Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2008; 4(3):184-199. doi:10.7150/ijbs.4.184 This issue Cite
Research Paper
STAT3 Activation in Pressure-Overloaded Feline Myocardium: Role for Integrins and the Tyrosine Kinase BMX
Christopher D. Willey1,*, Arun P. Palanisamy1,*, Rebecca K. Johnston1, Santhosh K. Mani1, Hirokazu Shiraishi1, William J. Tuxworth1, Michael R. Zile1,2, Sundaravadivel Balasubramanian1, Dhandapani Kuppuswamy1,2 ![]()
1. Cardiology Division of the Department of Medicine, Gazes Cardiac Research Institute, Medical University of South Carolina, Charleston, SC 29425-2221, USA
2. Ralph H. Johnson Department of Veterans Affairs Medical Center, Charleston, SC 29425-2221, USA
* Both authors contributed equally to this work
Received 2008-4-24; Accepted 2008-6-24; Published 2008-6-27
Abstract
Growth, survival and cytoskeletal rearrangement of cardiomyocytes are critical for cardiac hypertrophy. Signal transducer and activator of transcription-3 (STAT3) activation is an important cardioprotective factor associated with cardiac hypertrophy. Although STAT3 activation has been reported via signaling through Janus Kinase 2 (JAK2) in several cardiac models of hypertrophy, the importance of other nonreceptor tyrosine kinases (NTKs) has not been explored. Utilizing an in vivo feline right ventricular pressure-overload (RVPO) model of hypertrophy, we demonstrate that in 48 h pressure-overload (PO) myocardium, STAT3 becomes phosphorylated and redistributed to detergent-insoluble fractions with no accompanying JAK2 activation. PO also caused increased levels of phosphorylated STAT3 in both cytoplasmic and nuclear fractions. To investigate the role of other NTKs, we used our established in vitro cell culture model of hypertrophy where adult feline cardiomyocytes are embedded three-dimensionally (3D) in type-I collagen and stimulated with an integrin binding peptide containing an Arg-Gly-Asp (RGD) motif that we have previously shown to recapitulate the focal adhesion complex (FAC) formation of 48 h RVPO. RGD stimulation of adult cardiomyocytes in vitro caused both STAT3 redistribution and activation that were accompanied by the activation and redistribution of c-Src and the TEC family kinase, BMX, but not JAK2. However, infection with dominant negative c-Src adenovirus was unable to block RGD-stimulated changes on either STAT3 or BMX. Further analysis in vivo in 48 h PO myocardium showed the presence of both STAT3 and BMX in the detergent-insoluble fraction with their complex formation and phosphorylation. Therefore, these studies indicate a novel mechanism of BMX-mediated STAT3 activation within a PO model of cardiac hypertrophy that might contribute to cardiomyocyte growth and survival.
Keywords: STAT3, BMX, integrin, pressure-overload, cardiac hypertrophy
Introduction
Cardiac hypertrophy begins as a compensatory phenomenon when hemodynamic load to the heart increases. Terminally differentiated adult cardiac muscle cells, or cardiomyocytes, respond to mechanical load by increasing their mass through hypertrophy, although the mechanism that couples load to growth in the hypertrophied heart is not completely understood. Studies show that during mechanical loading of the heart, such as pressure-overload (PO), stretch-mediated changes[1] and adhesion-mediated changes[2] transduce biochemical signals into the cell. In this context, protein tyrosine phosphorylation of various intracellular substrates is critical for hypertrophic growth, and studies in various cell types have shown that integrin activation and focal adhesion complex (FAC) formation can mediate a transmembrane hierarchy of molecular responses,[2] including recruitment and activation of non-receptor tyrosine kinases (NTKs). Therefore, assembly and activation of NTKs at the FAC are critical for multiple downstream phenomena in virtually all cell types, including cardiomyocytes.
Our earlier studies,[3-5] using both in vivo pressure-overload (PO) and in vitro three-dimensional collagen matrix (3D) models, indicate the association of NTKs, including c-Src and FAK (focal adhesion kinase), ß3-integrin and several adaptor proteins with the detergent-insoluble actin-rich cardiac cytoskeletal (CSK) fraction. This process was found to be accompanied by tyrosine phosphorylation (PY) of several CSK-associated proteins. Although STATs are known to play critical roles as transcription factors for regulating cell proliferation and survival,[6] recent studies have identified other roles, including association at FAC and cell-cell junctions that may contribute to cell motility via alteration in adhesions and/or the cytoskeleton.[7-10] Of the six different STATs reported in humans, STAT3 is found to be widely expressed and also known to prevent apoptosis in different cell types.[11] STAT3 has become a recent focus of interest in cardiac research since its activation has been demonstrated during hypertrophy.[12, 13] Indeed, cardiac-restricted STAT3 knockout in mice[14, 15] implicated its importance to cardiac physiology as deletion leads to several detrimental effects, including increased sensitivity to injury, decreased LV capillary formation, increased fibrosis, and decreased contractile function. Moreover, there is evidence that activated STAT3 provides cardioprotection from ischemic reperfusion injury in vivo.[16] Therefore, studies on the activation of STAT3 in the stressed heart are important for understanding its crucial role to the normal functioning of the heart.
Since we have established activation of several NTKs both in our in vivo PO model and in vitro cell culture model, we explored in the present study both STAT3 activation and the importance of upstream NTKs using these models. Tyrosine phosphorylation of STAT3 by NTKs is critical for the activation of STAT3.[6, 17] Moreover, this phosphorylation may help direct STAT3 to particular subcellular locations including endosomal compartments,[18] cytoskeleton,[8, 9] microtubules,[10] and nucleus[19] which influences its function. A major upstream kinase known to phosphorylate STAT3 during cytokine/growth factor stimulation is Janus kinase-2 (JAK2).[6] However, c-Src can also mediate tyrosine phosphorylation of STAT3.[20] Similarly, another NTK, BMX (bone marrow tyrosine kinase in chromosome X), a member of the TEC family,[21, 22] has been shown to phosphorylate and activate STAT3 in multiple cell types.[21, 23-25] BMX has a wide expression profile but has a particularly high expression in the heart and plays a broad role in cell signaling.[21, 22] Specifically, during nitric oxide generation in the heart, BMX activation via PKCε provides cardioprotection.[26] Furthermore, both BMX[27] and STAT3[14, 15] also contribute to myocardial vascular growth following ischemic preconditioning of the heart. Our present studies indicate a novel mechanism of activation and redistribution of STAT3 in PO myocardium with the involvement of integrin-mediated BMX activation.
Materials and Methods
Animal Model
Adult male cats weighing 2.8-3.5 kg were used for right ventricular PO by partial occlusion of the pulmonary artery, as we described previously,[28, 29] which results in systemic arterial pressure remaining stable while the pulmonary arterial pressure at least doubles. As such, the left ventricles serve as internal controls. We relied on two methods for achieving PO depending on desired duration. For short term PO (4 h), cats underwent balloon-tipped catheter placement through the jugular vein under full surgical anesthesia. Long term PO (48 h and 1 wk) was achieved via placement of an external band using a 3.2 mm internal diameter band. Anesthesia was similar to that of the 4 h cats, except that the animals were allowed to recover following anesthesia. These cats were later anesthetized again and sacrificed at the specified times and processed as for short term PO.
Control cats (for both external banding and ballooning) were sham-operated by thoracotomy and pericardiotomy without arterial occlusion. The care of the animals and all experiments were conducted in accordance with the US National Institute of Health guidelines for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996) and the Institutional Animal Care and Use Committee of the Medical University of South Carolina.
Adult Cardiomyocyte Cell Culture Models
The isolation of adult feline cardiomyocytes and culture on laminin-coated plates have been described previously.[30] Three-dimensional (3D) embedding of adult cardiomyocytes and integrin stimulation with RGD peptide were carried out as we described previously.[3, 4] Briefly, freshly isolated adult feline cardiomyocytes (1.25 x 105) were suspended in Piper's medium prepared in M199 cell culture medium (GIBCO-BRL, Inc., Grand Island, NY) that contains the following: 2% bovine serum albumin (BSA), 5 mM creatine (Sigma), 2 mM L-carnitine (Sigma), 5 mM taurine (Sigma-Aldrich), 0.25 mM L-ascorbate (Sigma-Aldrich), 10 μM AraC, 40 nM insulin (GIBCO-BRL), 200 units/ml penicillin and 200 μg/ml streptomycin (GIBCO-BRL). While maintaining the pH at 7.4, suspended cells were mixed with soluble type-I collagen (Cohesion, Palo Alto, CA) at room temperature to a final volume of 2.5 ml/well in 8-well plastic dishes (Falcon) at a final collagen concentration of 0.1%. The cell-collagen mixture was allowed to polymerize by incubating at 37oC for 10 min in humidified 5% CO2 atmosphere, then covered with 0.5 ml of M199 and incubated for an additional 50 min. For experiments using RGD peptide (Sigma), stock solutions of RGD peptide (Gly-Arg-Gly-Asp-Ser) were prepared in M199 and added to the cell-collagen mixture prior to polymerization at concentrations as indicated in the figures. To recover cardiomyocytes from collagen gels, 7.5 ml of collagenase-D (Boehringer Mannheim GmbH, Germany) prepared in Krebs solution at a concentration of 4 mg/ml was added to the gel matrix and incubated for 12 min in a shaking 37oC water bath. The digested material was then centrifuged, and cells were subsequently washed two times with Krebs solution containing 0.1% BSA and once with phosphate buffered saline (PBS).
Generation of c-Src Adenoviral Constructs and their Expression in Adult Cardiomyocytes
The generation of adenoviral constructs was based on He's pAdEasy-1 system[31] as described previously.[3] Briefly, dominant negative c-Src cDNA (K295R, Y527F) was obtained from Upstate Biotechnology as a cloning vector and subcloned into the cloning vector, pSP72 (GIBCO-BRL). The c-Src restriction fragments were gel purified (Qiagen) and ligated into the adenoviral shuttle vector, pAdTrack-CMV, and cultured in LB agar with kanamycin. Positive clones were selected and screened by PCR using insert specific primers followed by propagation and DNA sequencing. The pAdTrack-CMV shuttle vectors were linearized using PmeI (New England Biolabs), gel purified, and recombined with the adenoviral genome plasmid with ampicillin resistance, pAdEasy-1, in competent BJ5183 cells and plated on kanamycin plates. Positive clones were selected by an initial PCR screen for the insert and propagated for restriction enzyme analysis. Selected clones were transformed into DH5α cells, maxiprepped, and linearized with PacI. Following phenol-chloroform extraction, the adenoviral plasmids were transfected into HEK293 cells for packaging of the replication-defective adenovirus. After two plaque purifications, the virus titers were calculated.
Adenoviruses were used to infect isolated adult feline cardiomyocytes at an MOI of 10. Approximately 5x105 cells were plated on laminin-coated 100 mm dishes with 10 ml of Piper's media. After 4 h of incubation at 37oC and 5% CO2, the media was changed to one containing a viral dose based on titration calculations. After 4-16 h of incubation, the virus media was removed and replaced with the same volume of media without virus. After 48 h of incubation, with one media change at 24 h, the cells were then washed with M199 twice and dead cells (if any) were removed with mild trypsinization (3.5 ml M199+600 μl of 0.25% trypsin/EDTA) for 5 minutes at 37oC. The viable cardiomyocytes were then isolated using 2.5 ml M199 and 1 ml of 0.25% trypsin/EDTA treatment for 5 min at 37oC and 5% CO2. An additional 1 ml of 0.25% trypsin/EDTA was then added every 5 min until all of the cells were suspended. 10 ml Piper's media was then added to quench the reaction. The cells were then pelleted with mild centrifugation and resuspended in 250 μl Piper's medium for subsequent collagen gel studies as described above.
Preparation of Tissue and Cell Lysates
For tissue lysates, the following preparation was performed. Felines that were surgically anesthetized had the hearts removed. The aortas underwent cannulation and retrograde flushing using ice-cold PBS. The heart was then separated into LV and RV tissue for immediate processing. For the preparation of tissue extracts, approximately 100 mg of tissue (~1.25 x 105) was homogenized in a Tekmar Tissuemizer (Tekmar Co., Cincinnatti, OH) in 2 ml of Triton X-100 extraction buffer (30 mM Tris-HCl, pH 7.4, 2% Triton X-100), containing Sigma protease inhibitor cocktail (P 8340) and phosphatase inhibitor cocktails I and II (P2850 and P5726). Triton X-100 soluble and insoluble components were prepared as described previously. [3-5] Briefly, the homogenate was centrifuged at 16,000 x g for 10 min to obtain a pellet and supernatant. The pellet was washed once by resuspending in 1 ml of Triton X-100 extraction buffer and then repeating the centrifugation (16,000 x g) step. The resulting pellet was mixed with 500 μl of SDS-sample buffer, boiled for 3 min and centrifuged briefly (16,000 x g) at room temperature to obtain the Triton X-100-insoluble cytoskeletal proteins (CSK). The supernatant from the first spin underwent an additional centrifugation at 100,000 x g for 2.5 h, and the pellet was resuspended in 1 ml of SDS-sample buffer and boiled for 3 min to obtain Triton X-100-insoluble membrane skeletal proteins (MSK). Finally, the 100,000 x g supernatant (detergent soluble fraction) was mixed with equal volume 2x SDS-sample buffer, boiled for 3 min and used as Triton X-100 soluble (Sol) fraction.
For lysates from collagen-embedded cells, a similar approach was taken but with slight modification because these cell lysates did not produce an MSK pellet on high-speed spin.[3-5] Harvested cells from collagen gels were homogenized in 0.3 ml of extraction buffer using a 26 G needle syringe and kept on ice for 10 min. This extract was centrifuged at 16,000 x g for 10 min to obtain a pellet and supernatant. The pellet was washed once by resuspending in 0.5 ml of Triton X-100 extraction buffer and then repeating the centrifugation (16,000 x g) step. The resulting Triton X-100-insoluble pellet was mixed with 300 μl of 2x SDS-sample buffer, boiled for 3 min and centrifuged briefly (16,000 x g) at room temperature to obtain Triton X-100-insoluble proteins. Our previous studies indicate that this fraction is found to contain both the CSK and most of the MSK proteins.[3-5] The supernatant from first spin (which does not yield any pellet at high-speed spin) represents the Triton X-100 soluble fraction and was prepared for Western blotting by adding equal volumes of 2x SDS-sample buffer, boiled for 3 min and used as detergent soluble (Sol) fraction.
Nuclear Isolation
Nuclear isolation for the detection of STAT3/P-STAT3 was performed per manufacturer's protocol with Nuclear and Cytoplasmic Extraction Kit (NE-PER, Pierce) with minor modifications. Briefly, 50 mg ventricular tissue was homogenized using Tekmar Tissuemizer for 45 s in 1 ml PBS with protease and phosphatase inhibitor cocktails (Sigma). The homogenate was centrifuged at 1000 x g for 8 min. To the pellet, 500 μl of cytoplasmic extraction buffer I (CERI) with inhibitors was added. After suspending the pellet, the solution was incubated on ice for 15 min, followed by the addition of 27 μl CERII. The contents were incubated on ice for 2 min, centrifuged at 16,000 x g for 15 min, and the supernatant (cytoplasmic proteins) was mixed with equal volume of 2X SDS-sample buffer. The pellet was washed with PBS, suspended in 250 μl of nuclear extraction buffer with inhibitors and incubated on ice for 40 min with occasional vortexing. After centrifugation at 16,000 x g for 15 min, the supernatant, consisting of nuclear proteins, was mixed with equal volume of 2X SDS sample buffer.
Immunoblotting
Subfractioned protein samples in SDS sample buffer were resolved by SDS-PAGE and immunoblotted as previously described.[3, 4] Primary antibodies used were: polyclonal antibodies for STAT3, phospho-STAT3, phospho-Etk (BMX) and monoclonal phosphotyrosine 416 Src (Cell Signaling Technology); monoclonal antibodies for BMX, and phosphotyrosine (BD Biosciences); monoclonal antibody for c-Src and polyclonal antibodies for JAK2, phospho-JAK2, and histone H4 (Upstate Biotechnology, Inc); polyclonal anti-actin antibody (Sigma); polyclonal antibody for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Research Diagnostics Inc). To normalize protein concentration between LV and RV pairs, GAPDH (Triton-soluble and cytoplasmic), actin (Triton-insoluble), and histone-H4 (nuclear) levels were measured. For denstitometric quantitation, NIH ImageJ v. 1.34r was used.
Immunoprecipitation of STAT3-BMX Complex
Membrane skeletal (MSK) pellets obtained from ventricular tissue were solubilized in modified radioimmunoprecipitation assay (mRIPA) buffer (containing: 50 mM Tris-HCl, pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 0.1% sodium dodecyl sulfate and protease and phosphatase inhibitors as mentioned previously) and briefly sonicated. The resulting solution was rotated with STAT3 antibody for 2 h at 4oC followed by rotation with Protein A/G beads (Santa Cruz) for 1 h at 4oC. The beads were then washed with mRIPA buffer thrice, boiled with 1X SDS sample buffer and used for Western blot analysis as described earlier.[3, 4] As controls, we rotated either the lysates with Protein A/G beads in the absence of STAT3 antibody or the buffer with Protein A/G beads in the presence of STAT3 antibody.
Two-dimensional (2D) Gel Electrophoresis
SDS-sample buffer boiled samples (Triton X-100 soluble and insoluble MSK proteins) were processed using Proteome Systems reagents/protocol except for minor modifications as follows. 250 μl of C7 detergent solution was added to 500 μl of Triton X-100 soluble or insoluble fractions. After mixing, 100 μl of this solution was added to 900 μl acetone, and placed on ice for 1 h. Samples were centrifuged for 10 min at 5000 rpm, and the pellet was washed twice in 75% acetone. The pellet was air-dried for 15 min and 1 ml of C7 reagent, and 40 μl of Tris-base solution were added to the protein pellet. Following sample solubilization, 50 μl of tributylphosphine (Sigma) and 20 μl of alkylation agent (Proteome Systems) were added and samples were incubated at room temperature for 90 min with occasional vortexing. 10 μl of quenching reagent was then added and samples were centrifuged for 5 min at 5000 rpm to pellet insoluble material. Supernatant was then aliquoted for subsequent 2D analysis.
For 2D gel electrophoresis, 11 cm IPG strips were re-hydrated with 200 μl of the aliquoted sample at 37ºC for 6 h. Strips were then subjected to 1st dimension electrophoresis using the following parameters: 100V-2 h; 100-1000V 3 h; 1000V-2 h; 1000-8000V-1 h; 8000V-10 h; 8000-2000V-1 h; 2000V-hold until processed. Strips were then placed on top of 6-12% Tris-acetate gels and electrophoresed for 2 h at 200V. Proteins from gels were transferred onto Immobilon-P membranes and immunoblotted as described above.
Statistics
Data are expressed as means +/-SEM. Statistical comparisons were made by one-way ANOVA followed by either Bonferroni or Tukey post hoc analysis. Failed normality or equal variance tests were followed by Kruskal-Wallis one way analysis of variance and Dunn's method for multiple comparisons to control groups (SigmaStat 3.5, Systat Software, Inc., San Jose, CA). A p-value < 0.05 was considered statistically significant.
Results
Temporal/spatial Relationship of STAT3 and Phosphorylated STAT3 within RVPO and RGD-stimulated 3D Collagen Models
We initially examined whether STAT3 becomes activated in our established feline right ventricular pressure-overload (RVPO) model[28-30, 32] in which the pulmonary artery is partially occluded to increase RV wall stress. The left ventricle (LV) remains normally loaded and is used as an internal control. Table 1 shows the hemodynamic data for this model. For biochemical analyses, we utilized two independent cell fractionation techniques: one fractionates proteins based on their detergent solubility, and the second fractionates nuclear proteins. In the first, we process tissue samples to obtain both Triton X-100 soluble fraction (which consists of all nonionic-detergent soluble proteins including those in the nucleus) and the insoluble fractions, obtained after low-speed (cytoskeleton pellet; CSK) and high-speed (membrane skeleton pellet; MSK) centrifugation. These Triton X-100 insoluble pellet samples (CSK and MSK) do not include Triton X-100 insoluble "raft-floatation" materials. The second protocol was primarily used to isolate nuclear proteins by lysing tissue or cell samples in a detergent-free buffer provided by the nuclear isolation kit.
Hemodynamic characteristics of RVPO felines
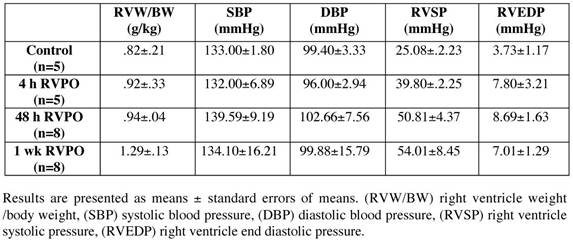
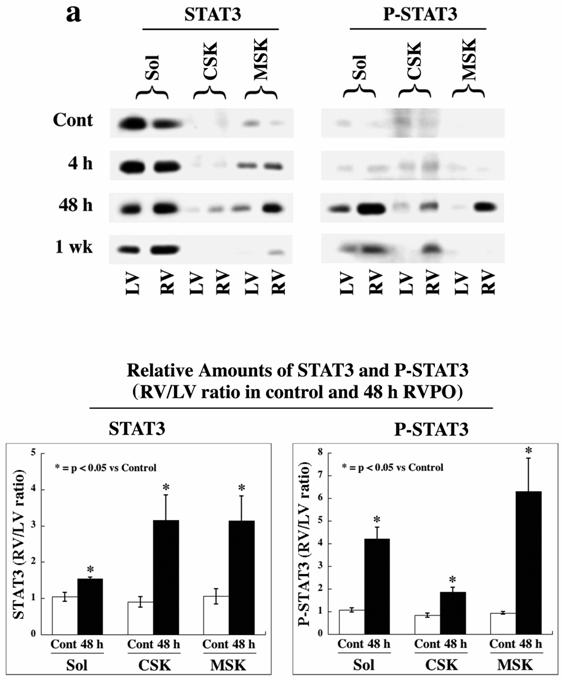
Analysis using detergent-lysed subfractions from both sham-operated control and RVPO cats for various time periods indicate STAT3 recruitment to MSK and to (a lesser extent) CSK fractions in 48 h pressure-overloaded RV (Figure 1a, left panel). Furthermore, increased tyrosine phosphorylation of STAT3 (P-STAT3) at Tyr705 (indicating activation) was detected in all three (soluble, CSK, and MSK) fractions of pressure-overloaded RV, when compared to normally loaded LV subfractions (right panel), although it was higher in Triton X-100 soluble and insoluble MSK fractions when compared to CSK fraction. These changes observed in 48 h RVPO cats were significant (Figure 1a, bottom panels) (p<0.05) and were not due to intrinsic differences between RV and LV based on the observation in sham control cats. By 1 wk RVPO, both STAT3 recruitment and phosphorylation were substantially reduced. Therefore, STAT3 undergoes a transient activation and recruitment to the detergent insoluble structures during PO.
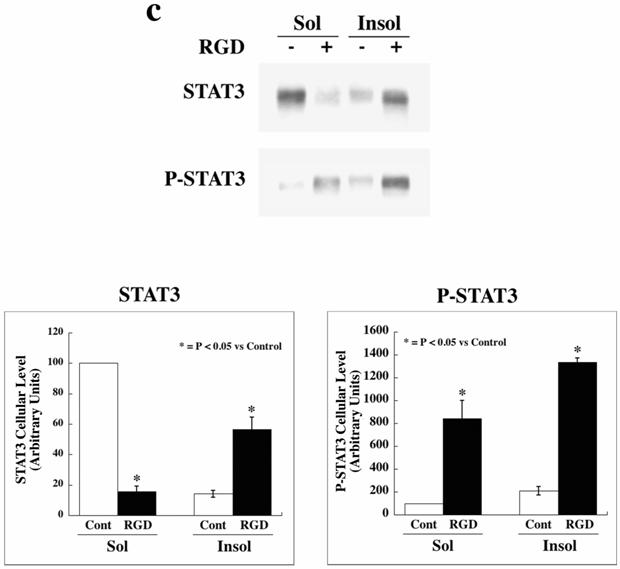
STAT3 localization and activation in RVPO felines and integrin-stimulated cardiomyocytes: (a) LV and RV from sham control and 4 h, 48 h and 1 wk RVPO felines were processed for cytoskeleton (CSK), membrane skeleton (MSK) and soluble (Sol) fractions. Protein concentrations between LV and RV pairs for each fraction were adjusted based on GAPDH (Soluble) and actin (Insoluble) contents (data not shown). The left panel shows immunoblots for total STAT3 while the right shows corresponding Tyr705 phosphorylated STAT3 (P-STAT3) blot. Quantitation of four sham control and five 48 h RVPO felines are shown (bottom panel). RV to LV densitometry ratios for each detergent-lysed subfraction was dilution-normalized and are shown. (b) LV and RV tissues from sham control and 48 h RVPO felines were processed for cytoplasmic and nuclear fractions. Immunoblots were performed using STAT3, P-STAT3, GAPDH (cytoplasmic marker), and histone-H4 (nuclear marker) antibodies. Four sham control and four 48 h RVPO feline samples were analyzed for STAT3 and P-STAT3 and normalized by GAPDH (cytoplasm samples) or histone-H4 (nuclear samples) levels. RV to LV ratios for each subfraction were plotted for control vs. 48 h RVPO (Middle Panels). In addition, percentage distribution of cytoplasmic vs. nuclear localization was plotted for each condition (Lower Panels). (c) Adult feline cardiomyocytes embedded within a 3D collagen matrix in the presence or absence of 9 mM RGD peptide for 1 h were extracted with Triton X-100 buffer and soluble (Sol) and insoluble (Insol) fractions were prepared for immunoblotting of STAT3 and P-STAT3. Densitometry from four independent experiments (bottom panel) was performed and ratios of RV/LV are plotted for (a) and (b) while for (c), STAT3 level in Sol fraction of control was arbitrarily set at 100. The mean +/- SEM is plotted for each. * p < 0.05 compared to corresponding controls.
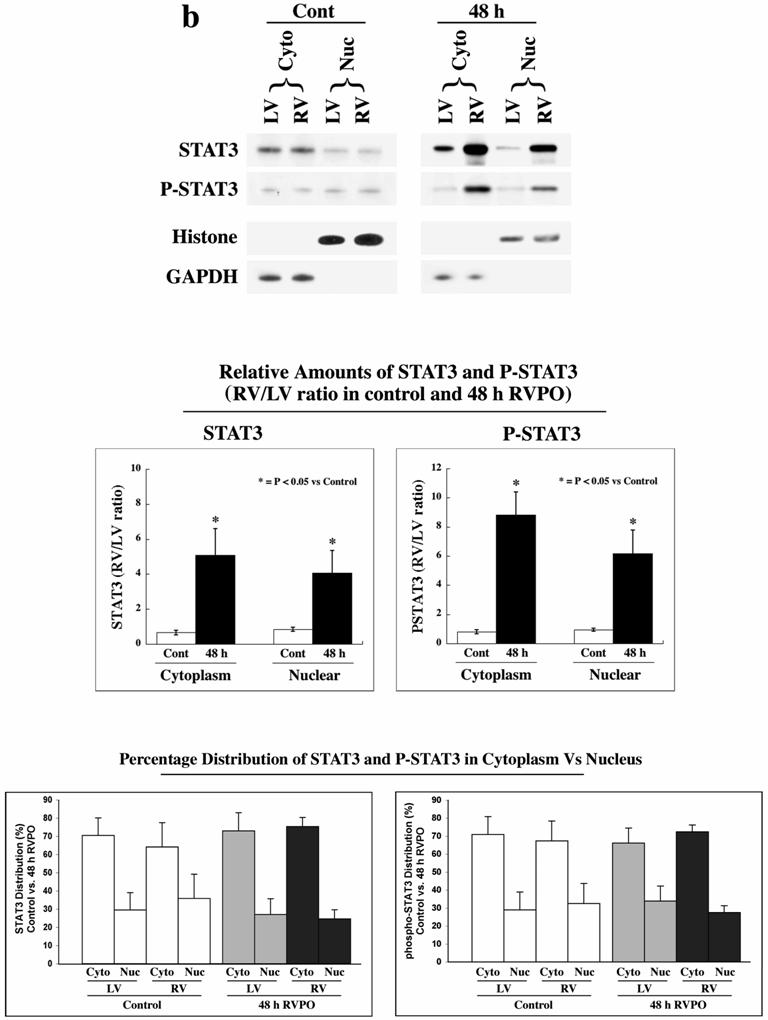
Since Tyr705 phosphorylation of STAT3 is indicative of its activation,[6, 8, 18, 19] we wanted to determine whether phosphorylated STAT3 is present in the nucleus for transcriptional activation. Therefore, we analyzed both sham control and 48 h RVPO felines for STAT3 translocation to the nucleus (Figure 1b). For this, LV and RV tissue from these cats were processed to obtain cytoplasmic and nuclear fractions (Figure 1b, top panels). The levels of GAPDH and histone-H4 show equal protein loading in each set of samples in the cytoplasmic and nuclear fractions, respectively. In sham control cat ventricles, STAT3 was predominantly cytoplasmic and P-STAT3 was barely detectable. In 48 RVPO cats, the levels of STAT3 and P-STAT3 in normally loaded LV control were similar to sham control cats, but the RV exhibited a substantial increase in STAT3 levels in both cytoplasmic and nuclear fractions. Interestingly, both cytoplasmic and nuclear STAT3 showed significant increases in phosphorylation showing activation in both locations. Quantification (Figure 1b, middle panels) revealed that the RV to LV ratios of both STAT3 and P-STAT3 in the cytosolic and nuclear fractions of 48 h RVPO cats are significantly (p<0.05) increased compared to sham controls. However, analysis of the distribution of STAT3 and P-STAT3 in the cytoplasmic/nuclear pools shows that 25-30% of STAT3 (total or phosphorylated) is present in the nucleus of control heart and that this distribution is not altered during pressure overload (Figure 1b, bottom panel).
We next determined whether STAT3 activation specifically occurs upon integrin activation. For this, adult feline cardiomyocytes were embedded in a 3D collagen matrix and treated with RGD peptide.[3, 4] In this model, our previous work[3] demonstrated that subfractionation of the detergent-lysed homogenate of cardiomyocytes during differential centrifugation yields only the low-spin pellet and corresponds to CSK, while additional high-speed centrifugation does not yield a second pellet (MSK) as seen in ventricular tissue samples. However, we do find many of the MSK proteins in the CSK fraction obtained during low-speed centrifugation.[3] In the present in vitro studies, analysis of the detergent soluble and insoluble-pellet (CSK+MSK) fractions revealed that STAT3 was predominantly present in the detergent soluble fraction under normal conditions (Figure 1c, top panel), similar to what was observed in in vivo control tissue. However, upon RGD stimulation in this 3D model, STAT3 was recruited to the insoluble pellet fraction and P-STAT3 levels were increased both in the insoluble and soluble fractions (p<0.05). Overall, experiments shown in Figure 1 indicate that in PO myocardium STAT3 and P-STAT3 levels are increased in the cytoplasmic and nuclear fractions, that P-STAT3 is recruited to the Triton X-100 insoluble pellet fractions and that this observation could be mimicked in adult cardiomyocytes via integrin stimulation with RGD.
STAT3 Activation in both RVPO in vivo and RGD Stimulation in vitro is not Accompanied by JAK2 Activation
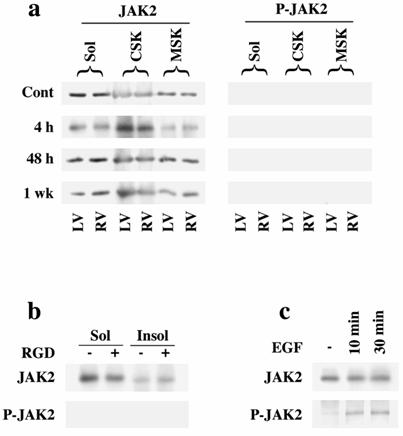
To ascertain whether the classic JAK2-mediated pathway is involved during STAT3 activation, we assayed for JAK2 recruitment to the insoluble fraction and phosphorylation at Tyr1007/1008 sites at all time points of PO (Figure 2a) and RGD stimulation of cardiomyocytes (Figure 2b). However, neither RVPO nor RGD stimulation of adult cardiomyocytes exhibited JAK2 recruitment or phosphorylation. We confirmed our data using an additional commercially available antibody for the detection of JAK2 phosphorylation at Tyr1007/1008 sites (data not shown). To test the antibody specificity for feline samples, we stimulated adult feline cardiomyocytes cultured on laminin-coated dishes with EGF. These experiments showed JAK2 phosphorylation upon EGF stimulation (Figure 2c), indicating the possibility that a JAK2-independent mechanism contributes to STAT3 phosphorylation/activation.
Analysis of JAK2 in PO myocardium and RGD-treated cardiomyocytes: Ventricular tissue samples and cardiomyocytes in 3D collagen matrix were processed to obtain detergent-lysed subfractions, and protein concentrations were adjusted. (a) Left column shows JAK2 immunoblots in the detergent-lysed subfractions at various times of RVPO while the right column shows the corresponding Tyr1007/1008-phosphorylated JAK2 (P-JAK2). (b) Top panel shows JAK2 immunoblots in soluble (Sol), and insoluble (CSK+MSK) fractions prepared from control and RGD-stimulated cardiomyocytes. Bottom panel shows the corresponding P-JAK2 immunoblot. Results of (a) and (b) were confirmed at least twice. (c) Positive control where adult feline cardiomyocytes cultured on laminin-coated plates were treated with 30 nM EGF for 10 and 30 min, and the soluble extract was used for JAK2 (top panel) and P-JAK2 (bottom panel) immunoblots.
STAT3 Activation is Independent of c-Src Activation
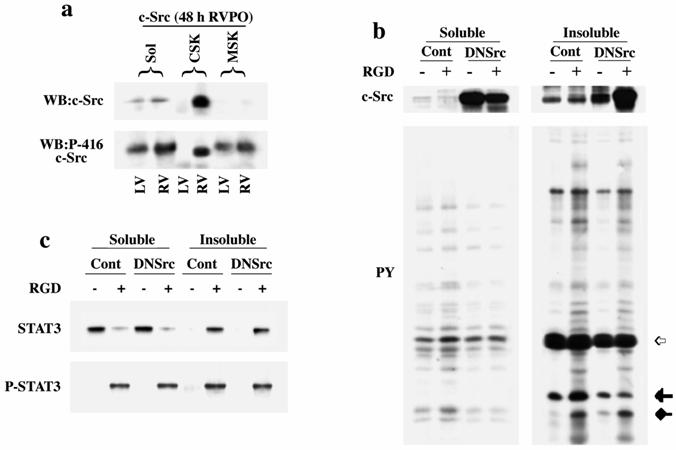
We next analyzed whether c-Src, a Src family NTK, was responsible for STAT3 phosphorylation/activation. In 48 h PO tissue samples, c-Src was active in all detergent-lysed subfractions, as indicated by its Tyr416 phosphorylation (Figure 3a, lower panel) and in addition, c-Src recruitment to the CSK (upper panel). We then sought to link c-Src and STAT3 in our 3D model through the adenoviral expression of a dominant negative form of c-Src (DNSrc). The DNSrc was created by double mutations (K295R/Y527F) where the lysine mutation causes loss of the ATP transfer site to render the kinase inactive, while the tyrosine mutation favors c-Src's open/inactive conformation. This well characterized mutant will retain adaptor function while losing kinase activity.[33, 34] At an MOI of 10, DNSrc expression was >20 fold higher over endogenous c-Src (Figure 3b, upper panels), and RGD stimulation of cardiomyocytes still resulted in recruitment of DNSrc to the detergent insoluble fraction (Figure 3b upper panels) but only minimally impacted overall tyrosine phosphorylation level of proteins compared to control (Figure 3b, lower panels). Indeed, the upper most arrow shows the position of actin. The middle arrow indicates a phosphorylated protein band that exhibited enhanced phosphorylation during RGD-stimulation and seems to be dependent on c-Src activation as it is greatly diminished upon inclusion of DNSrc. However, the lowest arrow exhibits a protein that has no dependence on c-Src activation, as its RGD-stimulated phosphorylation in the presence of DNSrc is unchanged from control cells. Together these data suggest that RGD stimulates c-Src dependent and independent tyrosine phosphorylation of cellular proteins. When we examined STAT3 under these conditions (Figure 3c), both RGD-stimulated recruitment to the insoluble fraction and phosphorylation of STAT3 were unaffected by DNSrc overexpression. These results indicate that the kinase function of c-Src does not contribute to STAT3 phosphorylation in PO myocardium in vivo or RGD-stimulated adult cardiomyocytes in in vitro.
The effect of DNSrc overexpression on integrin-stimulated STAT3 redistribution and phosphorylation in cardiomyocytes: (a) Detergent-lysed subfractions were prepared from 48 h RVPO felines as in Figure 1a and analyzed for c-Src distribution and Tyr416 phosphorylation. (b) Adult feline cardiomyocytes cultured on laminin-coated dishes were infected with DNSrc adenovirus at MOI=10 for 36 h. Uninfected (Cont) and infected (DNSrc) cells were harvested and embedded in type-I collagen (3D environment) with or without 9 mM RGD. Detergent soluble and insoluble fractions of cardiomyocytes were prepared as in Figure 1c. Immunoblots show c-Src (top panel) and tyrosine phosphorylated protein (bottom panel) levels. The upper most arrow (open) in the tyrosine phosphorylation blot shows the band corresponding to actin, which was used for normalization. The middle (solid) and bottom arrows (diamond) indicate phosphorylated protein bands that are dependent and independent on RGD-stimulated c-Src activation, respectively. (c) Detergent soluble and insoluble fractions prepared as in (b) were used for STAT3 and anti-P-STAT3 immunoblots. Results were confirmed thrice.
Membrane Skeletal Redistribution and Phosphorylation/activation of BMX in PO Myocardium
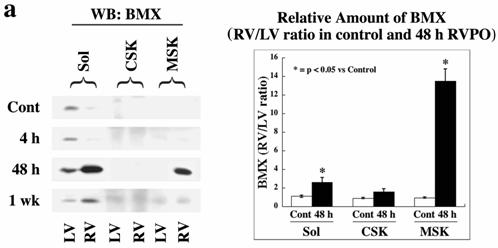
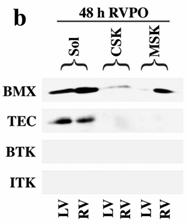
We next tested if members of the TEC family NTKs serve as upstream kinases for the STAT3 phosphorylation. Results shown in Figure 4a demonstrate that BMX levels in 48 h PO RV were markedly increased in the soluble fraction compared to same animal control LV. Importantly, a substantial amount of BMX was also recruited to the MSK, similar to STAT3 (Figure 1). However, by 1 wk PO myocardium, BMX level in the MSK was almost undetectable. The summary data, comparing BMX in control and 48 h RVPO felines (Figure 4a, right panel), show that BMX is predominantly present in the soluble fraction, and 48 h PO causes a dramatic increase (p < 0.05) both in the soluble (2.5 fold) and MSK (13 fold) fractions. Finally, we analyzed 48 h PO tissue for other TEC family members for their expression and distribution in PO myocardium (Figure 4b). These studies indicate that only BMX and TEC are expressed in the heart, although, unlike BMX, TEC did not show any change in expression or distribution during PO (Figure 4b).
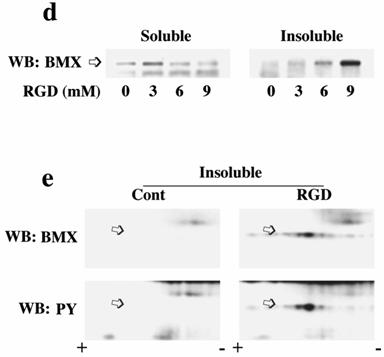
Localization and phosphorylation of BMX in Triton X-100 insoluble pellet fractions of both RVPO feline myocardium and integrin-stimulated cardiomyocytes: (a) Ventricular tissue samples from control and RVPO felines were processed to obtain detergent-lysed subfractions as in Figure 1a and used for anti-BMX immunoblotting. Six control and six 48 h RVPO felines were scanned and RV to LV ratios for each fraction of control and 48 h RVPO felines were determined and plotted. Values are the mean +/- SEM. * p < 0.05 compared to control. (b) Distribution of TEC family kinases in detergent-lysed subfraction of PO feline myocardium: LV and RV from a 48 h RVPO feline were processed to obtain Triton X-100 soluble (Sol) and insoluble cytoskeleton (CSK) and membrane skeleton (MSK) fractions. Samples were then used for Western blot analysis with anti-BMX, -TEC, -BTK and -ITK antibodies. (c) Two-dimensional gel electrophoresis of both soluble (Sol) and MSK fractions from 48 h RVPO felines was performed and immunoblotted with anti-BMX antibody (Top panel). To analyze tyrosine phosphorylation (PY) of BMX, blots were stripped and reprobed with phosphotyrosine antibody (bottom panel). Arrows indicate the position of phosphorylated BMX. Results were confirmed twice. (d) and (e) Adult cardiomyocytes were stimulated with RGD and detergent soluble and insoluble samples were prepared. (d) Immunoblot showing RGD dose response of BMX cellular redistribution. (e) Two-dimensional gel electrophoresis of control (Cont) and 9 mM RGD-stimulated detergent-insoluble (CSK+MSK) samples of collagen-embedded cardiomyocytes was performed with anti-BMX immunoblotting (Top panel). Blots were stripped and then reprobed for phosphotyrosine (bottom panel). Arrows indicate the position of phosphorylated BMX. The location of the + and – electrodes are indicated. Results were confirmed twice.
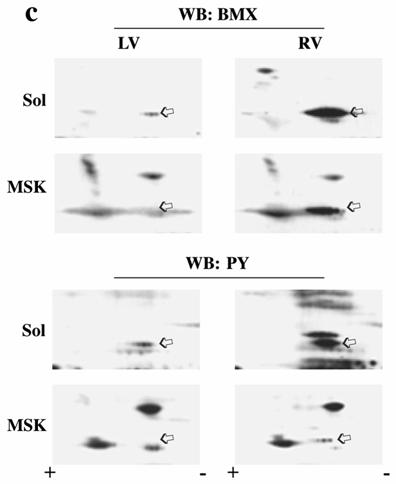
To confirm BMX activation, we explored its phosphorylation state in both detergent soluble and MSK fractions, since autophosphorylation on a specific tyrosine residue in the catalytic domain is critical for its kinase activity.[21] We performed two-dimensional (2D) gel electrophoresis (Figure 4c) of soluble and MSK fractions as commercial antibodies were not suitable for immunoprecipitation using feline detergent-insoluble fractions. BMX was found to migrate in multiple spots both in the soluble fraction of normally loaded LV and in the soluble and MSK fractions of pressure-overloaded RV. However, only pressure-overloaded RV (both Sol and MSK) showed an increase in overall levels, consistent with the RVPO time course (Figure 4a), and a significant migration towards the positive electrode during isoelectric focusing, indicating hyperphosphorylation.
To demonstrate whether these hyperphosphorylated spots have PY, we stripped and reprobed the membrane with phosphotyrosine antibody (Figure 4c bottom panels). These experiments indicate the presence of an increased level of phosphotyrosine in these hyperphosphorylated BMX spots in both soluble and MSK fractions of PO myocardium. To determine whether this process might be mediated via integrins, we analyzed our collagen model for BMX recruitment and activation. As shown in Figure 4d, BMX exhibits an RGD dose-dependent recruitment to the detergent insoluble fraction. Furthermore, 2D electrophoresis of the recruited BMX exhibited the characteristic pattern of hyperphosphorylation, including tyrosine phosphorylation (Figure 4e) as observed in PO tissue samples (Figure 4c).
STAT3 and BMX are Complexed in vivo During PO
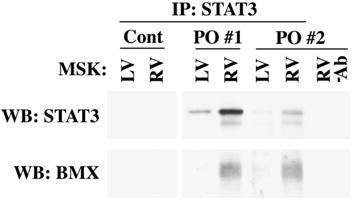
Finally, to demonstrate that PO induces BMX interaction with STAT3 in vivo and possibly serves as an upstream kinase, we gently solubilized the MSK fractions from control and PO felines with mRIPA buffer. Figure 5 shows STAT3 immunoprecipitation from MSK fractions of a control and two independent 48 h RVPO felines. As expected, STAT3 levels were much higher in the pressure-overloaded RV MSK samples. When STAT3 immunoprecipitates were analyzed, BMX was found to be complexed with STAT3 only in PO RV samples. For control purposes, a “no antibody control" was performed simultaneously where MSK sample from pressure overloaded RV was mixed with protein A/G beads and processed with other samples. This negative control did not show the presence of either STAT3 or BMX, although the MSK sample used for this IP has both these proteins. In addition, inclusion of 2 μg of nonimmune IgG (rabbit) in our immunoprecipitation protocol with MSK sample of pressure overloaded RV showed no STAT3 or BMX (data not shown). Finally, the bands seen in the Western blot for BMX and STAT3 could not be due to STAT3 antibody cross reaction, since normally loaded LV sample does not show such bands corresponding to BMX and STAT3.
Co-immunoprecipitation of STAT3 and BMX: Ventricular tissue samples from sham control (Cont) or 48 h RVPO felines (two independent felines. PO#1 and PO#2) were processed to obtain MSK pellet. The pellet was re-suspended in mRIPA and immunoprecipitated with STAT3 antibody. Immunoprecipitates were used for anti-STAT3 and anti-BMX immunoblotting. Non-specific binding to agarose beads was ruled out by omitting STAT3 antibody during immunoprecipitation from RV extract (RV-Ab lane).
Discussion
STAT3 is an important transcription factor for cellular proliferation, differentiation and survival. Phosphorylation of STAT3 activates gene expression affecting cell-cell adhesion, growth/proliferation and has also been implicated during activation of anti-apoptotic Bcl proteins.[6] Although STAT proteins exist as dimers and tetramers in unphosphorylated states, phosphorylation is critical for monomer conformational changes that will allow for effective nuclear import, in the case of STAT1, and DNA binding based on crystal structure studies. In the unphosphorylated state, STAT1 is not recognized by importin-α5 because the unphosphorylated dimers mask the nuclear-localization signal. In contrast, despite sharing 70% homology with STAT1, STAT3 does behave differently. STAT3 accumulates in the nucleus independent of phosphorylation state, mainly through direct nuclear translocation by importin-α3. However, this is a dynamic process in which STAT3 is shuttled into and out of the nucleus.[35] In fact, only a small fraction of non-phosphorylated STAT3 is found in the nucleus at any one time.[17] As such, it has been suggested that STAT3 phosphorylation can induce a STAT1/STAT3 heterodimer that has inducible nuclear transport allowing for increased nuclear accumulation.[19] Furthermore, recent studies[9, 35] show that prolonged phosphorylation of STAT3 results in more sustained nuclear localization. Also, although there is some evidence for unphosphorylated STAT3 to influence the transcription of a small number of genes, phosphorylation of STAT3 is very important for the vast majority of gene regulation by STAT3. Thus, tyrosine phosphorylation of STAT3 promotes a particular homodimerization conformation involving its SH2 (Src-homology 2) domain enabling STAT3 to bind target genes during transcriptional activation, including pro-survival genes.[6]
To study changes in STAT3 during cardiac hypertrophy, we initially utilized a RVPO model and analyzed both cellular distribution and phosphorylation of STAT3. Our studies demonstrate that STAT3 levels are significantly increased and redistributed to the MSK fraction in 48 h PO myocardium. In addition, STAT3 was significantly phosphorylated under these conditions in both detergent soluble and insoluble (CSK and MSK) fractions, and the level of P-STAT3 was increased in both nuclear and cytoplasmic fractions. Increased level of P-STAT3 in both the MSK and nucleus indicates a dual role of STAT3 in cytoskeletal remodeling and cell survival during PO hypertrophy, a model that has already been implicated in cancer.[8] Interestingly, a recent publication[9] has shown that STAT3 phosphorylation in multicellular aggregates of squamous cell cancer cell lines begins as a diffuse cytoplasmic distribution and at cell-cell junctions at first with very little nuclear localization, but at later time points (12-24 h), P-STAT3 accumulates in the nucleus. As such, there appears to be a difference in the length of time that STAT3 remains phosphorylated based on the type of stimulus responsible for the phosphorylation. Indeed, cytokine stimulation of STAT3 phosphorylation is transient (~30 minutes). However, v-Src-transformation of NIH 3T3 cells results in constitutive STAT3 phosphorylation which is unaffected by JAK inhibitors.[36] A recent study[35] has suggested that the mechanism by which this occurs is through more rapid cycling of “activated” STAT3 between the nucleus and cytoplasm than “non-activated” STAT3. However, our studies indicate that the tyrosine phosphorylation of STAT3 in 48 h PO myocardium does not appear to be required for the increased level of STAT3 in the nucleus, since the distribution of STAT3 or P-STAT3 between cytoplasm and nucleus was not significantly altered in 48 h PO myocardium.
Tyrosine kinases that mediate STAT3 phosphorylation include: (i) JAK kinases that function downstream of cytokines, (ii) other NTKs, such as c-Src and BMX, and (iii) growth factor receptor kinases, such as epidermal growth factor receptor. We have shown previously in vivo[4, 5] that PO can cause integrin activation, resulting in cytoskeletal assembly and activation of c-Src and FAK, and tyrosine phosphorylation of several cytoskeleton-bound proteins in 24-48 h PO myocardium. We were then able to reproduce this activation in 3D collagen-embedded cardiomyocytes stimulated with RGD peptide.[3, 4] Therefore, we utilized our 3D collagen model to demonstrate that integrin stimulation with RGD peptide causes STAT3 activation/phosphorylation and to characterize the responsible upstream NTK(s). These studies suggest that our previously established integrin-mediated NTK activation and FAC formation[3, 4] may contribute to STAT3 activation in PO myocardium. In support of this, other recent studies[37, 38] also demonstrate integrin-mediated c-Src activation and FAC formation can contribute to STAT3 phosphorylation/activation. Presence of STAT3 at FAC sites has been demonstrated previously[7] to enhance FAK activity.[39] Furthermore, evidence exists for STAT3 interaction with other components of the cytoskeleton, including paxillin, p130Cas, and stathmin.[10] Therefore, STAT3 presence in the MSK of 48 h PO myocardium is indicative of its possible role at the FAC for cytoskeletal remodeling, independent of its transcriptional role.
To identify the responsible kinases for STAT3 phosphorylation, we explored the activation of potential upstream NTKs. Our analysis using in vivo and in vitro models of hypertrophic stimulation did not show JAK2 phosphorylation, though JAK2 phosphorylation/activation could be observed in EGF-treated adult feline cardiomyocytes in vitro. An earlier study[40] in a murine transverse aortic constriction PO model demonstrates that STAT3 undergoes biphasic acute (3 h) and hypertrophic responses (48 h) via the gp130-mediated JAK2 signaling where the activated STAT3 plays an autoregulatory negative feedback role by promoting the expression of a JAK2 inhibitor protein, SOCS3 (suppressor of cytokine signaling-3). Therefore, our studies in PO feline myocardium cannot rule out JAK2's contribution to STAT3 phosphorylation in the early period of PO,[41] where it might be subsequently inactivated by the expression of SOCS, and under these conditions, other NTKs might sustain STAT3 activation. Indeed, it appears that STAT3 activation by non-JAK NTKs, such as v-Src, is not susceptible to SOCS overexpression.[36]
Although our previous work both in 48 h PO myocardium and in RGD-stimulated adult cardiomyocytes[3, 4] demonstrates c-Src recruitment and activation, expression of DNSrc in cardiomyocytes in the 3D environment did not affect the RGD-stimulated STAT3 phosphorylation. These data suggest that c-Src kinase activity is not critical for STAT3 phosphorylation, however, it is possible that c-Src may function as an adaptor to recruit other NTKs for STAT3 phosphorylation.
Finally, we focused on the possible role of a TEC family member, BMX, in STAT3 phosphorylation. BMX, which is highly expressed in the heart, may serve as an upstream kinase for STAT3 phosphorylation as reported previously both in c-Src dependent[25] and independent[24] manners. Our studies on BMX in 48 h PO myocardium include: (i) overall increase of BMX, (ii) recruitment of a significant amount of BMX to the MSK as in the case of STAT3, (iii) hyperphosphorylation of MSK-bound BMX and (iv) the presence of a BMX/STAT3 complex in MSK. These transient changes were observed in 48 h PO myocardium but were absent in both 4 h and 1 wk PO myocardium and correlate well with STAT3 activation and with our earlier observation in 48 h PO myocardium[4, 5] that showed integrin activation and FAC formation, consisting of NTKs. Whereas our previous studies show c-Src and FAK recruitment to the CSK fraction, we observe in the present study BMX recruitment to the MSK fraction similar to STAT3. Importantly, the increased level of BMX in Triton X-100 soluble and insoluble MSK fractions, relative to CSK fraction, correlates well with the increased levels of P-STAT3 in these fractions (Figure 1a). BMX is unique among tyrosine kinases in that it contains a pleckstrin homology (PH) domain that binds to specific phosphoinositides which may explain its MSK association. Although our studies[3-5] indicate that most focal adhesion proteins are found in the CSK fraction, it is not clear whether BMX and STAT3, which are complexed in the MSK fraction, are part of the focal adhesion or represent an independent localization in the cell. We were unable to resolve this issue confocally, since the commercially available BMX antibodies used in this study were not useful for confocal imaging in feline heart. Interestingly, several studies have localized STATs within lipid-rafts in connection with caveolins.[42] Furthermore, there is some evidence for BMX interacting with caveolin-1 in bone marrow cells.[43] It is therefore possible that the MSK-bound STAT3 and BMX in PO myocardium are localized to caveolae.
Our studies with adult cardiomyocytes in vitro demonstrate that in a 3D model, but not in a 2D model without FAC formation (data not shown), stimulation with an RGD peptide caused both BMX and STAT3 recruitment to the detergent insoluble fraction (CSK+MSK) and hyperphosphorylation. Since both STAT3 and BMX exhibit similar redistribution and phosphorylation, these data and our in vivo observations showing a complex formation between BMX and STAT3 in PO myocardium strongly indicate that integrin-mediated BMX activation might serve as an upstream kinase for STAT3 activation in hypertrophying myocardium.
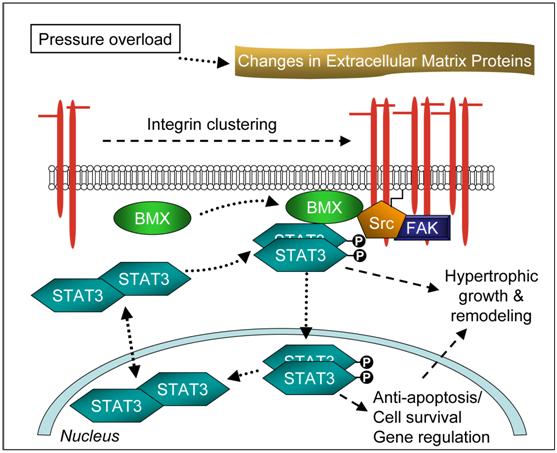
We therefore propose a novel mechanism of STAT3 activation during cardiac hypertrophy (summarized in Figure 6) where PO-induced integrin activation results in recruitment of several NTKs including BMX and STAT3 to the FAC. Subsequent activation of BMX can then mediate STAT3 phosphorylation. Since STAT3 phosphorylation was observed in both the nucleus and MSK of 48 h PO myocardium, even while JAK2 was inactive, these findings strongly suggest that BMX-mediated STAT3 activation not only contributes to cytoskeletal remodeling but also to the transcriptional activation in the nucleus. In this manner, STAT3 may control both FAC turnover for cytoskeletal remodeling and gene expression for cardioprotection during the early stages of cardiac hypertrophy.
Proposed Mechanism of STAT3 Activation in Cardiac Hypertrophy.
Acknowledgements
This study was supported by NIH PPG HL-48788, by Merit award from the Research Service of the Department of Veterans Affairs and by NRSA Student Fellowship GM08716. We would like to thank Dr. Paul McDermott for his adenoviral production help, Mary Barnes and Dr. Harinath Kasiganesan for adult cardiomyocyte isolation, Charlene Kerr for excellent technical assistance, Phillip Moschella for critical reading of the manuscript and Dr. Catalin Baicu for statistical analysis.
CONFLICT OF INTERESTS
The authors declare that no conflict of interest exists.
References
1. Ruwhof C, van der Laarse A. Mechanical stress-induced cardiac hypertrophy: mechanisms and signal transduction pathways. Cardiovasc Res. 2000;47(1):23-37
2. Howe A, Aplin AE, Alahari SK, Juliano RL. Integrin signaling and cell growth control. Curr Opin Cell Biol. 1998;10(2):220-231
3. Willey CD, Balasubramanian S, Rodriguez Rosas MC, Ross RS, Kuppuswamy D. Focal complex formation in adult cardiomyocytes is accompanied by the activation of beta3 integrin and c-Src. J Mol Cell Cardiol. 2003;35(6):671-683
4. Laser M, Willey CD, Jiang W, Cooper Gt, Menick DR, Zile MR. et al. Integrin activation and focal complex formation in cardiac hypertrophy. J Biol Chem. 2000;275(45):35624-35630
5. Kuppuswamy D, Kerr C, Narishige T, Kasi VS, Menick DR, Cooper Gt. Association of tyrosine-phosphorylated c-Src with the cytoskeleton of hypertrophying myocardium. J Biol Chem. 1997;272(7):4500-4508
6. Calo V, Migliavacca M, Bazan V, Macaluso M, Buscemi M, Gebbia N. et al. STAT proteins: from normal control of cellular events to tumorigenesis. J Cell Physiol. 2003;197(2):157-168
7. Silver DL, Naora H, Liu J, Cheng W, Montell DJ. Activated signal transducer and activator of transcription (STAT) 3: localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004;64(10):3550-3558
8. Germain D, Frank DA. Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin Cancer Res. 2007;13(19):5665-5669
9. Onishi A, Chen Q, Humtsoe JO, Kramer RH. STAT3 signaling is induced by intercellular adhesion in squamous cell carcinoma cells. Exp Cell Res. 2008;314(2):377-386
10. Gao SP, Bromberg JF. Touched and Moved by STAT3. Sci STKE. 2006;2006(343):pe30
11. Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R. et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10(1):105-115
12. Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross JJr. et al. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97(2):189-198
13. Hilfiker-Kleiner D, Hilfiker A, Drexler H. Many good reasons to have STAT3 in the heart. Pharmacol Ther. 2005;107(1):131-137
14. Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B. et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res. 2004;95(2):187-195
15. Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX. et al. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci U S A. 2003;100(22):12929-12934
16. Oshima Y, Fujio Y, Nakanishi T, Itoh N, Yamamoto Y, Negoro S. et al. STAT3 mediates cardioprotection against ischemia/reperfusion injury through metallothionein induction in the heart. Cardiovasc Res. 2005;65(2):428-435
17. Sehgal PB. STAT-signalling through the cytoplasmic compartment: consideration of a new paradigm. Cell Signal. 2000;12(8):525-535
18. Shah M, Patel K, Mukhopadhyay S, Xu F, Guo G, Sehgal PB. Membrane-associated STAT3 and PY-STAT3 in the cytoplasm. J Biol Chem. 2006;281(11):7302-7308
19. Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006;6(8):602-612
20. Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA. et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20(20):2499-2513
21. Qiu Y, Kung HJ. Signaling network of the Btk family kinases. Oncogene. 2000;19(49):5651-5661
22. Smith CI, Islam TC, Mattsson PT, Mohamed AJ, Nore BF, Vihinen M. The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays. 2001;23(5):436-446
23. Saharinen P, Ekman N, Sarvas K, Parker P, Alitalo K, Silvennoinen O. The Bmx tyrosine kinase induces activation of the Stat signaling pathway, which is specifically inhibited by protein kinase Cdelta. Blood. 1997;90(11):4341-4353
24. Wen X, Lin HH, Shih HM, Kung HJ, Ann DK. Kinase activation of the non-receptor tyrosine kinase Etk/BMX alone is sufficient to transactivate STAT-mediated gene expression in salivary and lung epithelial cells. J Biol Chem. 1999;274(53):38204-38210
25. Tsai YT, Su YH, Fang SS, Huang TN, Qiu Y, Jou YS. et al. Etk, a Btk family tyrosine kinase, mediates cellular transformation by linking Src to STAT3 activation. Mol Cell Biol. 2000;20(6):2043-2054
26. Zhang J, Ping P, Wang GW, Lu M, Pantaleon D, Tang XL. et al. Bmx, a member of the Tec family of nonreceptor tyrosine kinases, is a novel participant in pharmacological cardioprotection. Am J Physiol Heart Circ Physiol. 2004;287(5):H2364-2366
27. Mathur P, Kaga S, Zhan L, Das DK, Maulik N. Potential candidates for ischemic preconditioning-associated vascular growth pathways revealed by antibody array. Am J Physiol Heart Circ Physiol. 2005;288(6):H3006-3010
28. Rozich JD, Barnes MA, Schmid PG, Zile MR, McDermott PJ, Cooper Gt. Load effects on gene expression during cardiac hypertrophy. J Mol Cell Cardiol. 1995;27(1):485-499
29. Cooper Gt, Satava RMJr, Harrison CE, Coleman HNd. Mechanisms for the abnormal energetics of pressure-induced hypertrophy of cat myocardium. Circ Res. 1973;33(2):213-223
30. Mann DL, Urabe Y, Kent RL, Vinciguerra S, Cooper Gt. Cellular versus myocardial basis for the contractile dysfunction of hypertrophied myocardium. Circ Res. 1991;68(2):402-415
31. He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95(5):2509-2514
32. Cooper JA, Gould KL, Cartwright CA, Hunter T. Tyr527 is phosphorylated in pp60c-src: implications for regulation. Science. 1986;231(4744):1431-1434
33. Chaturvedi P, Reddy MV, Reddy EP. Src kinases and not JAKs activate STATs during IL-3 induced myeloid cell proliferation. Oncogene. 1998;16(13):1749-1758
34. Kaplan KB, Bibbins KB, Swedlow JR, Arnaud M, Morgan DO, Varmus HE. Association of the amino-terminal half of c-Src with focal adhesions alters their properties and is regulated by phosphorylation of tyrosine 527. Embo J. 1994;13(20):4745-4756
35. Herrmann A, Vogt M, Monnigmann M, Clahsen T, Sommer U, Haan S. et al. Nucleocytoplasmic shuttling of persistently activated STAT3. J Cell Sci. 2007;120(Pt 18):3249-3261
36. Iwamoto T, Senga T, Naito Y, Matsuda S, Miyake Y, Yoshimura A. et al. The JAK-inhibitor, JAB/SOCS-1 selectively inhibits cytokine-induced, but not v-Src induced JAK-STAT activation. Oncogene. 2000;19(41):4795-4801
37. Bhattacharya S, Ray RM, Johnson LR. Integrin beta3-mediated Src activation regulates apoptosis in IEC-6 cells via Akt and STAT3. Biochem J. 2006;397(3):437-447
38. Simon AR, Vikis HG, Stewart S, Fanburg BL, Cochran BH, Guan KL. Regulation of STAT3 by direct binding to the Rac1 GTPase. Science. 2000;290(5489):144-147
39. Schlessinger K, Levy DE. Malignant transformation but not normal cell growth depends on signal transducer and activator of transcription 3. Cancer Res. 2005;65(13):5828-5834
40. Yasukawa H, Hoshijima M, Gu Y, Nakamura T, Pradervand S, Hanada T. et al. Suppressor of cytokine signaling-3 is a biomechanical stress-inducible gene that suppresses gp130-mediated cardiac myocyte hypertrophy and survival pathways. J Clin Invest. 2001;108(10):1459-1467
41. Beckles DL, Mascareno E, Siddiqui MA. Inhibition of Jak2 phosphorylation attenuates pressure overload cardiac hypertrophy. Vascul Pharmacol. 2006;45(6):350-7
42. Sehgal PB, Guo GG, Shah M, Kumar V, Patel K. Cytokine signaling: STATS in plasma membrane rafts. J Biol Chem. 2002;277(14):12067-12074
43. Vargas L, Nore BF, Berglof A, Heinonen JE, Mattsson PT, Smith CI. et al. Functional interaction of caveolin-1 with Bruton's tyrosine kinase and Bmx. J Biol Chem. 2002;277(11):9351-9357
Author contact
![]() Correspondence to: Dhandapani Kuppuswamy, Ph.D., Gazes Cardiac Research Institute, 114 Doughty Street, Charleston, South Carolina 29425-2221. Telephone: 843-876 5067; Fax: 843-876 5068; E-mail: kuppusdedu
Correspondence to: Dhandapani Kuppuswamy, Ph.D., Gazes Cardiac Research Institute, 114 Doughty Street, Charleston, South Carolina 29425-2221. Telephone: 843-876 5067; Fax: 843-876 5068; E-mail: kuppusdedu