Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
MATERIALS AND METHODS
RESULTS
DISCUSSION
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2008; 4(5):291-299. doi:10.7150/ijbs.4.291 This issue Cite
Research Paper
SIRT3 interacts with the daf-16 homolog FOXO3a in the Mitochondria, as well as increases FOXO3a Dependent Gene expression
Kristi Muldoon Jacobs 1 *, J. Daniel Pennington 1 *, Kheem S. Bisht1, Nukhet Aykin-Burns 2, Hyun-Seok Kim 3, Mark Mishra1, Lunching Sun 1, Phuongmai Nguyen 1, Bong-Hyun Ahn 4, Jaime Leclerc 1, Chu-Xia Deng 3, Douglas R. Spitz 2, David Gius 1 ![]()
1. Molecular Radiation Oncology, Center for Cancer Research, Radiation Oncology Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD
2. Free Radical and Radiation Biology Program, Department of Radiation Oncology, University of Iowa, Iowa City, IA
3. Genetics of Development and Disease Branch, NIDDK, National Institutes of Health, Bethesda, MD
4. Cardiology Branch, National Heart, Lung, and Blood Institute, NIH, Bethesda, MD
* The first two authors contributed equally to this paper.
Received 2008-7-22; Accepted 2008-9-3; Published 2008-9-5
Abstract
Cellular longevity is a complex process relevant to age-related diseases including but not limited to chronic illness such as diabetes and metabolic syndromes. Two gene families have been shown to play a role in the genetic regulation of longevity; the Sirtuin and FOXO families. It is also established that nuclear Sirtuins interact with and under specific cellular conditions regulate the activity of FOXO gene family proteins. Thus, we hypothesize that a mitochondrial Sirtuin (SIRT3) might also interact with and regulate the activity of the FOXO proteins. To address this we used HCT116 cells overexpressing either wild-type or a catalytically inactive dominant negative SIRT3. For the first time we establish that FOXO3a is also a mitochondrial protein and forms a physical interaction with SIRT3 in mitochondria. Overexpression of a wild-type SIRT3 gene increase FOXO3a DNA-binding activity as well as FOXO3a dependent gene expression. Biochemical analysis of HCT116 cells over expressing the deacetylation mutant, as compared to wild-type SIRT3 gene, demonstrated an overall oxidized intracellular environment, as monitored by increase in intracellular superoxide and oxidized glutathione levels. As such, we propose that SIRT3 and FOXO3a comprise a potential mitochondrial signaling cascade response pathway.
Keywords: SIRT3, FOXO3a, gene expression, Superoxide
Introduction
One theme emerging over the last several years is that aging is a complex, genetically regulated cellular process [1]. Genes critical to longevity have been isolated and characterized in several model systems including yeast and C. elegans [1]. Sirtuin family genes first came to attention because their Saccharomyces cerevisiae homolog (Sir2) was shown to regulate both replicative and overall lifespan by caloric restriction (CR) [2]. In humans and mice there are seven Sirtuins, two that are located in the nucleus (SIRT1, 6, and 7), one in the cytoplasm and nucleus (SIRT2), and 3 are localized to the mitochondria (SIRT3, 4, and 5) [3]. While yeast Sir2 is a NAD+ dependent deacetylase, mammalian Sirtuins have more varied targets, including either deacetylase or ADP-ribosyltransferase activity [4].
It is clear that sirtuins regulate a series of essential intracellular processes that defend the cell against multiple types of cellular damage, including oxidative damage [5]. These observations imply that Sirtuin genes may be metabolic guardians that could play a central role in longevity by regulating cellular or organelle renewal. That is, as cells and critical intracellular organelles slowly degrade from age related oxidative stress, Sirtuin genes may respond to this by activating signaling pathways to repair damage.
When outlining a course of investigation for the potential role of Sirtuins as organelle surveillance genes, we chose to examine the mitochondrial inner membrane protein SIRT3 for several reasons. First, it has long been suggested that the mitochondria play a role in aging and oxidative stress [6]. Second, mitochondrial electron transport chains reduce of O2 to form superoxide (O2•-), leading to oxidative stress which plays a role in genomic instability and aging [7,8]. Third, SIRT3 localization to the inner mitochondrial membrane is consistent with a role in monitoring and responding to conditions harmful to electron transport, activating processes to minimize mitochondrial damage
The FOXO family of transcription factors: FOXO1, FOXO3a, FOXO4 and FOXO6 are the human homologs of the daf-16 C. elegans gene. Daf-16 is the master regulator of dauer formation [9] and contributes to the regulation of lifespan in nematodes [10]. FOXO transcription factors appear to regulate a wide range of intracellular processes including metabolism and cellular resistance to various forms of oxidative stress [11,12]. It is also established that sirtuins interact with, and under specific cellular conditions, regulate that activity of FOXO gene family proteins [13,14]. Thus, we chose to investigate if SIRT3 might also interact with and regulate the activity of any of the FOXO proteins.
MATERIALS AND METHODS
Cell lines and Plasmids
HCT-116 (human colon carcinoma) and Cos-7 cells were cultured in McCoy's 5A media, containing 10% heat-inactivated (56°C, 30min) fetal bovine serum (FBS) and. supplemented with penicillin (100 Units/mL) and streptomycin (100μg/mL). The expression vector pcDNA4 was obtained from Promega, Inc. pCMV-SIRT3-wt and pCMV-SIRT3-mt were a kind gift from Toren Finkel (National Institutes of Health). These expression plasmids express either the wild-type gene or an acetylation null gene where amino acid 248 has been changed from a histidine to tyrosine [15]. Permanent cell lines expressing wild-type SIRT3 (wt-SIRT3) or the deacetylase mutant (mt-SIRT3) (supplemental Fig. S1) were constructed by transfecting pcDNA4, pCMV-SIRT3-wt, or pCMV-SIRT3 in HCT116 cells using FuGene 6 and selected with zeosin (supplemental Fig. S2). For this work cell lines expressing the various SIRT3 genes are referred to as wt-SIRT3 or mt-SIRT3 while plasmids expressing these genes are denoted as pCMV-SIRT3-wt or pCMV-SIRT3-mt (supplemental Fig. S1). The FOXO3a expression plasmids (pCMV-wt-FOXO3a, pCMV-m-FOXO3a, and pCMV-N-FOXO3a) were obtained from Addgene (Cambridge, Ma).
IP and Mitochondrial Fractionation
Cell extracts were prepared using a modification of the previously described method [16,17] and protein concentrations were determined using the Bradford assay (BioRad Labs., Hercules, CA) on a Beckman DU-640 spectrophotometer. Mitochondria extracts were prepared using kit provided by Pierce (Rockford, Il). Briefly cells were lysed by dounce homogenization in lysis buffer followed by centrifugation at 700 x g to remove the nuclear fraction along with cell debris. The supernatant was then spun at 3,000 x g for 15 minuets to isolate the mitochondria fraction. Trypan blue exclusion analysis was performed to check for lysis efficiency following dounce homogenization. Western samples were loaded into denaturing SDS-polyacrylamide gels, membranes were probed with anti-SIRT3 (See supplement) or anti-FOXO3a antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) and incubated with horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
Co-transfections, Luciferase Assays, and Chromatin Immunoprecipitation
For transient assays, cells were plated at 2 ×106 cells per 100-mm plate, co-transfected, and harvested after 36 hrs. Cells were co-transfected with 1 µg of pCMV-β-gal and analysis for luciferase and β-galactosidase activity. Luciferase activity was normalized to β-galactosidase activity and the results are presented as luciferase/β-galactosidase relative units. The results are presented as relative-fold induction over the sham treated control.
Chromatin Immunoprecipitation
Cells were cultured for 24h and prepared using a ChIP assay kit from Upstate Biotechnology, Inc. (Lake Placid, NY) according to the manufacturer's recommendations and preformed as previously describe [28]. Primers for the FOXO3a and SCO2 promoter are shown (Supplemental Methods) and their location is shown in supplemental Fig. S3. IP and transient transfection IP westerns were done as previously described [28]. Bands for all IPs were detected using an ECL protocol (Santa Cruz Biotechnology, Santa Cruz, CA) and visualized with Fuji Las-3000 intelligent darkbox (FujiFilm Medical Systems, Stamford, CT).
Intracellular superoxide measurements and thiol analysis
Steady state levels of superoxide were estimated using the fluorescent dye, dihydroethidium (DHE) as described in Slane et al [8]. Glutathione (GSH) and glutathione disulfide (GSSG) contents were determined by the recycling method [18] and a yellow color change was detected spectrophotometrically at 412 nm as DTNB is converted to TNB by the conversion of 2 GSH to GSSG.”
RESULTS
The daf-16 homolog, FOXO3a, forms a complex with SIRT3
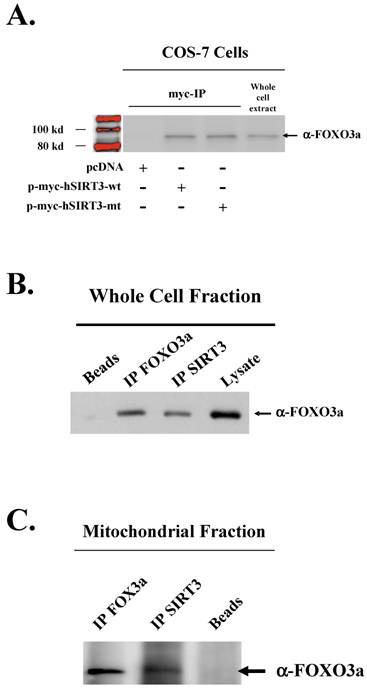
The FOXO family of transcription factors regulate metabolism and confer stress resistance [11,12]. It has also been shown that the well studied SIRT1 interacts with, and under specific cellular conditions, regulates the activity of FOXO gene family proteins [14,19]. We sought to establish whether the mitochondrial localized SIRT3 may also form a physical interaction with the FOXO family protein, FOXO3a, using co-immunoprecipitation (Co-IP) techniques. Carboxy-terminally myc tagged wild-type (p-myc-hSIRT3-wt), and mutant (p-myc-hSIRT3-mt) SIRT3 expression vectors were transfected into Cos-7 cells followed by Co-IP with an anti-myc antibody. Western analysis with an anti-FOXO3a antibody subsequently showed a physical interaction between both wild-type and mutant SIRT3 with FOXO3a (Fig. 1A). To ensure that this interaction was not an artifact of the COS-7 cell line, or the transient transfection conditions, HCT116 cells, which contain undetectable levels of endogenous SIRT3 by western analysis, were stably transfected with p-myc-hSIRT3-wt, and p-myc-hSIRT3-mt SIRT3 expression vectors (Fig S2). These cells were used to determine if the FOXO3a – SIRT3 interaction occurs in the mitochondria or the cytosol. Co-IP experiments were repeated using the HCT116 SIRT3 expressing stable cell lines. FOXO3a was found to interact with SIRT3 in both whole cell (Fig. 1B) and mitochondrial extracts (Fig. 1C). All fractions and samples in the Co-IP experiments were checked for the presence of SIRT3, using a SIRT3 specific antibody (Biomol, Plymouth Meeting , PA), as well as tubulin (Santa Cruz Biotechnology, Santa Cruz, CA), and Cytochrome C (Mitosciences, Eugene, OR) to ensure fraction purity (data not shown). These experiments demonstrate that both wild type and mutant SIRT3 form a physical interaction with FOXO3a in mitochondrial extracts.
The daf-16 homolog, FOX3a forms a complex with SIRT3. (A) FOXO3a binds to SIRT3 in vitro. Cos-7 cells were transfected with either SIRT3 wild-type (p-myc-hSIRT3-wt) or deacetylation mutant (p-myc-hSIRT3-mt) vectors and cell lysates were immunoprecipitated (IPd) with an anti-Myc antibody followed by Western analysis with an anti-FOXO3a antibody. (B) HCT116 cell lysates were IPd with either an anti-FOXO3a or anti-SIRT3 antibody, resolved by SDS-PAGE, and immunoblotted with anti-FOXO3a antibody. (C) Mitochondrial factions from HCT116 cells were IPd with either an anti-FOXO3a or anti-SIRT3 antibody and immunoblotted with anti-FOXO3a antibody.
FOX3a is a mitochondrial protein
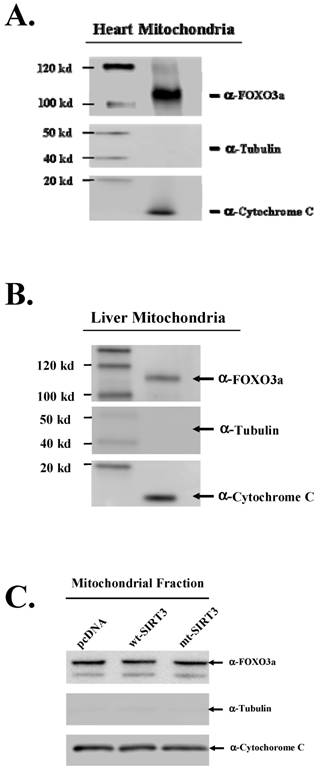
FOXO3a interacts with SIRT3 in mitochondrial extracts in vitro thus, we chose to investigate if any of the FOXO3a proteins might be localized to the mitochondrial tissue lysates as well. Western analysis of mitochondrial samples from human heart (Fig. 2A) and liver (Fig. 2B) (Microscience, Inc) lysates clearly shows that FOXO3a is present in the mitochondrial cellular fractions. Anti-tubulin and anti-cytochrome C antibodies were used as controls to determine mitochondrial and non-mitochondrial protein fractions. As expected, similar results were seen in HCT116 and HCT116 cells genetically altered to express either the wild-type or deacetylation null SIRT3 genes (Fig. 2C).
The daf-16 homolog, FOX3a is a mitochondrial protein. Mitochondrial protein fraction from heart (A) and liver (B) (DNA Technologies, ReadyWestern™) were analyzed by western analysis for the presence of the FOXO3a protein. The PVDF membrane was also probed with both and a-tubulin and a-cytochrome c antibodies that localize to the cytoplasm and mitochondria, respectively. (C) Mitochondrial protein fractions from of pcDNA, wt-SIRT3, or mt-SIRT3 cell lines were isolated and immunoreactive FOXO3a protein levels were determined.
SIRT3 activates FOXO3a dependent gene expression
To determine if FOXO3a activates promoter activity in our system, three FOXO3a expression vectors were employed that: (1) express the wild-type gene that is still inhibited by Akt (pCMV-FOXO3a-wt); (2) express a mutant gene that appears to be a dominant negative (pCMV-mt-FOXO3a); and (3) express a dominant positive (constitutively active) gene that cannot be phosphorylated and inhibited by Akt (pCMV-N-FOXO3a) [20]. Co-transfection of these three plasmids with p3400-MnSOD-Luc, which contains the MnSOD promoter and its two consensus FOXO binding sites (supplemental Fig. S3) cloned upstream of luciferase, demonstrated that pCMV-N-FOXO3a increased p3400-MnSOD-Luc luciferase activity (Fig. 3A, bar 3). In contrast, co-transfection with pCMV-mt-FOXO3a decreased MnSOD expression of the reporter (bar 2) as compared to the control, wild-type vector, pCMV-wt-FOXO3a (bar 1). These results clearly demonstrate that FOXO3a can activate the expression of the reporter gene.
SIRT3 activates FOXO3a dependent gene expression. (A) pMnSOD-Luc was transiently co-transfected with CMV based FOXO3a expression vectors encoding the wild-type (pCMV-wt-FOXO3a), dominant negative (pCMV-m-FOXO3a), or dominant positive (pCMV-N-FOXO3a) FOXO3a genes. Results for all the experiments presented in this figure were normalized to β-galactosidase activity and the results are presented as luciferase/β-galactosidase relative units. (B) SIRT3 activates FOXO3a dependent gene expression. A reporter plasmid containing three FOXO DNA-binding sites (p3x-FOXO-Luc) was transfected with either pCMV-SIRT3-wt or pCMV-SIRT3-mt and luciferase activity was determined as described above. (C) The p3x-FOXO-Luc reporter plasmid was transfected with wild type or mutant FOXO3a expression vectors (pCMV-wt-FOXO3a or pCMV-m-FOXO3a respectively) along with wild type or mutant SIRT3 expression vectors (pCMV-SIRT3-wt or pCMV-SIRT3-mt) and luciferase activity was determined and normalized to β-galactosidase as described above. All experiments are mean of at least three separate experiments and error bars for all data shown represent one standard deviation and statistical significance was established by Student's t-test. * Indicates P < 0.05 by t-test. (D) ChIP analysis of FOXO3a finding to the MnSOD and SCO2 promoters in HCT116 cells that overexpress either a wild-type or deacetylation null SIRT3 gene. Cells were fixed with 1% formaldehyde to crosslink protein-DNA interactions, sonicated, and fixed cells were immunoprecipitated with either an anti-FOXO3a antibody. DNA was eluted and purified before analysis specific primers and visualized by ethidium bromide staining.
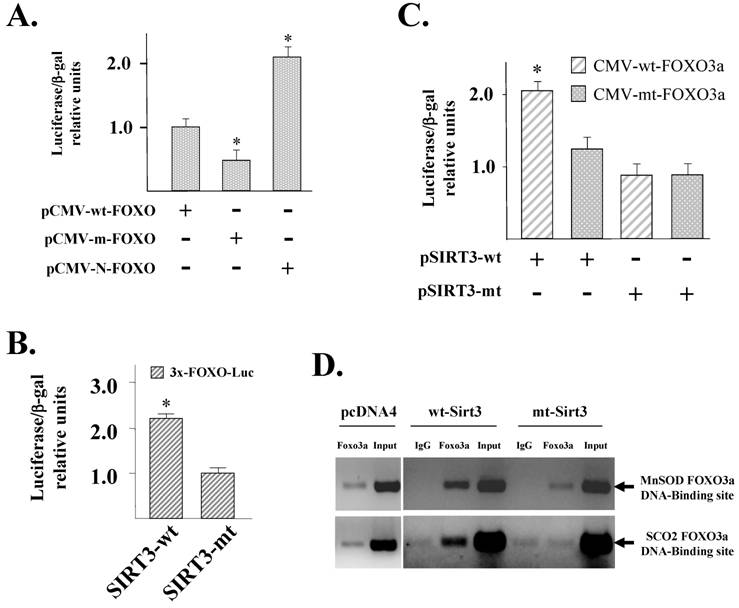
A FOXO3a luciferase reporter construct was made that contains three FOXO3a binding sites fused to the luciferase gene (p3x-FOXO-tk-Luc) and this reporter vector was used to determine FOXO3a dependent gene expression as measured by luciferase levels. p3x-FOXO-tk-Luc was subsequently co-transfected into HCT116 cells with either pCMV-SIRT3-wt or pCMV-SIRT3-mt expression vectors. Expression of the wild-type SIRT3 increases luciferase activity (Fig. 3B, bar 1 versus bar 2). These results show that SIRT3 activates FOXO3a dependent gene expression.
To determine if FOXO3a is downstream of SIRT3 in the activation of the p3x-FOXO-Luc the reporter plasmid was co-transfected with either the wild-type or mutant SIRT3 expression vector (as done in Fig. 3B), and either pCMV-wt-FOXO3a or pCMV-mt-FOXO3a. These experiments will determine if a mutant FOXO3a protein will change the ability of SIRT3 to activate p3x-FOXO-tk-Luc gene expression. Transfection of p3x-FOXO-tk-Luc or pCMV-SIRT3-wt (bar 2) is a positive control and is identical to Fig. 2B, bar 1. In contrast, co-transfection of pCMV-SIRT3-wt with a mutant FOXO3a expression vector (pCMV-mt-FOXO3a) decreased p3x-FOXO-tk-Luc driven luciferase activity (Fig. 3C, bar 2 versus 3. These results suggest that in transient assays FOXO3a dependent gene expression that is induced by SIRT3 is prevent by expression of a mutant FOXO3a protein.
ChIP analysis showed that wt-SIRT3 cells have an increase in FOXO3a binding to upstream regulatory regions of gene promoters that each contain two canonical FOXO3a binding sites roughly 1 kb upstream of the transcription start site (supplemental Fig. S3). FOXO3a binding to both the MnSOD (Fig.3D, upper panel) and SCO2 (lower panel) promoters is increased in the wt-SIRT3, as compared to mt-SIRT3 cells. These experiments imply that SIRT3 may increase FOXO3a DNA-binding.
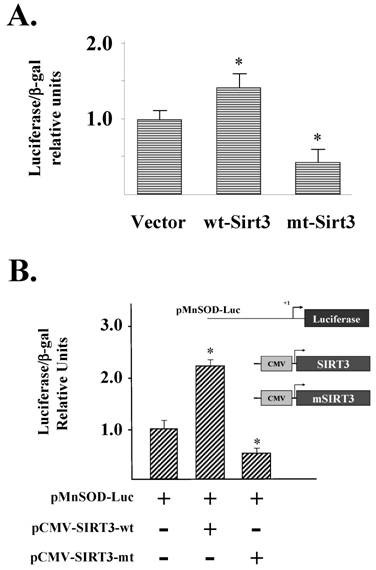
Overexpression of SIRT3 induced MnSOD
p3400-MnSOD-Luc was transfected into HCT116 cells genetically altered to over express either wild-type (wt-SIRT3) or a catalytically inactive deacetylation mutant (mt-SIRT3) SIRT3 gene (Supplemental Fig. S1). HCT116 cells were used since they have nearly undetectable SIRT3 protein (data not shown). These results showed an increase in luciferase activity in cells overexpressing the wild-type SIRT3 gene, as compared to the mutant gene (Fig. 4A). p3400-MnSOD-Luc was also co-transfected into HCT116 cells with either pCMV-SIRT3-wt or pCMV-SIRT3-mt SIRT3 expression vector. These transient transfection experiments showed that that SIRT3, but not the deacetylation mutant gene, induces MnSOD promoter activity (Fig. 4B).
Overexpression of SIRT3 induced MnSOD expression. (A) A MnSOD promoter luciferase reporter plasmid (pMnSOD-Luc) was transfected into wt-SIRT3, mt-SIRT, and vector control cell lines. Luciferase activity was normalized to β-galactosidase activity and presented as luciferase/β-galactosidase relative units. All results are the mean of at least three separate experiments. Error bars around data points represent one standard deviation about the arithmetic mean. (B) pMnSOD-Luc was co-transfected into HCT116 cells with either pCMV-SIRT3-wt or pCMV-SIRT3-mt expression vectors or luciferase assays were done as described above. * Indicates P < 0.05 by t-test.
SIRT3 alters intracellular superoxide levels
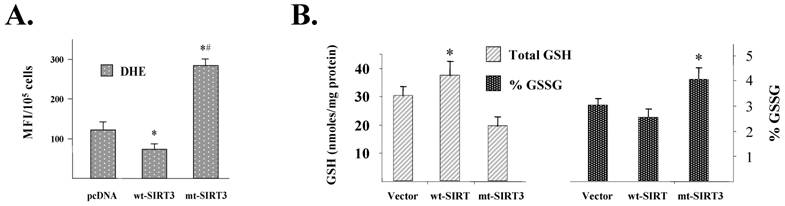
SIRT3 is localized in the inner mitochondrial membrane [21] along with the electron transport chain. The mitochondria are a major source of superoxide formation; and the accumulation of superoxide, from decreased election transport, is thought be a source of oxidative damage associated with degenerative diseases and aging [22]. Thus, it seemed logical to propose that one function of SIRT3 may be to mediate intracellular superoxide levels. HCT116 based wt-SIRT3 or mt-SIRT3 cells were isolated and intracellular superoxide levels were estimated by DHE oxidation [8]. These experiments showed that SIRT3 deacetylation mutant cells (mt-SIRT3) have significantly increased intracellular superoxide (O2●-) levels [8], relative to either vector control or wt-SIRT3 cells (Fig. 5A). Intracellular reduced (GSH) and oxidized glutathione (GSSG) concentrations were also altered in mutant mt-SIRT3 cells, relative to either vector control or wt-SIRT3 cells, consistent with a relatively oxidizing intracellular environment in the Sirt3 mutant expressing cells (Fig. 5B).
SIRT3 expression alters intracellular superoxide levels. (A) Altered steady-state levels of superoxide as shown by increased oxidation of DHE in HCT116 SIRT3 overexpressing cells. Control (pcDNA vector), wild-type SIRT3 (wt-SIRT3), or a deacetylation SIRT3 mutant (mut-SIRT3) cell lines were analyzed by flow cytometry for the amount of hydrolyzed DHE per 10,000 cells represented as Mean Florescent Intensity (MFI). Each bar represents the average of three separate experiments. Error bars indicate standard error. * Indicates P < 0.05 by t-test. (B) SIRT3 overexpressing cells contain increased GSH/GSSG redox ratios. Total levels of glutathione (GSH) was determined and normalized to total protein (left panel). GSSG/GSH ratios were determined and represented as % GSSG (right panel). Error bars represent of standard deviation of three independent experiments. * Indicates P < 0.05 by t-test.
DISCUSSION
In this study, we suggest that FOXO3a is a mitochondrial protein and its function may be regulated by a physical interaction with SIRT3 in the mitochondria. This idea is based on the observations that: (1) FOXO3a is present in the mitochondria and physically interacts with SIRT3; (2) SIRT3 increases the DNA-binding of FOXO3a at two promoters resulting in an increase in gene expression; and (3) altered SIRT3 function results in altered and increased intracellular O2●-. The results of these experiments all support the hypothesis that SIRT3 may be a mitochondrial surveillance factor that detects metabolic imbalances involving O2•- production and induces specific pathways to respond to these conditions.
While it is tempting to suggest that SIRT3 is initiating a signaling pathway in the mitochondria and sending it to the nucleus to activate gene expression via FOXO3a our results do not confirm this specific idea and the experimental assays to validate this idea do not exist. However, one possibility is that SIRT3 is both a mitochondrial and a cytoplasmic protein. Several manuscripts have been published suggest that SIRT3 is a mitochondrial protein [21,22] while others imply a more complex cellular localization pattern [24,25]. In our two model systems using HCT116 and SIRT3 knockout derived MEFs, SIRT3 appears present only in the mitochondria. However, this does not rule out a small amount of the protein might be in either the cytoplasm or nucleus.
Several previous publications suggest that SIRT3 may play a role in the regulation of mitochondrial metabolism [26] as well as acetyl-CoA synthetase acetylation [4]. It also appears that SIRT3 is the primary mammalian Sir2 homolog that regulates global mitochondrial lysine acetylation [1]. However, the mechanism and physiological function of SIRT3 has not been determined. The results presented here, when coupled with others, suggest that at least one function of SIRT3 serves as a sensor of small reactive oxygen molecules and initiates specific cellular signaling pathways. In this model the induction of SCO2 and MnSOD would be an appropriate response to detoxification O2•-.
The mitochondria are key organelles that maintain multiple cellular processes and decreases in function are critical to age-related diseases such as diabetes, neurodegeneration, and carcinogenesis [27]. SIRT3 is located near the electron transport chain [28] and is at an ideal location to function as a guardian gene against the accumulation of small reactive molecules that might induce cellular damages. Mitochondria are a major source of superoxide formed by the reduction of oxygen during electron transport. The oxidative damage caused by the reactive oxygen species derived from superoxide is believed to be a major cause of degenerative diseases associated with aging [22]. However, it has also been suggested that that physiological levels of O2•- and H2O2 production are essential for normal biological signaling processes leading to the attainment of a normal lifespan [26]. Thus, these results suggest a potential mechanism where the normal production of reactive oxygen species, such as O2●-, induces specific pathways that act to repair and, perhaps more importantly, to renew organelle functions.
On the other hand, the induction of proteins such as SCO2, should decrease O2•- levels indirectly by increasing the efficiency of the four electron reduction of O2. SCO2 is a mitochondrial membrane-bound protein involved in copper supply for the assembly of cytochrome c oxidase (CcO) [29]. CcO catalyzes the four-electron reduction of molecular oxygen to H2O and couples this reduction with proton translocation across the inner membrane to improve mitochondrial respiration efficiency [30]. CcO increases the efficiency of four electron transfer to O2 to 2H2O in Complex IV and as such, reduces the residence time of electrons at upstream sites capable of mediating one electron reductions of O2. Thus, the induction of SCO2 could theoretically decrease O2•- production at sites in the electron transport chains upstream of Complex IV (Complexes I, II, and III).
This work also suggests that there may be mitochondrial surveillance genes that sense organelle distress and pass signals to the nucleus to initiate the expression of nuclear genes to prevent or repair any functional damage to mitochondria. While no direct mechanism is presented for how this signaling is transduced out of the mitochondria, it does seem logical that such a reparative pathway would exist and has been proposed by others [27]. Finally, since SIRT3 is the primary deacetylase in the mitochondria [1] we would propose that deacetylation, perhaps induced by nutrient deprivation or oxidative stress, may activate families of evolutionary conserved reparative genes.
Supplementary Material
Supplemental Figures S1-S3
Acknowledgements
This research was supported (in part) by the Intramural Research Program of the NIH, NCI, and CCR and NIH R01-CA100045, NIH P01-CA66081, NIH F32-CA110611. DRS is supported by grants from the National Institutes of Health (R01 HL51469, P01 CA66081, and P20 CA91709). We thank Melissa Stauffer, PhD, of Scientific Editing Solutions, for editorial assistance.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Lombard DB, Alt FW, Cheng HL. et al. Mammalian Sir2 Homolog SIRT3 Regulates Global Mitochondrial Lysine Acetylation. Mol Cell Biol. 2007;27:8807-8814
2. Kaeberlein M, Jegalian B, McVey M. AGEID: a database of aging genes and interventions. Mechanisms of ageing and development. 2002;123(8):1115-9
3. Guarente L. Calorie restriction and SIR2 genes--towards a mechanism. Mechanisms of ageing and development. 2005;126(9):923-8
4. Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006;103(27):10224-9
5. Okawara M, Katsuki H, Kurimoto E, Shibata H, Kume T, Akaike A. Resveratrol protects dopaminergic neurons in midbrain slice culture from multiple insults. Biochem Pharmacol. 2007;73(4):550-60
6. Hall DM, Sattler GL, Sattler CA. et al. Aging lowers steady-state antioxidant enzyme and stress protein expression in primary hepatocytes. The journals of gerontology. 2001;56(6):B259-67
7. Ishii N, Fujii M, Hartman PS. et al. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature. 1998;394(6694):694-7
8. Slane BG, Aykin-Burns N, Smith BJ. et al. Mutation of succinate dehydrogenase subunit C results in increased O2•-, oxidative stress, and genomic instability. Cancer Res. 2006;66(15):7615-20
9. Lin K, Hsin H, Libina N, Kenyon C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nature genetics. 2001;28(2):139-45
10. Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003;115(4):489-502
11. Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem Sci. 2002;27(7):352-60
12. Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117(4):421-6
13. Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2006;5:630-634
14. Guarente L. Mitochondria--a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132(2):171-6
15. Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial Sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. 2005;280(14):13560-7
16. Karimpour S, Lou J, Lin LL. et al. Thioredoxin reductase regulates AP-1 activity as well as thioredoxin nuclear localization via active cysteines in response to ionizing radiation. Oncogene. 2002;21(41):6317-27
17. Gius D, Cui H, Bradbury CM. et al. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer cell. 2004;6(4):361-71
18. Spitz DR, Sim JE, Ridnour LA, Galoforo SS, Lee YJ. Glucose deprivation-induced oxidative stress in human tumor cells. A fundamental defect in metabolism? Ann N Y Acad Sci. 2000;899:349-62
19. Pinkston-Gosse J, Kenyon C. DAF-16/FOXO targets genes that regulate tumor growth in Caenorhabditis elegans. Nature genetics. 2002;1:299-305
20. Kops GJ, Dansen TB, Polderman PE. et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419(6904):316-21
21. Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci U S A. 2002;99(21):13653-8
22. Brand MD, Buckingham JA, Esteves TC. et al. Mitochondrial superoxide and aging: uncoupling-protein activity and superoxide production. Biochemical Society symposium. 2004;71:203-13
23. Hallows WC, Albaugh BN, Denu JM. Where in the cell is SIRT3?--functional localization of an NAD+-dependent protein deacetylase. Biochem J. 2008;411(2):e11-3
24. Scher MB, Vaquero A, Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007;21(8):920-8
25. Nakamura Y, Ogura M, Tanaka D, Inagaki N. Localization of mouse mitochondrial SIRT proteins: shift of SIRT3 to nucleus by co-expression with SIRT5. Biochem Biophys Res Commun. 2008;366(1):174-9
26. Afanas'Ev IB. Mechanism of superoxide-mediated damage relevance to mitochondrial aging. Ann N Y Acad Sci. 2004;1019:343-5
27. Singh KK. Mitochondria damage checkpoint, aging, and cancer. Ann N Y Acad Sci. 2006;1067:182-90
28. Cui H, Onyango P, Brandenburg S, Wu Y, Hsieh CL, Feinberg AP. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 2002;62(22):6442-6
29. Leary SC, Cobine PA, Kaufman BA. et al. The human cytochrome c oxidase assembly factors SCO1 and SCO2 have regulatory roles in the maintenance of cellular copper homeostasis. Cell metabolism. 2007;5(1):9-20
30. Matoba S, Kang JG, Patino WD. et al. p53 regulates mitochondrial respiration. Science. 2006;312(5780):1650-3
Author contact
![]() Correspondence to: David Gius, M.D., Ph.D., Chief, Molecular Radiation Oncology, National Cancer Institute, National Institutes of Health, 9000 Rockville Pike, Bethesda, MD 20892. Tel: (301) 435-9411; E-mail: giusdnih.gov
Correspondence to: David Gius, M.D., Ph.D., Chief, Molecular Radiation Oncology, National Cancer Institute, National Institutes of Health, 9000 Rockville Pike, Bethesda, MD 20892. Tel: (301) 435-9411; E-mail: giusdnih.gov