Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
BRCA1 in transcriptional...
BRCA1 and Estrogen Receptor
BRCA1 and Progesterone Receptor...
BRCA1 and Estrogen Biosynthesis
The Interplay of BRCA1 and...
An Added Note
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(1):20-27. doi:10.7150/ijbs.5.20 This issue Cite
Review
BRCA1, Hormone, and Tissue-Specific Tumor Suppression
Yanfen Hu ![]()
Department of Molecular Medicine/Institute of Biotechnology, University of Texas Health Science Center at San Antonio, San Antonio, Texas 78245, USA
Received 2008-11-3; Accepted 2008-12-9; Published 2008-12-13
Abstract
Germline mutations of BRCA1 predispose women to breast and ovarian cancers. Elucidating molecular mechanism of tissue- and gender-specific phenomena in BRCA1-related tumors is a key to our understanding of BRCA1 function in tumor suppression. This review summarizes studies in recent years on the link between BRCA1 and estrogen/progesterone signaling pathways, as well as discusses various models underscoring a triangle relationship among BRCA1, estrogen and genome instability.
Keywords: BRCA1, tumor suppression, estrogen, genome instability.
Introduction
Breast and ovarian cancer susceptibility gene BRCA1 was initially proposed by Mary-Claire King through linkage analysis in large cohorts of hereditary breast cancer families [1] and was subsequently cloned in 1994 by Mark Skolnick and his colleagues[2]. Mutations in BRCA1 account for about half of the hereditary form of breast cancer and 80-90% of hereditary breast-ovarian cancers [2, 3]. Genetic testing in existing clinical samples and newly registered breast cancer families quickly revealed several distinct features about BRCA1. First, BRCA1 is clearly a tumor suppressor as loss of heterozygosity (LOH) in tumor samples from BRCA1-related breast/ovarian cancer invariably resulted in the loss of the wild type (WT) copy of BRCA1 and retention of the inherited mutant copy. Second, BRCA1 exerts its tumor suppression function in a gender- and tissue-specific manner. Germline mutations of BRCA1 predominantly lead to breast and/or ovarian cancer in women. Third, unlike mutations of the classic tumor suppressor p53 that are associated with both hereditary and sporadic cancers, somatic mutations in the BRCA1 coding region are rarely associated with sporadic breast or ovarian cancer.
A wealth of information from the research in the last two decades on the classical tumor suppressors such as p53 and Rb has facilitated mechanistic studies of BRCA1 in tumor suppression. Indeed, there is a striking functional similarity between BRCA1 and p53. A large body of evidence indicates that BRCA1 plays a pivotal role in DNA damage response including DNA repair and cell cycle checkpoint control [4-7]. Although the exact role of BRCA1 in DNA repair remains to be elucidated, the overwhelming evidence in the literature supports the notion that BRCA1 contributes to the maintenance of genome stability mainly through its function in DNA repair. The function of BRCA1 in DNA damage response provides a reasonable molecular explanation for its role as a tumor suppressor. Compromised functions of BRCA1 in DNA repair and cell cycle checkpoint likely contribute in a significant manner to cancer susceptibility. However, BRCA1 is ubiquitously expressed; and loss of BRCA1 function in maintenance of genetic stability, a cellular function that is fundamental and universally important to all cell types in both genders, cannot easily explain why it would increase cancer risks in such a gender- and tissue-specific manner. Furthermore, it is not clear why somatic mutations of BRCA1, which are expected to lead to genome instability, are rarely found in sporadic cancers [8].
The studies of BRCA1 functions in DNA repair and genome stability have dominated the BRCA1 field since its discovery. In comparison, understanding of the intriguing tissue- and gender-specificity associated with BRCA1 tumor suppression is a less traveled road. While the role of estrogen in sporadic breast cancer development has been widely studied, the potential interplay between BRCA1 and the hormone synthesis and actions had been surprisingly under-appreciated and under-investigated in BRCA1 field. Nevertheless, the striking restriction of BRCA1-related tumors to the major hormone-responsive tissues begs the question as to whether the tumor suppressor function of BRCA1 is linked to the hormone homeostasis in the breast and ovaries. In addition, epidemiological studies have demonstrated that prophylactic oophorectomy in women who carry BRCA1 mutations prevents occurrence and reoccurrence of breast cancer by 75%[9, 10], further supporting the involvement of hormone factors in BRCA1-related tumor development. In this review, I will briefly summarize the published studies that attempted to address the BRCA1 function in hormone signaling and regulation. I will discuss the challenges in tackling tissue-specific tumor suppressors and speculate on the underlying mechanisms for tissue-specific tumor suppression in general.
BRCA1 in transcriptional regulation and tumor suppression
BRCA1 is a multi-functional protein. BRCA1 has been implicated in transcriptional regulation in addition to DNA repair and cell cycle checkpoint [11-13]. When fused to a heterologous DNA binding domain, the BRCA1 C-terminus confers transcriptional activity in reporter assays [14-16]. BRCA1 has also been shown to complex with RNA polymerase II holo-enzyme and a number of well-characterized transcription factors [13, 17]. When ectopically expressed, BRCA1 has been shown to regulate expression of multiple endogenous genes. In a cell-free system, BRCA1 is capable of both stimulating and repressing transcription in an in vitro transcription assay[18, 19]. Although the fact that BRCA1 is not a sequence-specific binding protein compounded the challenge of studying its function in transcription, circumstantial evidence supports that BRCA1 is involved in transcriptional regulation. The critical question is: Is transcription function of BRCA1 related to its tumor-suppressor function, or is change in gene transcription just an innocent bystander in BRCA1-initiated tumor development?
An inclusive view is that both DNA repair and transcriptional functions of BRCA1 contribute to its role in tumor suppression. Loss of BRCA1 function in DNA repair leads to genomic instability and, eventually, cancer. However, such effect alone would unlikely be tissue-specific. On the other hand, transcriptional regulation by BRCA1 could be tissue- or cell type-specific, which could exacerbate the genomic instability in certain tissues and, therefore, increase the penetrance of tumor development in these tissues. In other words, the combined loss of function in both DNA repair and transcription could lead to tissue-specific tumors. While deregulation of multiple BRCA1 target genes identified so far [20-25] could contribute to tumor development, I will focus on the published results that shed light on a link between BRCA1 and hormone-dependent transcription.
BRCA1 and Estrogen Receptor
The connection between BRCA1 and the hormone signaling pathway was first brought to light by Eliot Rosen in 1999[26], whose group reported that ectopically expressed BRCA1 inhibits signaling by the ligand-activated estrogen receptor (ER-α) in an estrogen responsive reporter assay. Their follow-up studies demonstrated that BRCA1 also blocks the expression of two endogenous estrogen-responsive genes, pS2 and cathepsin D [27]. Furthermore, genome-wide microarray analyses revealed a large number of estrogen-responsive genes whose expression can be modulated by exogenous BRCA1[28]. The negative regulation of ER-α activity by BRCA1 results from either direct interaction between the BRCA1 N-terminus (aa 67-100 and 101-133) and the ligand-binding domain/activation function-2 (LBD/AF-2) region of ER-α, or BRCA1 down-regulation of p300, a nuclear receptor cofactor [27, 29]. The initial observation is supported by findings from other investigators as well [30]. It has been shown that BRCA1 also mediates the ligand-independent repression of ER-α [31].
The BRCA1 function in repression of ER-α transcriptional activity provides a compelling explanation for the tissue- and gender-specific phenomenon in BRCA1-related tumors. A conundrum of this model is that BRCA1-derived breast cancer cells are ER-α negative. It has been suggested (model #1) that regulation of ER-α function by BRCA1 exerts its anti-proliferation effect indirectly on potential ERα- tumor cells through paracrine pathways from neighboring ERα+ cells. Alternatively (model #2), the ERα- breast cancer cells may derive from ERα+ cells. In support of this notion, Chu-Xia Deng's group reported that mice carrying conditional Brca1 knockout in their mammary gland developed mammary gland tumors that were ERα+ at early stages, but became ERα- at later stages [32]. It is conceivable that genomic instability of human breast cancer cells in earlier stages would eventually lead to estrogen-independent proliferation of the cancer cells. These ERα- cancer cells would ultimately comprise the entire tumor population, as they have a significant growth advantage over hormone-dependent cells. Deng's mouse model explains why ER-α could still play a role in ERα- breast cancer initiation and progression. It remains to be addressed why the tumor cells are almost exclusively ERα- cells and why very few ERα+ tumors derive from this pathway. This is in stark contrast to sporadic breast cancers, most of which are ERα+, where estrogen also plays a pivotal role. In fact, animal studies showed that activation of the estrogen signaling pathway collaborates with loss of Brca1 to promote both ERα- and ERα+ mammary tumor development [33], suggesting that ERα+ tumor cells would not be excluded by loss of mouse Brca1. The answer to the ERα- nature of BRCA1-related breast cancer may lie in another interplay of BRCA1 and hormone action. It has been reported that ER-α activates BRCA1 expression [34-36](Fig.1). Therefore, in ERα+ cells, the elevated BRCA1 would provide additional protection in DNA damage response and genome stability maintenance. The benefits from ER-α associated genome stability cancels the slight risk from ER-α mediated proliferation. In contrast, ERα- cells have no added genome protection from ERα-mediated BRCA1 elevation. This model suggests that ERα- cells have the advantage in cancer initiation. The third model is that BRCA1 activates ER-α expression, proposed by Hosey et al. [37](Fig.1). As a result, loss of BRCA1 in precancerous cells would lead to genome instability as well as lack of ER-α expression. Combined with Deng's model, Hosey's result suggests a possibility that ERα- is a result of cancer progression, rather than the cause for initiation. Interestingly, Max Wicha's group reported that BRCA1 is required for the differentiation of ERα- stem/progenitor cells to ERα+ luminal cells [38]. They hypothesize that BRCA1-mediated genome instability in mammary stem cells ultimately leads to breast cancer in women carrying germline mutations of BRCA1. In this context, absence of ERα could be a byproduct of cells lacking functional BRCA1 (consistent with Hosey's finding that BRCA1 is a positive regulator of ERα expression), but not a pre-requisite for tumor development.
The interplay between ERα and BRCA1 pathways and its impact on cell proliferation (1) and genome stability maintenance (2). Solid arrows indicate a direct role while the dashed arrow indicates an indirect pathway (e.g. via a paracrine pathway etc.)
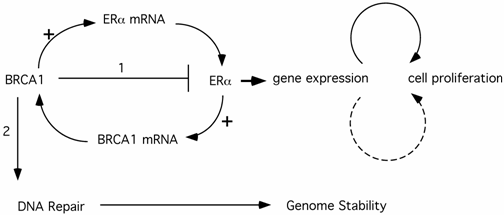
The emerging picture shows that the crosstalk between BRCA1 and ER-α pathways exerts a complicated network on cell proliferation and genome stability. Although the complete picture is lacking, it is likely that the combination of agonistic and antagonistic actions of hormone somehow results in preferential development of breast cancer in ERα- cells.
BRCA1 and Progesterone Receptor (PR), Androgen Receptor (AR) Pathways
In addition to estrogen, the progesterone-mediated signaling pathway also contributes to breast cancer development. The crosstalk between estrogen and progesterone pathways may have synergistic effect on breast cancer initiation and progression, as hormone replacement therapy (HRT) with combined estrogen and progesterone increased breast cancer risk in postmenopausal women [39-41]. Research from both cell culture and animal models suggests that BRCA1 negatively modulates the progesterone receptor signaling pathway. Since progesterone receptors (PRA and PRB) are also target genes that are up-regulated by ER-α, one might predict that BRCA1 represses the PR pathway by inhibiting ER-α activity. This indirect mechanism might contribute to the inverse relationship between BRCA1 expression and PR activity in tissue culture and animal models. However, there is more to the story. Eliot Rosen's group reported that BRCA1 directly interacts with PRA or PRB to repress their transcription activity on luciferase reporters as well as endogenous progesterone-responsive genes [42]. They show that absence of functional BRCA1 sensitizes progesterone signaling in both T47D cells and mouse mammary gland, presumably due to removal of repression normally mediated by BRCA1. In an independent study, Eva Lee's group demonstrated that BRCA1 down-regulates PR protein level by affecting PR protein stability [43]. This is due to the presence of wild type BRCA1 that leads to polyubiquitination and degradation of PR. More importantly, treatment of Brca1/p53-deficient mice with progesterone antagonist mifepristone (RU 486) prevented mammary tumorigenesis, providing additional in vivo evidence of the involvement of the progesterone signaling pathway in BRCA1-related mammary tumor development.
The epidemiological data regarding PR expression and BRCA1 status is limited and controversial at this point. One study reported that BRCA1 mutation carriers have reduced PR expression and, even strikingly, lack of expression of the PRB isoform in prophylactically removed normal breast tissue [44]. Another study reported that PR expression is increased in the benign epithelium of BRCA1-related breast cancers [45]. The discrepancy could be due to the intrinsic difference between normal tissue and benign cells surrounding the cancer cells. Alternatively, it could be due to the fact that each study used relatively small sample numbers.
The molecular mechanism of BRCA1 on PR activity shares some similarities to that of BRCA1 on ER-α activity. This mechanism also shares the same conundrum that BRCA1-related cancers tend to be both ERα- and PR-negative. While the same argument that helped explain ERα action can be used to explain PR action, this is certainly a paradox remained to be experimentally explored in the future.
Intriguingly, there has been one report that BRCA1 increases AR transcription activity, although its significance to breast cancer development remains to be elucidated [46].
BRCA1 and Estrogen Biosynthesis
Estrogen signaling plays a significant role in the development and progression of breast cancer. The research on estrogen receptors has provided tremendous insight into our understanding of estrogen signaling and functions. Ablation of ER function by selective ER modulators such as tamoxifen has proven to be highly efficacious in breast cancer treatment. At the same time, the clinical importance of estrogen biosynthesis is underscored by the great success of aromatase inhibitors (AI) in breast cancer treatment. Aromatase, a key enzyme in estrogen biosynthesis, catalyzes the last and rate-limiting step in estrogen biosynthesis that converts androgen to estrogen [47] [48]. In recent years, aromatase inhibitors have demonstrated greater clinical efficacy in treating ERα+ postmenopausal breast cancer. Interestingly, several groups made unexpected connection between BRCA1 and aromatase.
It has been well established that circulating estrogen level in pre-menopausal women is dictated by aromatase expressed in ovarian granulosa cells, whereas in many post-menopausal breast cancer and endometrial malignancy tissues, the aromatase produced in situ is significantly elevated [49, 50]. Expression of aromatase in multiple tissues is controlled by tissue-specific promoters and alternative splicing [51](Fig.2). Aromatase in ovarian granulosa cells is expressed mainly via the action of an ovary-specific promoter (PII). Intriguingly, the utilization of the aromatase promoter in many post-menopausal breast cancer and endometrial malignancy tissues is switched from the relatively weak adipose type promoter (I.4) to the more potent ovarian type promoter (PII). Thus, the ovary-specific PII promoter of aromatase is the primary determinant for both the circulating level of estrogen in pre-menopausal women and intratumoral aromatase expression in post-menopausal women.
The genomic locations of several tissue-specific promoters of the aromatase gene (CYP 19) and tissue-specific alternative splicing patterns of the aromatase transcripts.
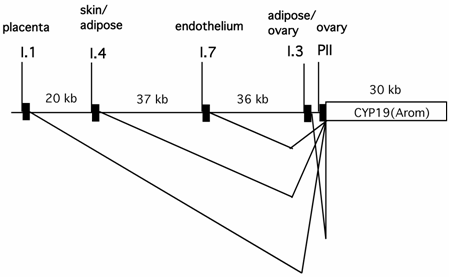
Ovarian expression of aromatase occurs in a cell type-specific and developmental stage-dependent manner. In response to the pituitary-released follicle stimulating hormone (FSH), aromatase expression is significantly increased in ovarian granulosa cells from follicles at the large antral/preovulatory (PO) stage [52]. FSH increases the intracellular cyclic AMP (cAMP) level, which ultimately activates the aromatase PII promoter via the combined action of the CREB family of transcription factors and steroidogenic factor 1 (SF-1).
In situ hybridization of ovaries from adult mice has detected very low levels of Brca1 mRNA in ovarian epithelial cells, the cell type from which BRCA1-associated ovarian cancer is thought to derive[34, 53]. Instead, Brca1 expression is predominantly follicular, with the highest levels of Brca1 mRNA in granulosa cells of developing follicles. Interestingly, ovarian expression of Brca1 was significantly decreased at the large antral/preovulatory (PO) stage [54], a follicular stage where aromatase expression is significantly increased[55]. Therefore, there appears to be an inverse correlation between Brca1 and aromatase expression under this physiological context, raising the possibility that there may be a causal relationship between these two events. These observations from mouse in situ hybridization raised an intriguing possibility of potential functional link between BRCA1 and aromatase expression in the same ovarian cell type.
Based on the above-mentioned observation, we hypothesized that BRCA1 negatively regulates aromatase expression from ovary-specific promoter PII, and ovarian granulosa cells containing inherited mutant BRCA1 would have elevated basal aromatase expression [56]. Using a human granulosa cell line that maintains physiological response to cAMP-mediated aromatase expression, we were able to recapitulate the inverse correlation between BRCA1 and aromatase expression level in a tissue culture model. When aromatase expression is induced in response to cAMP activation, the BRCA1 level is significantly reduced in granulosa cells cultured in vitro. Furthermore, knockdown of endogenous BRCA1 by small interfering RNA (siRNA) leads to elevated aromatase basal expression. More importantly, the increased aromatase expression results from elevated ovary-specific promoter activity. Although the molecular mechanism of BRCA1 regulation of the aromatase promoter is not fully understood, these data raised the possibility that BRCA1 mutation carriers might have increased basal circulating estrogen levels throughout their reproductive years, while their peak estrogen level during the menstrual cycle may not be affected, as there is no detectable BRCA1 expression (whether wild type or mutant) in granulosa cells at the preovulatory stage. It is this long-term elevated estrogen basal level that could potentially increase the breast cancer risk for women with germline mutations in BRCA1.
The notion that BRCA1 down-regulates the aromatase expression in ovarian granulosa cells is consistent with the clinical observation that prophylactic oophorectomy reduces the occurrence and re-occurrence of breast cancers in BRCA1 mutation carriers. However, the benefits of oophorectomy on breast cancer reduction could also be explained solely by deprivation of estrogen/progesterone on inhibiting breast cancer cell proliferation, as in the case of sporadic breast cancer that does not involve BRCA1 mutations. An independent study using conditional knockout mouse model provided strong in vivo evidence for a cell non-autonomous function of BRCA1 in ovarian granulosa cells in tumor suppression [57]. Using ovarian granulosa-specific Cre driven by the FSH receptor promoter, Dubeau's group showed that female mice with BRCA1 knockout specifically in ovarian granulosa cells developed benign tumors on the uterine horn and the ovaries. Importantly, these tumors all had wild type BRCA1, suggesting that lack of BRCA1 in ovarian granulosa cells leads to tumorigenesis in a paracrine manner.
It appears that BRCA1 not only regulates aromatase expression in ovarian granulosa cells, it also modulates the ovary-specific aromatase promoter in adipose tissues, as shown in primary adipose fibroblasts derived from either breast or abdominal tissues [58, 59]. It is conceivable that mutant BRCA1 contributes to elevated circulating estrogen level as well as in situ estrogen expression in breast tissues.
The notion that BRCA1 negatively regulates estrogen biosynthesis in non-epithelial cells also provides reasonable explanation for the gender-specificity of BRCA1 cancer and the rare association of somatic mutations of BRCA1 with sporadic breast cancer. If elevated estrogen production due to BRCA1 mutations in ovarian granulosa cells and adipose stromal cells constitutes a significant contributing factor in BRCA1-associated tumor development, male BRCA1 mutation carriers would obviously be exempt from such hormonal influence, despite presumably the same degree of predisposition to genetic instability as their female counterparts due to the BRCA1 mutation status. Likewise, somatic BRCA1 mutations within sporadic breast tumor cells, albeit increasing genetic instability of the tumor cells, are unlikely to enhance aromatase expression in ovaries or adipose tissue. This could explain why somatic mutations of BRCA1 are rarely found in sporadic breast cancer. In contrast, women with germline mutations of BRCA1 would have the elevated genetic instability in breast epithelial cells and increased estrogen production in non-epithelial cells from estrogen-producing tissues/organs, the combination of which could lead to tissue- and gender-specific cancer in BRCA1 mutation carriers.
The linkage between BRCA1 and estrogen biosynthesis also provides another angle with which to address the conundrum that BRCA1-related tumors are generally ER-α negative. In addition to stimulating cell proliferation mediated by ER-α, it has been suggested that estrogen exerts a direct tumorigenic effect due to the fact that estrogen metabolic products could serve as carcinogens that induce free radical-mediated DNA damage and genetic instability [60]. In other words, estrogen has an ER-independent role in tumorigenesis. This could render ERα-negative cells more vulnerable as they do not have the benefit of elevated BRCA1 to protect genome stability as do ERα-positive cells.
The Interplay of BRCA1 and Hormone Signaling
The emerging picture of BRCA1 in hormone signaling and action indicates that the pathway is not simple, linear, and unidirectional. As mentioned above, the estrogen/ER pathway induces BRCA1 expression. While BRCA1 represses ERα signaling and aromatase expression, it is also a positive regulator for ERα expression. The interplay between BRCA1 and hormone pathways likely maintain the balance of two opposing effects: tumorigenic or tumor suppression. When BRCA1 is mutated, the balance is tilted towards tumorigenic, especially in ERα-negative cells, which lack the beneficial outcome from ERα-mediated BRCA1 expression and subsequent genome stability protection.
The effect of hormone on BRCA1-related cancer development is certainly more complicated than we would like to believe. On one hand, oophorectomy significantly reduces the breast cancer risk in BRCA1 mutation carriers, strongly suggesting that hormones play a pivotal role in BRCA1-related tumor development, as in sporadic breast cancer. On the other hand, hormone (especially estrogen) levels seem to have the opposite effect on breast cancer occurrence in BRCA1 mutation carriers and in sporadic breast cancers. For instance, early pregnancy is known to reduce breast cancer risk in the general population, while BRCA1 mutation carriers are especially susceptible to breast cancer during pregnancy [61, 62]. This is consistent with the observation that increased exposure to estrogen has both beneficial and harmful effects on the breast in the general population depending on age. It has been proposed that elevated circulating estrogen levels play a duel role [63]. Estrogen increases breast cancer risk in postmenopausal women, because it stimulates the proliferation of precancerous cells that have accumulated genomic mutations in the breast tissues over years. In contrast, young women have not had the time to accumulate mutations in their cells and therefore, the protection from estrogen-induced BRCA1 expression and genome stability out-weighs the potential harm from estrogen-induced cell proliferation. In BRCA1 mutation carriers, however, the beneficial effect of estrogen is partially lost due to the presence of mutant BRCA1. As a result, elevated estrogen will only increase breast cancer risk, even in young women. Once again, this could be explained by tilting the balance of agonistic and antagonistic estrogen effects. [63]
The multi-functional role of BRCA1 in hormone expression and signaling has provided a starting point to address the tissue- and gender-specificity in BRCA1 related cancer, but, at the same time, it raises more questions: Why do BRCA1-associated tumors develop in some estrogen-responsive tissues (breast and ovary), but not others (endometrium etc)? Why do only about 70% of the BRCA1 mutation carriers develop breast and/or ovarian cancer by age of 70 years, while the rest (30%) are tumor free [3, 64, 65]? Why is the penetrance of BRCA1 mutation in this generation much higher than that in the pre-war generation [66]? Clearly, other factors in addition to hormone pathways influence the tumor development in BRCA1 mutation carriers. These factors may include tissue microenvironment, other variations/modifiers such as the genetic makeup and life-style/environment impact on the individual.
It is intrinsically more challenging to study tumor development in an endocrine/paracrine system, as compared to studying DNA repair function, which can be conducted in cells taken out of the context of organs/tissues. Tissue-culture-based mechanistic studies of BRCA1 function in breast tumor cells have provided tremendous insight into BRCA1 functions in the maintenance of genomic stability and modulation of ER activity. The intriguing link between BRCA1 and estrogen synthesis in non-epithelial cells highlights importance of understanding the tissue- and gender-specific tumor suppressor function of BRCA1 in a physiologically relevant context. Hopefully, with infusion of fresh ideas from inter-disciplinary fields and innovative research approaches, more light will be shed on this clinically important and intellectually interesting question.
An Added Note
Tissue-specific tumor suppressors are not limited to BRCA1. Other tumor suppressors, such as BRCA2, RB and APC, also display certain degree of tissue specificity or preference in tumor spectrum when LOH occurs. For example, LOH of APC leads to familial adenomatous polyposis coli, RB leads to retinoblastoma [67], while loss of these tumor suppressors in somatic cells lead to broader tumor spectra. In these cases, the difference in familial and sporadic tumor spectra may well be attributed to their transcription impact in vivo. i.e., loss of genome stability would lead to tumor occurrence in multiple tissues, as in the case of sporadic tumors resulting from somatic mutations of these genes. However, in germline mutation carriers, every cell carries the mutation and its impact on transcription varies tissue to tissues and cell type to cell type. This could render certain tissues more susceptible to tumor development. The transcription regulation could take place in situ (cell-specific gene expression in the same cells from which cancer derives) or in different cells (affect cancer cells via endocrine or paracrine pathways). One would predict that, in both cases, it would lead to tumor in preferred tissues, and in the second case, predominantly to familial cancer. In other words, the same concept of dual functions in both epithelial and non-epithelial cells could be potentially applied to the understanding of the tissue specificity or preference of tumors that arise from the loss of other tumor suppressors such as APC and Rb.
Acknowledgements
I thank Rong Li, Ahsan Choudary, Brian Allen and Sagar Ghosh for critical reading of the manuscript. I sincerely wish to apologize to the many colleagues whose important contributions were not able to be cited due to space constraints. YH is supported by R01CA118578.
Conflict of Interest
The author has declared that no conflict of interest exists.
References
1. Hall J.M. et al. Linkage of Early-Onset Familial Breast Cancer to Chromosome 17q21. Science. 1990;250:1684-1689
2. Miki Y. et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66-71
3. Ford D. et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am. J. Hum. Genet. 1998;62:676-689
4. Scully R, Livingston D.M. In search of the tumor-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408:429-432
5. Zheng L. et al. Lessons learned from BRCA1 and BRCA2. Oncogene. 2000;19(53):6159-75
6. Venkitaraman A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171-182
7. Jasin M. Homologous repair of DNA damage and tumorigenesis: The BRCA connection. Oncogene. 2002;21:8981-8993
8. Futreal P.A. et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994;266:120-122
9. Rebbeck T.R. et al. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. New Engl. J. Med. 2002;346:1616-1622
10. Kauff N.D. et al. Risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 mutation. N. Engl. J. Med. 2002;346:1609-1615
11. Monteiro A.N.A. BRCA1: exploring the links to transcription. TIBS. 2000;25:469-474
12. Starita L.M, Parvin J.D. The multiple nuclear functions of BRCA1:transcription, ubiquitination and DNA repair. Curr. Opin. Cell Biol. 2003;15:345-350
13. Lane T.F. BRCA1 and transcription. Cancer Biol. and Ther. 2004;3:75-80
14. Chapman M.S, Verma I.M. Transcriptional activation by BRCA1 [letter; comment]. Nature. 1996;382(6593):678-9
15. Monteiro A.N, August A, Hanafusa H. Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc Natl Acad Sci U S A. 1996;93(24):13595-9
16. Hu Y.-F. et al. Characterization of a novel trans-activation domain of BRCA1 that functions in concert with the BRCA1 C-terminal (BRCT) domain. J. Biol. Chem. 2000;275:40910-40915
17. Scully R. et al. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc Natl Acad Sci U S A. 1997;94(11):5605-10
18. Horwitz A.A, Sankaran S, Parvin J.D. Direct Stimulation of Transcription Initiation by BRCA1 Requires Both Its Amino and Carboxyl Termini. J Biol Chem. 2006;281(13):8317-8320
19. Horwitz A.A. et al. A mechanism for transcriptional repression dependent on the BRCA1 E3 ubiquitin ligase. Proc Natl Acad Sci U S A. 2007;104(16):6614-6619
20. Harkin D.P. et al. Induction of GADD45 and JNK/SAPK-dependent apoptosis following inducible expression of BRCA1. Cell. 1999;97:575-586
21. Ouchi T. et al. BRCA1 regulates p53-dependent gene expression. Proc Natl Acad Sci U S A. 1998;95:2302-2306
22. Zhang H. et al. BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene. 1998;16:1713-1721
23. Welcsh P.L. et al. BRCA1 transcriptionally regulates genes involved in breast tumorigenesis. Proc Natl Acad Sci U S A. 2002;99(11):7560-5
24. Wang R.H, Yu H, Deng C.X. A requirement for breast-cancer-associated gene 1 (BRCA1) in the spindle checkpoint. Proc Natl Acad Sci U S A. 2004;101(49):17108-13
25. Furuta S. et al. Removal of BRCA1/CtIP/ZBRK1 repressor complex on ANG1 promoter leads to accelerated mammary tumor growth contributed by prominent vasculature. Cancer Cell. 2006;10:13-24
26. Fan S. et al. BRCA1 inhibition of estrogen receptor signaling in transfected cells. Science. 1999;284(5418):1354-6
27. Fan S. et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene. 2001;20:77-87
28. Xu J, Fan S, Rosen E.M. Regulation of the Estrogen-Inducible Gene Expression Profile by the Breast Cancer Susceptibility Gene BRCA1. Endocrinology. 2005;146(4):2031-2047
29. Fan S. et al. p300 Modulates the BRCA1 Inhibition of Estrogen Receptor Activity. Cancer Research. 2002;62:141-151
30. Wang C. et al. Cyclin D1 Antagonizes BRCA1 Repression of Estrogen Receptor alpha Activity. Cancer Res. 2005;65(15):6557-6567
31. Zheng L. et al. BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc. Natl. Acad. Sci. USA. 2001;98:9587-9592
32. Li W. et al. A role of estrogen/ERalpha signaling in BRCA1-associated tissue-specific tumor formation. Oncogene. 2007;26:7204-7212
33. Jones L.P. et al. Activation of estrogen signaling pathways collaborates with loss of Brca1 to promote development of ERalpha-negative and ERalpha-positive mammary preneoplasia and cancer. Oncogene. 2008;27:794-802
34. Marquis S.T. et al. The developmental pattern of Brca1 expression implies a role in differentiation of the breast and other tissues. Nat Genet. 1995;11:17-26
35. Gudas J.M. et al. Hormone-dependent regulation of BRCA1 in human breast cancer cells. Cancer Res. 1995;55:4561-4565
36. Spillman M.A, Bowcock A. BRCA1 and BRCA2 mRNA levels are coordinately elevated in human breast cancer cells in response to estrogen. Oncogene. 1996;13:1639-1645
37. Hosey A.M. et al. Molecuar Basis for Estrogen Receptor alpha Deficiency in BRCA1-Linked Breast Cancer. J Natl Cancer Inst. 2007;99(22):1683-94
38. Liu S. et al. BRCA1 regulates human mammary stem/progenitor cell fate. Proc Natl Acad Sci U S A. 2008;105(5):1680-5
39. Rossouw J.E. et al. Risks and Benefits of Estrogen Plus Progestin in Healthy Postmenopausal Women. JAMA. 2002;288(3):321
40. Beral V, Banks E, Reeves G. Evidence from randomised trials on the long-term effects of hormone replacement therapy. the Lancet. 2002;360:942-944
41. Lee S. et al. Postmenopausal hormone therapy and breast cancer risk: The Multiethnic Cohort. Int. J. Cancer. 2006;118:1285-1291
42. Ma Y. et al. The Breast Cancer Susceptibility Gene BRCA1 Regulates Progesterone Receptor Signaling in Mammary Epithelial Cells. Molecuar Endocrinology. 2006;20(1):14-34
43. Poole A.J. et al. Prevention of Brca1-Mediated Mammary Tumorigenesis in Mice by a Progesterone Antagonist. Science. 2006;314:1467-1470
44. Mote P.A. et al. Germ-Line Mutations in BRCA1 or BRCA2 in the Normal Breast Are Associated with Altered Expression of Estrogen-Responsive Proteins and the Predominance of Progesterone Receptor A. Genes, Chromosomes & Cancer. 2004;39:236-248
45. King T.A. et al. Increased Progesterone Receptor Expression in Benign Epithelium of BRCA1-Related Breast Cancers. Cancer Res. 2004;64:5051-5053
46. Park J.J. et al. Breast Cancer Susceptibility Gene 1 (BRCA1) Is a Coactivator of the Androgen Receptor. Cancer Res. 2000;60:5946-5949
47. Simpson E.R, Davis S.R. Minireview: aromatase and the regulation of estrogen biosynthesis-some new perspectives. Endocrinol. 2001;142:4589-4594
48. Kamat A. et al. Mechanisms in tissue-specific regulation of estrogen biosynthesis in humans. Trends Endo. Metab. 2002;13:122-128
49. Sasano H, Harada N. Intratumoral aromatase in human breast, endometrial, and ovarian malignacies. Endocrine Rev. 1998;19:583-607
50. Chen S. et al. Modulation of aromatase expression in human breast tissue. J. Steroid Bioc. & Mol. Biol. 2001;79:35-40
51. Simpson E.R. et al. Aromatase-a brief overview. Annu. Rev. Physiol. 2002;64:93-127
52. Richards J.S. Hormonal Control of Gene Expression in the Ovary. Endo. Reviews. 1994;15:725-751
53. Rajan J.V. et al. Developmental expression of Brca2 colocalizes with Brca1 and is associated with proliferation and differentiation in multiple tissues. Dev Biol. 1997;184(2):385-401
54. Phillips K.W. et al. Brca1 is expressed independently of hormonal stimulation in the mouse ovary. Lab. Inves. 1997;76:419-425
55. Turner K.J. et al. Development and validation of a new monoclonal antibody to mammalian aromatase. J. Endocrinol. 2002;172:21-30
56. Hu Y. et al. Modulation of aromatase expression by BRCA1: a possible link to tissue-specific tumor suppression. Oncogene. 2005;24:8343-8348
57. Chodankar R. et al. Cell-nonautonomous induction of ovarian and uterine serous cystadenomas in mice lacking a functional Brca1 in ovarian granulosa cells. Curr Biol. 2005;15(6):561-5
58. Lu M. et al. BRCA1 negatively regulates the cancer-associated aromatase promoters I. 3 and II in breast adipose fibroblasts and malignant epithelial cells. J. Clin Endocrinol. Metab. 2006;91(11):4514-9
59. Ghosh S. et al. Tumor suppressor BRCA1 inhibits a breast cancer-associated promoter of the aromatase gene (CYP19) in human adipose stromal cells. Am. J. Physiol. Endocrinol. Metab. 2007;292:246-252
60. Liehr J. Is estradiol a genotoxic mutagenic carcinogen? Endocr. Rev. 2000;21:40-54
61. Johannsson O. et al. Pregnancy-associated breast cancer in BRCA1 and BRCA2 germline mutation carriers. Lancet. 1998;352:1359-1360
62. Jernstrom H. et al. Pregnancy and risk of breast cancer in carriers of BRCA1 and BRCA2. Lancet. 1999;354:1846-1850
63. Hilakivi-Clarke L. Estrogens, BRCA1, and Breast Cancer. Cancer Res. 2000;60:4993-5001
64. King M. et al. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643-6
65. Easton D.F. et al. Breast cancer risks for BRCA1/2 carriers. Science. 2004;306:2187-2191
66. King M.C, Marks J.H, Mandell J.B. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302(5645):643-6
67. Weinberg R.A. The Biology of Cancer. US: Garland Science. 2006
Author contact
![]() Correspondence to: Dr. Yanfen Hu, Fax: 210-567-7324; Tel: 210-567-7216; Email: huy3edu
Correspondence to: Dr. Yanfen Hu, Fax: 210-567-7324; Tel: 210-567-7216; Email: huy3edu