Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Regulation of the cell cycle...
p38 MAPK and survival
Remarks
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(1):44-52. doi:10.7150/ijbs.5.44 This issue Cite
Review
Non-Classical P38 Map Kinase Functions: Cell Cycle Checkpoints and Survival
Tina M. Thornton, Mercedes Rincon ![]()
Department of Medicine/Immunobiology Program, University of Vermont, Burlington, Vermont 05405-0068, USA.
Received 2008-11-7; Accepted 2008-12-15; Published 2008-12-19
Abstract
The p38 MAPK kinase pathway is activated in response to a wide range of cellular stress stimuli and cytokines. Our understanding of the important functions of p38 MAPK in the process of differentiation and cell death has grown considerably in the recent years and is now relatively established. Here we discuss the role of p38 MAPK in the mediation of cell cycle checkpoints and cell survival, processes that have received less attention. We describe how p38 MAPK regulates both the G2/M as well as a G1/S cell cycle checkpoint in response to cellular stress such as DNA damage. While p38 MAPK has classically been associated with the induction of apoptosis, we discuss that p38 MAPK can also mediate cell survival in specific situations, such as in response to DNA damage. It is important to recognize these less appreciated functions of p38 MAPK when considering the potential use of pharmacological inhibitors of p38 MAPK in therapeutic treatments for disease.
Keywords: Cell Cycle Checkpoint, Survival, p38 MAPK
Introduction
The family of mitogen activated protein kinases (MAPKs) is composed of three major groups: the extracellular regulated kinases (ERKs), the C-Jun N-terminal Kinases (JNKs) and the p38 MAPKs [1]. The p38 MAPK group consists of four members: p38α, p38β, p38γ and p38δ. These proteins are encoded by separate genes and are approximately 60% identical at the amino acid level. Based on their expression pattern, substrate specificity and sensitivity to pharmalogical inhibitors, they can be divided into two groups: p38α and p38β on the one hand and p38γ and p38δ on the other [2]. While p38α and p38β are universally expressed, p38γ and p38δ appear to have a more tissue specific expression pattern. p38γ is most abundant in skeletal muscle while p38δ is highly expressed in testes, pancreas, kidney and small intestine. All isoforms phosphorylate the Ser-Pro or Thr-Pro MAPK consensus motifs, but some substrate selectivity has been reported. For example, the microtubule associated protein Tau is a better substrate for p38γ and p38δ, while MAPKAP kinase-2 (MK2) and MK3 are better substrates for p38α and p38β. The commonly used p38 MAPK inhibitor SB203580 specifically inhibits p38α and p38β, but does inhibit p38γ and p38δ activities.
Loss of p38β, p38γ or p38δ does not perturb normal development indicating that there is some redundancy in function for the family members. However disruption of the p38α gene results in embryonic death because of placental defects demonstrating that some functions cannot be compensated by other family members[3-6]. p38α is the best characterized family member and will be referred to as p38 MAPK and other family members will be specifically designated when discussed.
In response to cellular stress and cytokines p38 MAPK is activated by phosphorylation of the Thr-Gly-Tyr (TGY) dual phosphorylation motif. MAPK kinases (MKK) 3 and MKK6 are the primary upstream activators of p38 MAPK. In response to certain stimuli, MKK4 has also been shown to activate p38 MAPK but only in specific situations. A variety of upstream MAPK Kinase Kinases phosphorylate (MKKKs) are known to phosphorylate and activate the more specific MKKs [7]. p38 MAPK can also be activated independently of the MKKK/MKK cascade by autophosphorylation [8-10].
The role of the p38 MAPK pathway in the cell differentiation, growth inhibition and apoptosis is well established and has been previously reviewed [11, 12]. Based on the role of p38 MAPK in these processes, p38 MAPK has been defined as a tumor suppressor. The focus of this review is on the less characterized functions of the p38 MAPK pathway, such as its role in the regulation of cell cycle checkpoints and cell survival.
Regulation of the cell cycle checkpoints by p38 MAPK
In eukaryotic cells, there are evolutionarily conserved pathways that sense and respond to various types of cellular stress via a series of checkpoints that delay the progression to the next phase of the cell cycle. In response to stress stimuli that cause DNA damage, cells undergo cell cycle checkpoints to allow time for DNA repair. Two major cell cycle checkpoints have been characterized, a G1/S checkpoint including both prior to and during DNA synthesis and a G2/M checkpoint prior to cell division [13]. The p38 MAPK pathway is involved in the induction of both checkpoints although its role in the G2/M checkpoint is better established.
The role of p38 MAPK in the G2/M cell cycle checkpoint
Exposure to ultraviolet light (UV), γ-irradiation and chemotherapeutic drugs such as etoposide results in the generation of DNA double strand breaks (DSBs). In response to DSBs, p38 MAPK is activated and leads to the establishment of a G2/M cell cycle checkpoint [14, 15]. The mechanism by which DNA DSBs activate p38 MAPK is not completely clear. p38 MAPK activation in response to DNA DSBs has been reported to be dependent on the activation of Ser/Thr protein kinases that serve as DNA damage sensors, specifically the ataxia telangiectasia-mutated (ATM) and ataxia telangiectasia-mutated and Rad3-related (ATR) kinases [16]. However while the activation of ATM may be required for the activation of p38 MAPK, ATM does not appear to be able to directly phosphorylate p38 MAPK. Indeed the consensus motif recognized by ATM is not present in the p38 family members. Instead activation of p38 MAPK appears to be mediated by the Thousand and one (Tao) kinases which are directly activated by ATM. Tao kinases are one of the multiple MAPKKK that can activate MKK3/6 [17]. It should be noted that alternative mechanisms to the ATM/ATR pathway can also activate p38 MAPK because activation of p38 MAPK can occur in the presence of the ATM inhibitor caffeine and in ATM knockout cell lines in response to topoisomerase II inhibitors and UV [15, 16]. Thus, it appears that in addition to the ATM-dependent pathway other not yet established mechanisms activate p38 MAPK in response to DNA damage. The specific pathways connecting DNA damage to p38 MAPK activation may vary by cell type, the specific DNA damage stimuli and the extent of DNA damage.
Regarding the mechanism by which p38 MAPK contributes to the induction of the G2/M cell cycle checkpoint two pathways have been described (Fig.1). p38 MAPK phosphorylates and activates p53 and this leads to the induction of a p53-dependent G2/M checkpoint [18-21]. p38 MAPK can phosphorylate mouse p53 at Ser18 and Ser389 (human Ser15 and Ser392, respectively). Phosphorylation of these residues results in the dissociation of p53 from Murine double minute 2 (Mdm2), a protein that binds to the transactivation domain of p53 and promotes p53 ubiquitination and degradation. Disruption of this interaction promotes the accumulation of p53. p38 MAPK also associates with Growth arrest and DNA damage inducible 45α (Gadd45α) proteins and promotes their association with p53 further increasing p53 stability [22, 23]. The activation and stabilization of p53 by p38 MAPK leads to the transcription of p53-target genes such as Gadd45α, p21 and 14-3-3 [24-26]. These proteins enforce a G2/M checkpoint by either directly or indirectly inactivating cdc2/cyclinB complex which is the major engine driving the transition from G2 to M.
The role of p38 MAPK in the G2/M Checkpoint.
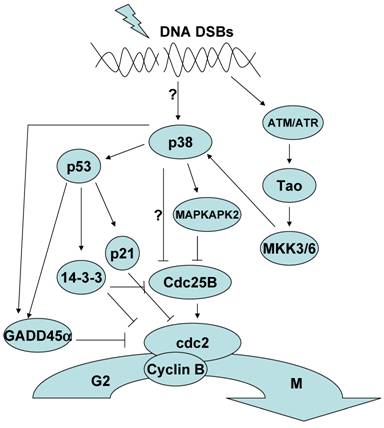
p38 MAPK can also induce a G2/M checkpoint through the phosphorylation and inhibition of the phosphatase Cdc25B [27]. Cdc25B dephosphorylates cdc2 and activates the CyclinB/cdc2 complex driving progression of the cell cycle. It has been proposed that phosphorylation of Cdc25B inhibits its activity by promoting its association with 14-3-3 proteins and sequestering it in the cytoplasm [28, 29]. While it was initially reported that p38 MAPK could directly phosphorylate Cdc25B in vitro, more recent studies have shown that p38 MAPK promoted the phosphorylation of Cdc25B indirectly through the activation of MK2 one of the well known substrates of p38 MAPK [30, 31]. Activation of MK2 was also shown to be required for the induction of a G2/M checkpoint in response to UV in these studies.
As mentioned above, although similar in substrate specificity, the different p38 MAPK family members can also mediate distinct functions. Thus all p38 MAPK isoforms appear to be activated in an ATM-dependent manner in response to γ-irradiation, but only p38γ seems to be required for the induction of the G2/M checkpoint [32]. Specific p38 MAPK isoforms therefore may have different roles in the induction of a G2/M checkpoint depending on the experimental system.
The function of p38 MAPK in the G1/S cell cycle checkpoint
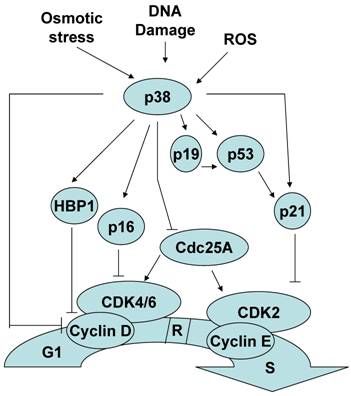
Although less established, p38 MAPK activation can also contribute to the induction of a G1/S checkpoint in response to stimuli such as osmotic stress, reactive oxygen species and cellular senescence. Similar to the G2/M checkpoint regulation, several mechanisms by which p38 MAPK mediates a G1/S checkpoint have been described. p38 MAPK can induce a G1/S checkpoint through the activation of p53. Consistent with its role as a master regulator of the cell cycle, in addition to controlling the G2 to M progression, p53 also regulates G1 to S progression. p38 MAPK activation of p53 results in the accumulation of p21, one of the p53 targets[33, 34]. While p21 promotes a G2/M checkpoint by inactivating cdc2, to establish a G1/S checkpoint p21 inactivates cdk2. In addition, it has been reported that p38 MAPK can directly phosphorylate and stabilize p21 in vivo [33].
Cyclin D binds to and actives cyclin dependent kinase 4 and 6 (Cdk4/6) and this active complex is essential for the transition to S phase and further cell proliferation. p38 MAPK had been shown to reduce the levels of cyclin D1 by two independent mechanisms. p38 MAPK can negatively regulate cyclin D1 at the level of transcription [35]. This negative effect could be explained by the phosphorylation and stabilization of HMG-box protein 1 (HBP1) by p38 MAPK. HBP1 is a transcriptional repressor that inhibits cyclin D1 gene expression [36]. p38 MAPK can also directly phosphorylate cyclin D1 and resulting in cyclin D1 ubiquitination and proteosomal degradation [37].
Unlike other members of the Cdc25 family that only regulate the G2/M transition, Cdc25A is a cyclin-dependent protein kinase phosphatase that can regulate the G1/S transition as well [38-40]. p38 MAPK can phosphorylate and promote the degradation of Cdc25A contributing to establishment of a G1/S checkpoint [41].
p16INK4a and p19ARF are two different proteins encoded in the INK4a/ARF locus that regulate the G1/S checkpoint by two different pathways. p16 promotes a G1/S checkpoint by inhibiting cyclin dependent protein kinase Cdk4/6 activation [42], and p19 promotes a G1/S checkpoint by regulating p53 function [43, 44]. p38 MAPK can also mediate a G1/S checkpoint by upregulating p16INK4a and p19ARF gene expression [45-47]. Thus, p38 MAPK can regulate the induction of a G1/S checkpoint by multiple distinct mechanisms (Figure 2).
The role of p38 MAPK in the G1/S checkpoint.
p38 MAPK and survival
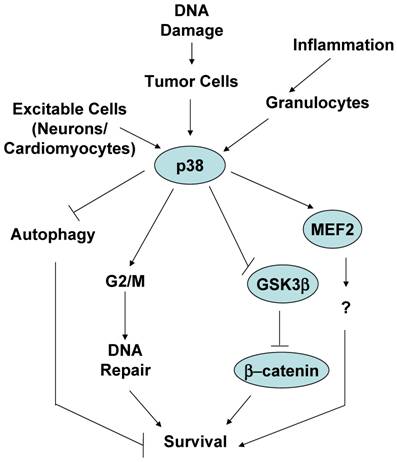
As mentioned above, p38 MAPK was identified as a stress kinase because it is activated by stimuli that cause some kind of stress to cells, eventually leading to cell death. As a consequence, p38 MAPK is generally believed to be a kinase that mediates cell death. However, while this assumption is correct in most cases, cause-effect studies have also found that activation of p38 MAPK by stress stimuli may not necessarily promote death, instead it enhances cell survival. Here we will discuss this alternative and less characterized function of the p38 MAPK signaling pathway, and provide potential scenarios where survival instead of death is the main goal of p38 MAPK activation (Figure 3).
The role of p38 MAPK in mediating survival.
One of the common circumstances where activation of p38 MAPK has been shown to be essential for survival is in response to stimuli that cause a type of DNA damage. As discussed above, UV, γ-irradiation and chemotherapeutic cancer drugs are stimuli that damage DNA and result in cell death. These stimuli are also strong activators of the p38 MAPK pathway. Thus, activation of p38 MAPK was observed in Jurkat T cells when treated with a combination of UV and 8-methoxypsoralen (8-MOP), a tricyclic aromatic compound that intercalates into DNA and crosslinks the two strands of DNA in the presence of UV leading DNA damage and cell death [48]. However, instead of preventing death, inhibition of p38 MAPK with the pharmacological inhibitor increased death of these cells in a dose dependent manner [48]. Similarly, treatment of human B cell lymphoma cells with etoposide, a DNA-damaging chemotherapeutic drug, activates p38 MAPK, but pharmacological and genetic inhibition of this pathway enhances the apoptotic effect of etoposide [49]. Doxorubicin (topoisomerase II inhibitor) and cisplatin (DNA crosslinker) activate p38 MAPK and induce death of osteosarcoma cells. Nevertheless, downregulation of MK2 enhances death triggered by cisplatin and doxorubicin [16]. Since MK2 is one of the major substrates of p38 MAPK in response to irradiation [30], it is likely that p38 MAPK is also involved in the survival process of these cells through activation of MK2. Thus, while there is no strong evidence that p38 MAPK by itself induces cell proliferation and malignancies, in response to DNA damage stimuli activation of p38 MAPK in cancer cells increases cell survival. This could be a defense mechanism that cells develop to overcome the effect of cytotoxic drugs affecting DNA integrity. Indeed, p38 MAPK may not be directly activated in response to damage, but it can be activated by the counterpart survival response. Simultaneously to DNA damage, ionizing irradiation also causes the activation of tyrosine kinase receptors, such as the insulin-like growth factor 1 receptor (IGF-1R) that can provide survival signals. Activation of p38 MAPK by IGFR1 in response to ionizing irradiation appears to contribute to the radioresistance effect provided by IGFR1 [50]. Considering the role of p38 MAPK in the induction of a G2/M cell cycle checkpoint, the survival effect of p38 MAPK in response to DNA damage could be explained by its effect in facilitating DNA repair prior to the continuation through mitotsis, helping tumor cells to escape the mitotic catastrophe. In this regard, it has been shown that p38 MAPK contributes to resistance to chemotherapeutic DNA-methylating drugs by promoting DNA mismatch repair and G2/M cell cycle checkpoint [51]. Alternatively, an active survival signal may provide advantage for cells to survive through the G2/M cell cycle checkpoint, properly repair and further expand (see below).
It is well known that DNA DSBs are normally generated in response to DNA damaging agents (e.g. ionizing irradiation, chemotherapeutic drugs). However, less known is the fact that DSBs are naturally generated in specific cells in the absence of danger stimuli. The expression of antigen specific T cell receptor (TCR) and B cell receptor (BCR) genes is unique and different to any other type of gene expression since it requires the rearrangement of one of the multiple V, D and J DNA segments to form a coding gene. This process is defined as V(D)J recombination and it is mediated by the T and B cell specific endonucleases, RAG1 and RAG2, that recognize the recombination signal sequences (RSSs). During the recombination process, RAG proteins bind to these sequences, bring them together and cleave DNA immediately adjacent to the RSSs through the endonuclease activity to generate nicked DNA. The 3' hydroxyl groups of these two nicks attack the other DNA strands to form two independent hairpins (coding ends) within the genomic DNA, while the double stranded breaks (DSBs) from excised fragments (signal ends) remain as DSBs. Thus, DSBs are continuously generating in developing T and B cells. During the development of T cells in the thymus, recombination of the germ line TCRβ gene occurs at the double negative (DN)3 stage of thymocyte differentiation. It has been shown that p38 MAPK is activated at this stage of T cell development, and its activation is dependent on the presence of RAG-mediated DSBs [52]. In vivo activation of p38 MAPK in these cells, induces a G2/M cell cycle checkpoint, but it also provides survival signals [52], and evidence that p38 MAPK can mediate survival in the context of DNA damage in vivo.
Although less frequently reported, some studies have also involved p38 MAPK in survival of tumor cells independently of DNA damage, and suggested that p38 MAPK contributes to metastasis. However, this effect appears to be indirectly mediated by p38 MAPK through the regulation of the secretion of factors that promote survival or migration of cells. Thus, basal activation of p38 MAPK in B-cell chronic lymphocytic leukaemia (B-CLL) is required for the expression of the MMP-9 metalloprotease that is turn is required for survival of these cells when cultured in the presence of stroma cells [53]. Decreased basal as well as TGFβ1-induced MMP-9 activity has also been found in breast cancer cells when p38 MAPK pathway was inhibited by genetic approaches and pharmacological compounds, leading to decreased bone metastases in vivo [54].
p38 MAPK can also mediate cell survival in non-malignant cells. Early studies in PC12 neuronal cell line proposed that p38 MAPK mediates death in these cells [55]. However, it was later shown that genetic inhibition of p38 MAPK promotes death of primary cerebellar granule neurons grown in vitro in survival conditions by the presence of a depolarizing dose of KCL [56]. Similar to neurons, primary cardiac myocytes are also excitable when treated with specific growth factors. Activation of p38 MAPK by expression of a constitutive active MKK6 in primary cardiomyocytes seems to protect these cells against apoptosis triggered by the treatment with anisomycin (a protein synthesis inhibitor) [57].
Interestingly, several studies have reported the role of p38 MAPK in survival of different type of mature granulocytes. Granulocytes (e.g. neutrophils, eosinophils, basophils) have in common their terminal differentiation stage. These cells have fragmented nuclei and have an accumulation of granuli containing preformed secretion factors. It has been proposed that p38 MAPK is required for survival of neutrophils during inflammation, and inactivation of p38 MAPK is essential for death and the elimination of these cells during the resolution of the inflammatory response [58, 59]. Similarly, inhibition of p38 MAPK appears to enhance constitutive apoptosis of eosinophils upon cytokine-deprivation (e.g. IL-5), suggesting that activation of p38 MAPK during the inflammatory response is critical for the survival of these short life cells, and inactivation of this pathway is also important for resolution of inflammation [60].
What are the mechanisms involved in p38 MAPK-mediated cell survival? We already described above that p38 MAPK can indirectly regulate survival by promoting the expression of survival secreted factors such as MMP-9. Although it has been proposed that p38 MAPK can mediate survival by phosphorylation of caspase 3 and caspase 8 in neutrophils [58], it is unlikely to be due to a direct phosphorylation by p38 MAPK since the identified phosphorylation site motifs (SL) in caspases are not consensus MAP kinase recognized motifs (SP or TP). In response to DNA damage, an obvious mechanism by which p38 MAPK may indirectly increase survival is by inducing the G2/M cell cycle checkpoint and increasing the chance for DNA repair prior to a mitotic catastrophe. p38 MAPK could also directly favor the DNA repair process since pharmacological inhibition of p38 MAPK appears to decrease binding of the repair Ku proteins to DNA [50], but the mechanism remains unclear.
A recent study has shown a novel mechanism by which p38 MAPK can directly mediate cell survival. β-catenin is a transcription factor known to promote survival by regulating the expression of other survival genes such as c-myc [61]. β-catenin levels are regulated by a multiprotein complex containing the tumor suppressor adenomatous polyposis coli (APC), the scaffolding protein axin and GSK3β . Phosphorylation of β-catenin by GSK3β targets β-catenin for ubiquitination and subsequent degradation [62]. Signals that prevent GSK3-mediated β-catenin phosphorylation, cause an accumulation of β-catenin in the cytosol and its subsequent translocation to the nucleus [63]. Previous studies have indicated that p38 MAPK can indirectly modulate GSK3β activity through either AKT or Gadd45α [64-66]. While the inactivation of GSK3β is normally mediated via phosphorylation of its N-terminus at S9 by AKT [67], a recent study has shown that p38 MAPK directly inactivates GSK3β by phosphorylation at a novel site (Ser389) in the C-terminus of GSK3β [68]. Phosphorylation of GSK3β at Ser389 by p38 MAPK inhibits GSK3β probably through a mechanism similar to the mechanism reported for the phosphorylation at the Ser9. Thus, phosphorylation and inactivation of GSK3β followed by the accumulation of β-catenin could be a mechanism by which p38 MAPK mediates survival in different cell types and in response to different stimuli. In contrast to the broadly distributed phosphorylation of GSK3β at Ser9, p38 MAPK-mediated phosphorylation of GSK3β at Ser389 appears to be tissue specific (predominantly in brain). Thus, although it has been proposed that p38 MAPK mediates neuronal survival by phosphorylation of the transcription factor MEF2 [56], inactivation of GSK3β could be an alternative mechanism. It remains to be determine whether phosphorylation of GSK3β on Ser389 participate in those scenarios where p38 MAPK has already been shown to mediate survival, such as in tumor cells in response to DNA damage-inducing stimuli.
Another proposed mechanism by which p38 MAPK can promote survival of tumor cells is by inhibition of autophagy, such in the case of colon cancer cells [69]. Autophagy was initially identified as an alternative pathway for cell death other than apoptosis because it is a mechanism to eliminate parts of intracellular compartments including mitochondria [70]. Autophagy was shown to occur primarily in tumor cells, but we now know that it is a widely distributed phenomenon in primary cells as well. Some studies have reported that p38 MAPK inhibits the initiation of autophagy and, thereby, inhibits cell death [70]. Inhibition of p38 MAPK appears to induce gene expression of GABARAP, a component of the vacuoles formed during autophagy. However, it is becoming more clear that autophagy does not always represents a mechanism for cell death, but it can also be a mechanism for cell survival since it mediates the clearance of unhealthy intracellular compartments (diminishing the intracellular damage) and it can minimize the metabolism of cells when they are under stress conditions. Thus, it remains to be clarified whether the role of p38 MAPK in autophagy is indeed associated with cell death or cell survival.
As an important note, most studies cited above were performed using pharmacological inhibitors of p38 MAPK to address the role of this pathway in cell survival or cell cycle checkpoint. While these compounds are relatively specific for p38 MAPK, this specificity is strictly determined by the used concentration. Studies exclusively based in the use of high concentrations (≤ 10 μM) of these compounds may be misleading since at high concentrations the p38 MAK inhibitors also interfere with other MAPKs such as JNK, and the JNK signaling pathway can also provide survival in specific situations [71, 72]. Only studies using different acceptable doses of the p38 MAPK inhibitors, or in combination with genetic studies have been considered in this review. Future studies using conditional knockout mice for the different p38 MAPK family members will be important to further corroborate the current conclusions.
Remarks
In summary, in addition to the well-characterized role of the p38 MAPK in cell death and differentiation, we have described here the contribution that this signaling pathway also has to cell survival and the initiation/maintenance of cell cycle checkpoints in response to specific stimuli. It is important to be aware of these other alternative functions of p38 MAPK considering that pharmacological inhibitors of p38 MAPK have been and are still being developed by a large number of corporations with a therapeutic purpose. While these compounds are being designed primarily for treatment of autoimmune disease such as rheumatoid arthritis, the role of p38 MAPK providing survival to tumor cells suggests that these drugs could also be used as anti-tumor agents in combination with other classical chemotherapeutic drugs. As mentioned above, additional studies using genetic approaches are urgently needed to confirm the conclusions obtained from the use of inhibitors in vitro. It also remains to be defined the specific mechanisms used by p38 MAPK to mediate survival and the cell cycle checkpoints, and how p38 MAPK discriminates its targets to mediate cell death from those to mediate cell survival. Although most studies have focused on p38α MAPK, it is important to better understand the contribution of the other p38 MAPK family members, and how these complement or compensate each other.
Acknowledgements
We thank Dr. David Wood for careful reading of the manuscript and helpful suggestions. This work was supported by NIH grants R01 A1051454 and NCRR P20 RR15557 (M.R.).
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Raman M, Chen W, Cobb M.H. Differential regulation and properties of MAPKs. Oncogene. 2007;26(22):3100
2. Iñesta-Vaquera F, Sabio G, Kuma Y, Cuenda A. Alternative p38 Pathways MAPK. Stress-Activated Protein Kinases. Heidelberg: Springer Berlin. 2008:17
3. Adams R.H. et al. Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular development. Mol Cell. 2000;6(1):109
4. Allen M. et al. Deficiency of the stress kinase p38alpha results in embryonic lethality: characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. J Exp Med. 2000;191(5):859
5. Mudgett J.S. et al. Essential role for p38alpha mitogen-activated protein kinase in placental angiogenesis. Proc Natl Acad Sci U S A. 2000;97(19):10454
6. Tamura K. et al. Requirement for p38alpha in erythropoietin expression: a role for stress kinases in erythropoiesis. Cell. 2000;102(2):221
7. Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15(1):11
8. Ashwell J.D. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat Rev Immunol. 2006;6(7):532
9. Salvador J.M. et al. Alternative p38 activation pathway mediated by T cell receptor-proximal tyrosine kinases. Nat Immunol. 2005;6(4):390
10. Ge B. et al. MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science. 2002;295(5558):1291
11. Han J, Sun P. The pathways to tumor suppression via route p38. Trends Biochem Sci. 2007;32(8):364
12. Hui L, Bakiri L, Stepniak E, Wagner E.F. p38alpha: a suppressor of cell proliferation and tumorigenesis. Cell Cycle. 2007;6(20):2429
13. Zhou B.B, Elledge S.J. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408(6811):433
14. Bulavin D.V, Amundson S.A, Fornace A.J. p38 and Chk1 kinases: different conductors for the G(2)/M checkpoint symphony. Curr Opin Genet Dev. 2002;12(1):92
15. Mikhailov A, Shinohara M, Rieder C.L. Topoisomerase II and histone deacetylase inhibitors delay the G2/M transition by triggering the p38 MAPK checkpoint pathway. J Cell Biol. 2004;166(4):517
16. Reinhardt H.C, Aslanian A.S, Lees J.A, Yaffe M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11(2):175
17. Raman M. et al. TAO kinases mediate activation of p38 in response to DNA damage. Embo J. 2007;26(8):2005
18. Bulavin D.V. et al. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. Embo J. 1999;18(23):6845
19. She Q.B. et al. Resveratrol-induced activation of p53 and apoptosis is mediated by extracellular-signal-regulated protein kinases and p38 kinase. Cancer Res. 2001;61(4):1604
20. She Q.B, Chen N, Dong Z. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J Biol Chem. 2000;275(27):20444
21. Huang C. et al. p38 kinase mediates UV-induced phosphorylation of p53 protein at serine 389. J Biol Chem. 1999;274(18):12229
22. Jin S. et al. Gadd45a contributes to p53 stabilization in response to DNA damage. Oncogene. 2003;22(52):8536
23. Bulavin D.V, Kovalsky O, Hollander M.C, Fornace A.J. Jr, Loss of oncogenic H-ras-induced cell cycle arrest and p38 mitogen-activated protein kinase activation by disruption of Gadd45a. Mol Cell Biol. 2003;23(11):3859
24. Zhan Q. et al. Association with Cdc2 and inhibition of Cdc2/Cyclin B1 kinase activity by the p53-regulated protein Gadd45. Oncogene. 1999;18(18):2892
25. el-Deiry W.S. et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75(4):817
26. Hermeking H. et al. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell. 1997;1(1):3
27. Bulavin D.V. et al. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature. 2001;411(6833):102
28. Morris M.C. et al. An essential phosphorylation-site domain of human cdc25C interacts with both 14-3-3 and cyclins. J Biol Chem. 2000;275(37):28849
29. Lopez-Girona A, Furnari B, Mondesert O, Russell P. Nuclear localization of Cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature. 1999;397(6715):172
30. Manke I.A. et al. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol Cell. 2005;17(1):37
31. Lemaire M. et al. CDC25B phosphorylation by p38 and MK-2. Cell Cycle. 2006;5(15):1649
32. Wang X. et al. Involvement of the MKK6-p38gamma cascade in gamma-radiation-induced cell cycle arrest. Mol Cell Biol. 2000;20(13):4543
33. Kim G.Y. et al. The stress-activated protein kinases p38 alpha and JNK1 stabilize p21(Cip1) by phosphorylation. J Biol Chem. 2002;277(33):29792
34. Kishi H. et al. Osmotic shock induces G1 arrest through p53 phosphorylation at Ser33 by activated p38MAPK without phosphorylation at Ser15 and Ser20. J Biol Chem. 2001;276(42):39115
35. Lavoie J.N. et al. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996;271(34):20608
36. Yee A.S. et al. The HBP1 transcriptional repressor and the p38 MAP kinase: unlikely partners in G1 regulation and tumor suppression. Gene. 2004;336(1):1
37. Casanovas O. et al. Osmotic stress regulates the stability of cyclin D1 in a p38SAPK2-dependent manner. J Biol Chem. 2000;275(45):35091
38. Zhao H, Watkins J.L, Piwnica-Worms H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci U S A. 2002;99(23):14795
39. Jinno S. et al. Cdc25A is a novel phosphatase functioning early in the cell cycle. Embo J. 1994;13(7):1549
40. Blomberg I, Hoffmann I. Ectopic expression of Cdc25A accelerates the G(1)/S transition and leads to premature activation of cyclin E- and cyclin A-dependent kinases. Mol Cell Biol. 1999;19(9):6183
41. Goloudina A. et al. Regulation of human Cdc25A stability by Serine 75 phosphorylation is not sufficient to activate a S phase checkpoint. Cell Cycle. 2003;2(5):473
42. Roussel M.F. The INK4 family of cell cycle inhibitors in cancer. Oncogene. 1999;18(38):5311
43. Weber J.D. et al. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1(1):20
44. Tao W, Levine A.J. P19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc Natl Acad Sci U S A. 1999;96(12):6937
45. Faust D. et al. p38alpha MAPK is required for contact inhibition. Oncogene. 2005;24(53):7941
46. Bulavin D.V. et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36(4):343
47. Ito K. et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12(4):446
48. Cappellini A. et al. Antiapoptotic role of p38 mitogen activated protein kinase in Jurkat T cells and normal human T lymphocytes treated with 8-methoxypsoralen and ultraviolet-A radiation. Apoptosis. 2005;10(1):141
49. Kurosu T. et al. p38 MAP kinase plays a role in G2 checkpoint activation and inhibits apoptosis of human B cell lymphoma cells treated with etoposide. Apoptosis. 2005;10(5):1111
50. Cosaceanu D. et al. Ionizing radiation activates IGF-1R triggering a cytoprotective signaling by interfering with Ku-DNA binding and by modulating Ku86 expression via a p38 kinase-dependent mechanism. Oncogene. 2007;26(17):2423
51. Hirose Y. et al. The p38 mitogen-activated protein kinase pathway links the DNA mismatch repair system to the G2 checkpoint and to resistance to chemotherapeutic DNA-methylating agents. Mol Cell Biol. 2003;23(22):8306
52. Pedraza-Alva G. et al. Activation of p38 MAP kinase by DNA double-strand breaks in V(D)J recombination induces a G2/M cell cycle checkpoint. Embo J. 2006;25(4):763
53. Ringshausen I. et al. Constitutive activation of the MAPkinase p38 is critical for MMP-9 production and survival of B-CLL cells on bone marrow stromal cells. Leukemia. 2004;18(12):1964
54. Suarez-Cuervo C. et al. Breast cancer cells with inhibition of p38alpha have decreased MMP-9 activity and exhibit decreased bone metastasis in mice. Clin Exp Metastasis. 2004;21(6):525
55. Xia Z. et al. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270(5240):1326
56. Mao Z, Wiedmann M. Calcineurin enhances MEF2 DNA binding activity in calcium-dependent survival of cerebellar granule neurons. J Biol Chem. 1999;274(43):31102
57. Zechner D. et al. MKK6 activates myocardial cell NF-kappaB and inhibits apoptosis in a p38 mitogen-activated protein kinase-dependent manner. J Biol Chem. 1998;273(14):8232
58. Alvarado-Kristensson M. et al. p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J Exp Med. 2004;199(4):449
59. Alvarado-Kristensson M. et al. p38 Mitogen-activated protein kinase and phosphatidylinositol 3-kinase activities have opposite effects on human neutrophil apoptosis. Faseb J. 2002;16(1):129
60. Kankaanranta H. et al. SB 203580, an inhibitor of p38 mitogen-activated protein kinase, enhances constitutive apoptosis of cytokine-deprived human eosinophils. J Pharmacol Exp Ther. 1999;290(2):621
61. He T.C. et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509
62. Liu C. et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108(6):837
63. Doble B.W, Woodgett J.R. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116(Pt 7):1175
64. Hildesheim J. et al. Gadd45a regulates matrix metalloproteinases by suppressing DeltaNp63alpha and beta-catenin via p38 MAP kinase and APC complex activation. Oncogene. 2004;23(10):1829
65. Rane M.J. et al. p38 Kinase-dependent MAPKAPK-2 activation functions as 3-phosphoinositide-dependent kinase-2 for Akt in human neutrophils. J Biol Chem. 2001;276(5):3517
66. Wang Y. et al. The role of the NADPH oxidase complex, p38 MAPK, and Akt in regulating human monocyte/macrophage survival. Am J Respir Cell Mol Biol. 2007;36(1):68
67. Cross D.A. et al. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785
68. Thornton T.M. et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science. 2008;320(5876):667
69. Comes F. et al. A novel cell type-specific role of p38alpha in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ. 2007;14(4):693
70. Simone C. Signal-dependent control of autophagy and cell death in colorectal cancer cell: the role of the p38 pathway. Autophagy. 2007;3(5):468
71. Heasley L.E, Han S.Y. JNK regulation of oncogenesis. Mol Cells. 2006;21(2):167
72. Weston C.R, Davis R.J. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19(2):142
Author contact
![]() Correspondence to: Dr. Mercedes Rincon, Immunology Division, Department of Medicine, University of Vermont College of Medicine, Given Medical Building Room D-305, 89 Beaumont Ave., Burlington, VT 05405-0068, USA. Phone: 802-656-0937; Fax: 802-656-3854; Email: mrinconedu
Correspondence to: Dr. Mercedes Rincon, Immunology Division, Department of Medicine, University of Vermont College of Medicine, Given Medical Building Room D-305, 89 Beaumont Ave., Burlington, VT 05405-0068, USA. Phone: 802-656-0937; Fax: 802-656-3854; Email: mrinconedu