per 10000 cells), while no tumorsphere was found in the double negative cells (Fig. 6B).
Nevertheless, the most important feature of cancer stem cells is their ability to form tumors. For this reason we sorted out double positive and double negative cells from primary BRCA1 tumors developed in Brca1Co/Co;p53+/-;MMTV-Cre mice [28] and tested their tumor forming ability. When as low as 500 CD24+CD29+ cells were injected into the mammary fat pad immediately after sorting, these cells were able to form tumors (4/5 injections), whereas no tumor formation was seen in mammary fat pads injected with 500 double negative cells (Fig. 6C). Notably, tumors grown from CD24+CD29+ cells showed a phenotypical heterogeneity when they were checked by flow cytometry, since both double positive and double negative cells were present in these tumors resembling the profile of the primary tumors (data not shown). These results suggest that double positive cells share some of the characteristics of cancer stem cells, since they exhibit the capacity for self-renewal, differentiation and the ability to form tumors in vivo.
Enhanced self-renewal capacity and tumor growth ability of CD24+CD29+ cells compared to double negative cells. (A) morphology of tumorspheres grown in ultra-low attachment plates from W0069 cells, CD24+CD29+, and CD24-CD29- cells in culture. CD24-CD29- cells can form small cell aggregates but they cannot form tumorspheres. (B) Quantification of tumorsphere initiating cells was done by performing limiting dilution assay after plating 1 cell/well in 96 well plates. (C) CD24+CD29+ and CD24-CD29- cells were sorted out from primary BRCA1 tumors. Immediately after sorting, 500 cells from each group were injected into the fourth mammary fat pad of nude mice. Denominators in the table represent the number of injections, and numerators represent the number of resultant tumors. Tumor growth rates were monitored as described in Materials and Methods and average tumor size is shown.
4. Discussion
Recently a subpopulation (Lin-CD49fhiCD24med) was identified that is highly enriched for normal mammary stem cells by transplantation [15, 16]. After replacing CD49f with CD29 (β1-integrin), which forms heterodimers with α6-integrin (CD49f), a similar subpopulation (Lin-CD29hiCD24+) was also purified [15, 16]. Actually, a single cell with this phenotype can reconstitute a complete mammary gland in vivo, thus generating luminal and myoepithelial lineages as well as lobuloalveolar structures during pregnancy [15, 16]. If tumors originate from stem cells since normal stem cells may be the targets for transformation, it was hypothesized that it is possible that markers used for the isolation of normal mammary stem cells may also work for the isolation of mammary cancer stem cells [36]. In the present study, we tested this hypothesis and identified a subpopulation of mammary tumor initiating cells using markers such as CD24/CD29 or CD24/CD49f, which have been used to purify normal mammary stem cells. Thus, our study provides evidence for the first time that BRCA1-associated mammary cancer stem cells might originate from normal mammary stem cells.
It was previously shown that mammary stem cells are Hoechst 33342 negative and Sca-1 positive [37]. However, recently it was found that a single cell with the phenotype CD24+CD29+ can reconstitute a complete mammary gland in vivo [15, 16]. Interestingly, neither high Sca-1 expression nor Hoechst33342 dye exclusion were enriched in this CD24 and CD29/CD49f population. Moreover, in contrast to a general concept that mammary stem cells divide slowly [38], the CD49fhiCD24med cells were found to be in G1 or S/G2/M fractions [16]. indicating that they are in a cycling, instead of quiescent, stage. Here we show that the amount of double positive cells in BRCA1 mutated cell lines correlates with the proliferative capacity, the ability to grow in anchorage-independent conditions and the tumor-forming ability of either these cell lines, or different subclones derived from the same cell lines. Furthermore, we proved that CD24+CD29+ cells retain their ability to proliferate and drive tumor formation by sorting out these cells from double negative cells. Indeed, we showed that CD24+CD29+ cells could repopulate the heterogeneity found in the parental cells, since different cell populations were found after 2-3 passages in culture. Even more interestingly, the CD24+CD29+ cells could grow in three-dimensional culture systems, where differentiated structures were visible in contrast to double negative cells, suggesting that double positive cells might be less differentiated than CD24-CD29- cells. Thus, we conclude that CD24+CD29+ cells are enriched for cancer stem cells in BRCA1 deficient tumors.
With the expansion of the cancer stem cell field many different markers for stem cells have been identified. Focusing on mammary/breast tumors, different markers have been described to enrich for cancer stem cells. For example, in MMTV-cNeu oncogene induced mammary tumors, cancer-initiating cells were found exclusively in either Sca1+CD24+ or Sca1-CD24+ populations [39], whereas in MMTV-Wnt1 mammary tumors, Thy+CD24+ were highly enriched for cells capable of regenerating new tumors [40]. On the other hand, in breast tumors, it has been shown that CD44+CD24Low cells share characteristics of cancer stem cells [17]. These differences may imply that in different kind of tumors, different cells are the targets for transformation, thus different markers are needed for the isolation of the specific tumorigenic subpopulation. Of note, even in tumors caused by a mutation of the same tumor suppressor gene, there is a discrepancy in the phenotype of this specific subpopulation, as it was recently shown that BRCA1 mutant tumors contain distinct CD44+CD24- and CD133+ populations with cancer stem cell characteristics [41]. Given that in our study we showed that CD24+CD29+ cells are enriched for cancer initiating cells, it is possible that different markers may be expressed in stem cells at different stages of differentiation within the same tumor. Thus, by using different markers we may isolate primitive cancer stem cells, early progenitors, or differentiated progenitors. All of these cells may drive tumor formation, but they can differ in other aspects, such as the capacity for self-renewal, differentiation potential, or metastatic ability. Thus, although there has been dramatic progress in the breast cancer stem cell field, future studies are needed to find out which markers stain what types of cells, in order to further define the heterogeneity exhibited by cancer stem/precursor cells.
Our data suggest that during BRCA1 related tumorigenesis, normal stem cells may be the targets for transformation and tumor formation as it has been shown recently for MMTV-Wnt1 mammary tumors [40]. The self-renewal and differentiation capacity of these cells in non-adherent conditions supports the idea that although these cells have been transformed to cancer cells they still maintain many of the characteristics of normal stem cells. We propose that our data provides evidence of the cancer stem cell origin of BRCA1 tumors. Although recent data from human samples suggest that BRCA1 plays a role in the differentiation of normal breast stem cells [21, 23], its exact role in cancer stem cells remains to be elucidated. In this context we believe that Brca1Co/Co;p53+/-;MMTV-Cre mice should serve as a powerful tool to dissect the role of BRCA1 in the cancer initiating cells and provide insights into development and treatment of breast cancer specifically in BRCA1 mutation carriers.
Acknowledgements
We gratefully acknowledge members of the Deng laboratory for helpful discussion, and Drs. J. Tyler Lahusen and C. Xiao for critical reading of the manuscript. This work was supported by the Intramural Research Program of the National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, USA.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Alberg AJ, Lam AP, Helzlsouer KJ. Epidemiology, prevention, and early detection of breast cancer. Curr Opin Oncol. 1999;11:435-441
2. Brody LC, Biesecker BB. Breast cancer susceptibility genes. BRCA1 and BRCA2. Medicine (Baltimore). 1998;77:208-226
3. Eccles DM, Pichert G. Familial non-BRCA1/BRCA2-associated breast cancer. Lancet Oncol. 2005;6:705-711
4. Jemal A, Siegel R, Ward E. et al. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43-66
5. Zhang J, Powell SN. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol Cancer Res. 2005;3:531-539
6. Kang SP, Martel M, Harris LN. Triple negative breast cancer: current understanding of biology and treatment options. Curr Opin Obstet Gynecol. 2008;20:40-46
7. Rodriguez-Pinilla SM, Sarrio D, Honrado E. et al. Prognostic significance of basal-like phenotype and fascin expression in node-negative invasive breast carcinomas. Clin Cancer Res. 2006;12:1533-1539
8. Banerjee S, Reis-Filho JS, Ashley S. et al. Basal-like breast carcinomas: clinical outcome and response to chemotherapy. J Clin Pathol. 2006;59:729-735
9. Song LL, Miele L. Cancer stem cells--an old idea that's new again: implications for the diagnosis and treatment of breast cancer. Expert Opin Biol Ther. 2007;7:431-438
10. Pina C, Enver T. Differential contributions of haematopoietic stem cells to foetal and adult haematopoiesis: insights from functional analysis of transcriptional regulators. Oncogene. 2007;26:6750-6765
11. Reya T, Morrison SJ, Clarke MF. et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-111
12. Hennighausen L, Robinson GW. Think globally, act locally: the making of a mouse mammary gland. Genes Dev. 1998;12:449-455
13. Villadsen R, Fridriksdottir AJ, Ronnov-Jessen L. et al. Evidence for a stem cell hierarchy in the adult human breast. J Cell Biol. 2007;177:87-101
14. Kordon EC, Smith GH. An entire functional mammary gland may comprise the progeny from a single cell. Development. 1998;125:1921-1930
15. Shackleton M, Vaillant F, Simpson KJ. et al. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84-88
16. Stingl J, Eirew P, Ricketson I. et al. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993-997
17. Al-Hajj M, Wicha MS, Benito-Hernandez A. et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-3988
18. Miki Y, Swensen J, Shattuck-Eidens D. et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66-71
19. Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response, and cancer evolution. Nucleic Acids Res. 2006; 34: 1416-1426, Deng CX Tumor formation in Brca1 conditional mutant mice. Environ Mol Mutagen. 2002;39:171-177
20. Asselin-Labat ML, Shackleton M, Stingl J. et al. Steroid hormone receptor status of mouse mammary stem cells. J Natl Cancer Inst. 2006;98:1011-1014
21. Foulkes WD. BRCA1 functions as a breast stem cell regulator. J Med Genet. 2004;41:1-5
22. Li W, Xiao C, Vonderhaar BK. et al. A role of estrogen/ERalpha signaling in BRCA1-associated tissue-specific tumor formation. Oncogene. 2007;26:7204-7212
23. Liu S, Ginestier C, Charafe-Jauffret E. et al. BRCA1 regulates human mammary stem/progenitor cell fate. Proc Natl Acad Sci U S A. 2008;105:1680-1685
24. Antoniou A, Pharoah PD, Narod S. et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72:1117-1130
25. Jernstrom H, Lubinski J, Lynch HT. et al. Breast-feeding and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst. 2004;96:1094-1098
26. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730-737
27. Lapidot T, Sirard C, Vormoor J. et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645-648
28. Xu X, Wagner KU, Larson D. et al. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation [see comments]. Nat Genet. 1999;22:37-43
29. Deng CX, Xu X. Generation and analysis of Brca1 conditional knockout mice. Methods Mol Biol. 2004;280:185-200
30. Brodie SG, Xu X, Qiao W. et al. Multiple genetic changes are associated with mammary tumorigenesis in Brca1 conditional knockout mice. Oncogene. 2001;20:7514-7523
31. Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895-902
32. Petersen OW, Ronnov-Jessen L, Howlett AR. et al. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci U S A. 1992;89:9064-9068
33. Gudjonsson T, Ronnov-Jessen L, Villadsen R. et al. Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. J Cell Sci. 2002;115:39-50
34. Zucchi I, Sanzone S, Astigiano S. et al. The properties of a mammary gland cancer stem cell. Proc Natl Acad Sci U S A. 2007;104:10476-10481
35. Dontu G, Abdallah WM, Foley JM. et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253-1270
36. Wang RH. The new portrait of mammary gland stem cells. Int J Biol Sci. 2006;2:186-187
37. Welm B, Behbod F, Goodell MA. et al. Isolation and characterization of functional mammary gland stem cells. Cell Proliferation. 2003;36(Suppl 1):17-32
38. Welm BE, Tepera SB, Venezia T. et al. Sca-1(pos) cells in the mouse mammary gland represent an enriched progenitor cell population. Dev Biol. 2002;245:42-56
39. Liu JC, Deng T, Lehal RS. et al. Identification of tumorsphere- and tumor-initiating cells in HER2/Neu-induced mammary tumors. Cancer Res. 2007;67:8671-8681
40. Cho RW, Wang X, Diehn M. et al. Isolation and molecular characterization of cancer stem cells in MMTV-Wnt-1 murine breast tumors. Stem Cells. 2008;26:364-371
41. Wright MH, Calcagno AM, Salcido CD. et al. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008;10:R10
Author contact
![]() Correspondence to: Dr. Chu-Xia Deng, Phone: 301-402-7225, Fax: 301-480-1135; Email: chuxiadniddk.nih.gov
Correspondence to: Dr. Chu-Xia Deng, Phone: 301-402-7225, Fax: 301-480-1135; Email: chuxiadniddk.nih.gov
Received 2008-4-28
Accepted 2008-5-4
Published 2008-5-4