Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(5):444-450. doi:10.7150/ijbs.5.444 This issue Cite
Research Paper
Essential Roles of mTOR/Akt Pathway in Aurora-A Cell Transformation
Makoto Taga1, Eiji Hirooka1, Toru Ouchi1,2 ![]()
1. ENH, Department of Medicine, Feinberg School of Medicine, Northwestern University, Evanston, IL 60201, USA
2. The Robert H. Lurie Comprehensive Cancer Center, Basic Science Division, Northwestern University, Evanston, IL 60201, USA
Received 2009-5-15; Accepted 2009-6-15; Published 2009-6-19
Abstract
We have recently demonstrated that Aurora-A kinase is a potential oncogene to develop mammary gland tumors in mice, when expressed under MMTV promoter. These tumors contain phosphorylated forms of Akt and mTOR, suggesting that Akt-mTOR pathway is involved in transformed phenotype induced by Aurora-A. In the present studies, we discovered that stable cell lines expressing Aurora-A contain phosphorylation of Akt Ser473 after prolonged passages of cell culture, not in cells of the early period of cell culture. Levels of PTEN tumor suppressor are significantly reduced in these late passage cells at least in part due to increased poly ubiquitination of the protein. Akt-activated Aurora-A cells formed larger colonies in soft agar and are resistant to UV-induced apoptosis. Aurora-A inhibitor, VX-680, can cause cell death of Aurora-A cells in which Akt is not activated. siRNA-mediated depletion of mTOR in those cells resulted in decreased phosphorylation of Akt Ser473, suggesting that TORC2 complex phosphorylates Akt in Aurora-A cells. Treatment of late-passage Aurora-A cells with mTOR inhibitor reduced colony formation in soft agar. These results strongly suggest that commitment of cell transformation by Aurora-A is determined by at least co-activation of Akt/mTOR pathway.
Keywords: Aurora-A kinase, Akt-mTOR pathway, oncogene
INTRODUCTION
The Aurora family of kinase consists of three related proteins (Aurora-A, B and C) that share the high sequence homology in their catalytic domains (1,2). Among them is Aurora-A that regulates mitotic progression in various organisms (1). It has been well documented that activation of Aurora-A is required for mitotic entry, centrosome maturation and separation, and G2 to M transition (1,3). Overexpression of Aurora-A is frequently observed in varieties of human cancer, including breast, colorectal, bladder, pancreatic, gastric, ovarian and esophageal cancer (4-9). Significantly, overexpression of Aurora-A in fibroblasts resulted in cell transformation, supporting a notion that high levels of this protein are correlated to cell malignancy.
Potential roles of Aurora-A in cell transformation were also demonstrated from recent studies that this kinase phosphorylates a breast cancer tumor suppressor BRCA1 at Ser308 (10). This phosphorylation occurs at G2 to M transition and is necessary for the entry into mitosis. Interestingly, BRCA1-negative fibroblasts re-expressing phospho-deficient BRCA1 are arrested in G2 phase without DNA damage. In studies using mice, we demonstrated that, when Aurora-A is expressed under control of MMTV promoter, mammary tumors are developed, providing direct evidence that this kinase can function as an oncogene in vivo, although latency is quite long (1,5 to 2 years) (11). Immunohistochemical analysis showed that mTOR and Akt are highly phosphorylated in these tumors (11). These results strongly suggest that Aurora-A activates mTOR/Akt pathway in the process of cell transformation.
Many small chemical compounds of Aurora kinase inhibitors are currently undergoing preclinical and clinical assessment. VX-680 is one of the pioneering chemicals in these trials (12). It is a pyrimidine derivative with high affinity for Aurora-A, B and C at nanomolar concentrations. VX-680 prevents cytokinesis but allows cells to progress through the other stages of mitosis, which leads to polyploidy and, in some cancer cell lines, massive cell death. In preclinical models, VX-680 blocked tumor xenograft growth and induced regression of the tumors (12).
In this study, we investigated the biological and biochemical phenotype of cells overexpressing Aurora-A using established cell lines of different passage numbers. We discovered that more malignant phenotype is observed in Aurora-A cells containing activated mTOR/Akt pathway, which occurred only when Aurora-A cells are maintained for longer passage. These results suggest that overexpression of Aurora-A does not induce malignant phenotypes immediately, and that it cooperates with other oncogenic pathway for the full transformation.
EXPERIMENTAL PROCEDURES
Cell culture and western blotting
U2OS human osteosarcoma cell line was grown in DMEM containing 10% fetal bovine serum and 100U of penicillin-streptomycin/ml. Cells were transfected with FLAG-Aurora-A/pcDNA3 and maintained in the presence of G418 (700μg/ml) for 7 days. Three independent clones (clone 1, 2 and 3) were isolated. For Western blot analysis, cells were lysed in EBC buffer (50 mM Tris-HCl, pH 8.0, 120 nM NaCl, 0.5% Nonidet P-40, 100 mM NaF, 200 μM sodium orthovanadate, 100 μg/ml phenylmethylsulfonyl fluoride, 2 μg/ml leupeptin, 2 μg/ml aprotinin). Protein concentration in cell lysates were determined using the Bio-Rad protein assay kit. Sixty micrograms of whole cell extract were loaded per lane and separated by 6, 7.5% or 12% SDS-PAGE. Transfer to Immobilon-P membrane (Milipore, Billercia, MA) was performed using a semidry transfer method (Trans-Blot; Bio-Rad, Hercules, CA) in 25 mM Tris base, 192 mM glycine and 10% methanol for 1.5 h at 15V. Membranes were blocked in 1% nonfat dried milk in phosphate-buffered saline/0.05% Tween and incubated with primary antibodies and horseradish peroxidase-conjugated secondary antibodies (Jackson Laboratories, Westgrove, PA) followed by ECL detection. For immunoprecipitation, 1 mg of total cell lysate was incubated with the specific antibody over night at 4ºC followed by either protein A- or protein-G Sepharose beads (Sigma, St. Louis, MO). The samples were washed with NET-N buffer and subjected to SDS-PAGE.
siRNA Assay
Synthesized siRNA (#6381 and #6556) were purchased from Cell Signaling and tested for suppression. Briefly, U2OS cells were seeded at 30% confluence the day prior to transfection and cells were transfected with FuGENE (Roche Applied Science, Indianapolis, IN). Twenty-four hours later, transfections were repeated as before. Lysates were collected after a further 48hr and subjected to SDS-PAGE. #6381 were used for the experiments since it showed better suppression.
Plasmids and antibodies
Expression vectors for HA-ubiquitin and PTEN in pcDNA3 were from Dr. A. Chan at the Mt. Sinai School of Medicine. FLAG-tagged Aurora-A cDNA was expressed with pcDNA3. The antibodies for HA tag, PTEN (A2B1), p53 (DO-1), PI3K (D-4) and Akt (B-1) were purchased from Santa Cruz. The antibodies for Akt phospho-Ser473, p53 phospho-Ser15, and ATR, DNA-PK were purchased from Cell Signaling. The antibody for ATM was purchased from Rockland.
Annexin V/PI staining
Cells were washed with PBS and harvested by trypsinization and the concentration adjusted to 1x106 cells/ml. Annexin V was detected using an Annexin V-FITC Apoptosis Detection Kit (Oncogene Research) as recommended by the manufacturer. VX-680 was provided by Vertex/Merck.
Assays for colony formation in soft agar and cell growth
Six well plates were filled with 2 ml of 0.6% noble agar (Invitrogen) in DMEM supplemented with 10% calf serum as a bottom layer and allowed to solidify at 4oC overnight. 104 cells were suspended in 1 ml of 0.4% agarose. Cell culture plates were maintained for 2 weeks in a CO2 incubator. For cell growth, 106 cells were seeded on the plates and cell numbers were counted every day in duplicate.
RESULTS
Elevated Phosphorylation of Akt Ser473 in Aurora-A expressing cell culture
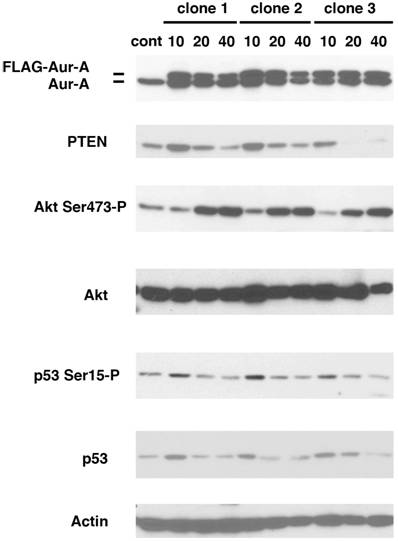
We have previously shown that MMTV-Aurora-A transgene developed mammary gland tumors that expressed phosphorylated mTOR and Akt (11). To study the mechanism of how Akt pathway is activated by Aurora-A, we established stable cell lines overexpressing Aurora-A. U2OS cells were chosen because p53 is wild type and highly transfectable. Cells were transfected with a plasmid encoding FLAG-tagged Aurora-A cDNA regulated by CMV promoter. After maintained in the presence of G418 for seven days (passage-number = 0), we started to count the passage numbers every time we seeded cell culture. Aurora-A cells at passage number 10, 20 and 40 (termed A-10, A-20 and A-40 cells, respectively) were studied as below. Levels of Flag-Aurora-A, phosphorylation of Akt at Ser473, PTEN and p53 were immunoblotted using lysates of A-10, A-20 and A-40 cells.
Three independent clones were isolated and immunoblotted with the indicated antibodies (Fig.1). Levels of Flag-Aurora-A were slightly decreased in A-40 cells, compared to those in A-10 and A-20 cells. Constitutive phosphorylation of Akt at Ser473 was similar in both control cells and A-10 cells, however, it increased significantly in A-20 and A-40 cells. It has been well documented that PTEN functions upstream of Akt, and negatively regulates Akt activity (13,14). Levels of PTEN increased in A-10 cells, but became lower in A-40 cells than those of control cells. Levels of both p53 and phosphorylation of p53 Ser15 were also increased in A-10 cells, and decreased in A-20 and A-40 cells. These results suggest that p53-mediated checkpoint is transiently activated in Aurora-A cells of early passage number. Since it has been shown that p53 directly regulates the genomic promoter of PTEN (15), it is suggested that activated p53 increased levels of PTEN in A-10 cells.
Increased phosphorylation of Akt Ser473 in Aurora-A overexpressing cells. U2OS cells were transfected with FLAG-Aurora-A and maintained with G418 (700μg/ml) for 7 days. Isolated clones (clone 1, 2 and 3) were expanded and immunoblotted with the indicated antibodies.
To further explore the mechanism of lowered levels of PTEN in Aurora-A cells, poly-ubiquitination of PTEN was studied in both vector cells of passage-number of 40 and A-40 cells. Cells were transfected with PTEN and HA-ubiquititn under treatment with MG132, a proteasome inhibitor. As demonstrated in Fig. 2, poly-ubiquitination of PTEN was much increased in A-40 cells compared to those in vector control cells. These results indicate that levels of PTEN are down-regulated in ubiquitin-dependent manner in A-40 cells.
Increased poly-ubiquitination of PTEN in A-40 cells. A-40 cells and control cells transfected with a vector (passage number 40) were transfected with PTEN and HA-ubiquitin for 2 days. Cells were treated with MG-132 (SIGMA, 5 μM, 12 hr) PTEN was immunoprecipitated and samples were immunoblotted with anti HA antibody.

Colony Formation Ability of Aurora-A Cells is Enhanced After Increased Passage
Next, we characterized colony formation and cell growth of Aurora-A expressing cells. We used vector-transfected cells of passage-number of 40, A-10 and A-40 cells. Same numbers of these cells (1 x 104 cells) were plated in soft agar, and colony forming ability was determined after 14 days. Although U2OS cells are originally human cancer cells, the size of the colonies in soft agar was small. However interestingly, A-10 cells could form slightly larger colonies than control cells, and A-40 cells formed much larger colonies (Fig. 3A).
Colony formation and cell growth assay. (A) Control cells, A-10 cells and A-40 cells were seeded in agar plate and colony formation was studied after 14 days. A scale bar is shown. (B) Cells were seeded in cell culture plates in duplicate and numbers were counted every day for 7 day.
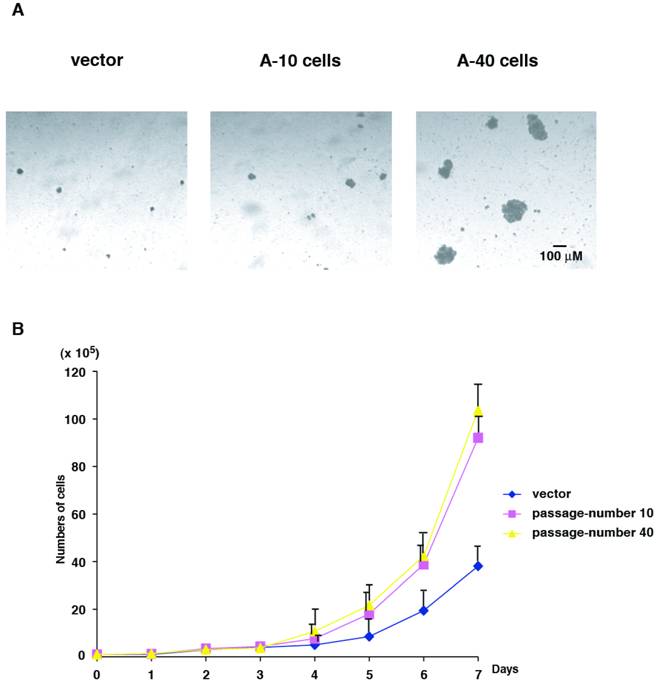
Cell growth was determined for 7 days using the same set of cells that were used in colony formation assay. Compared to the control cells, cell proliferation of both A-10 and A-40 cells were faster, however no significant differences in A-10 and A-40 cells were found (Fig. 3B). These results suggest that cells expressing high levels of Aurora-A immediately promote enhanced advantage of cell growth, but malignant phenotype of anchorage-independency is acquired later, resulting from after increased cell division. Colony formation assay and cell growth assay were examined using other Aurora-A clones described in Fig.1, and all clones showed the similar phenotypes (data not shown).
Suppression of Cell Death of Aurora-A Cells
We examined expression of Aurora-A affects induction of cell death under conditions of apoptotic stimuli. Vector control U2OS cells of passage-number of 40, A-10 and A-40 cells were treated with UV damage (100mJ/cm2), and cell death was measured with Annexin V staining after 12 hr (Fig. 4A). Although UV treatment induced significant cell death of control cells (35.2%), A-10 and A-40 cells showed resistance to cell death under the same conditions, although A-10 cells showed slightly more resistance (6.46% and 15.37%, respectively). Basal levels of apoptosis of both A-10 and A-40 cells without UV stimulation were similar to those of control cells (0.49%, and data not shown).
A-40 cells are resistant to both UV and VX-680. (A) Control, A-10 and A-40 cells were treated with UV (100mJ/cm2) and apoptosis was studied with Annexin V stain. (B) Indicated cells were treated with VX-680 for 12 hr and apoptosis was determined by Annexin V stain.
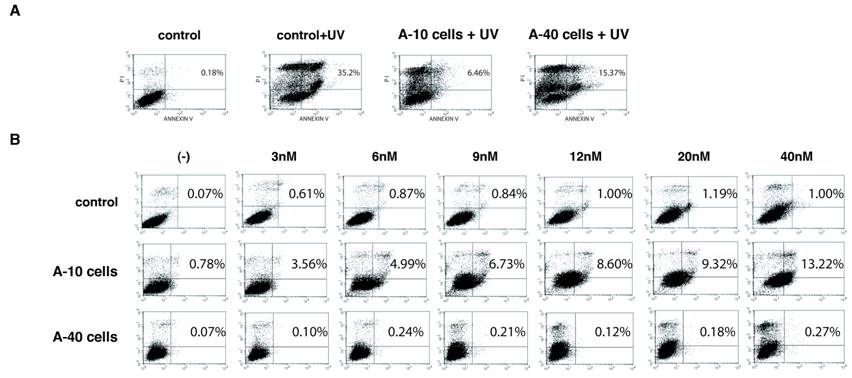
Next, sensitivity of Aurora-A cells to VX-680, an Aurora-A kinase inhibitor (12), was studied. Vector control cells, A-10 and A-40 were treated with different concentration of VX-680 for 12 hr, and cell death was measured by Annexin V staining (Fig. 4B). Although control cells did not show significant cell death in these conditions, higher levels of cell death of A-10 cells were observed in dose dependent manner. Interestingly, A-40 cells were resistant to VX-680-induced cell death in these experiments. We examined different clones of Aurora-A cells (see Fig.1), but all of long-passaged cells showed similar resistance to VX-680.
mTOR is Responsible for Phosphorylation of Akt Ser473 in Aurora-A Cells
Roles of Akt in cell transformation have been well illustrated. Recent studies have demonstrated that TORC2 complex that contains mTOR, mLST8, Rictor, mSin1 and Protor phosphorylates Akt at Ser473 (16-18). To understand the mechanism of Aurora-A's cell transformation, we studied how Akt phosphorylation is regulated in these cells. First, vector control cells of passage-number of 40 and three independent clones (clone 1, 2 and 3, see Fig.1) of A-40 cells were treated with wortmannin, a general PI3K (phosphoinositide 3 kinase) inhibitor (Fig. 5A). Phosphorylation of Akt Ser473 was significantly inhibited with this treatment in all of three clones, suggesting that PI3K members of proteins are involved in this phosphorylation of Akt in Aurora-A cells.
Akt Ser473 is phosphorylated by mTOR in Aurora-A cells. (A) Control and Aurora-A cells of passage number of 40 (clone 1, 2 and 3, see Fig.1) were treated with wortmannin (4 μM, 2 hr), and immunoblotted with the indicated antibodies. (B) A-40 cells were transfected with siRNA for mTOR for 48 hr. Cell lysates were immunoblotted with the indicated antibodies. (C) A-40 cells were treated with rapamycin (1 μM, 12 hr) and apoptosis was determined by Annexiv V stain.
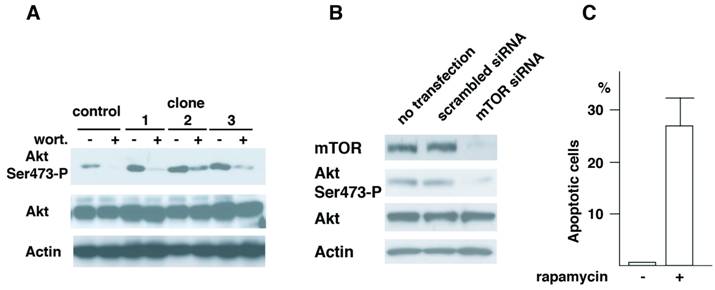
There are several protein kinases that belong to PI3K family. These include, ATM (ataxia telangiectasia mutated), ATR (ATM-related), DNA-PK, PI3K and mTOR. Among the kinases tested, depletion of mTOR but not other kinases resulted in inhibition of phosphorylation of Akt at Ser473 (Fig.5B and data not shown). Cells transfected with scrambled siRNA did not show the decreased phosphorylation of Akt Ser473. When A-40 cells were treated with rapamycin for 12 hr, increased cell death was observed (Fig.5C). These results strongly suggest that Akt Ser473 in Aurora-A cells is phosphorylated by mTOR, and that mTOR-Akt pathway activated in Aurora-A cells is essential for cell transformation induced by Aurora-A.
DISCUSSION
Our previous studies have illustrated that tissue specific expression of Aurora-A kinase in mammary gland can develop mammary tumors in mice, providing direct evidence that this kinase is oncogenic in animal model (11). Tumor incidence in these mice are however low (30 to 40%) and their latency is significantly long (more than 1 year), suggesting that oncogenic potential of Aurora-A is not as high as other oncogenes, such as MMTV-neu mice (19). In our immunohistochemical studies, we discovered that mTOR/Akt pathway was activated in tumors developed in these mice, although the role and mechanisms of this activation were not clear.
In the present studies, we explored how Aurora-A and mTOR/Akt pathway collaborate for cell transformation. We discovered that activation of mTOR/Akt does not occur immediately after overexpression of Aurora-A in U2OS cells. Consistent with these results, we found that transient expression of Aurora-A in human embryonic kidney cell line, 293 cells, does not cause phosphorylation of Akt and mTOR (data not shown). We are in the midst of investigating how Aurora-A activates mTOR/Akt pathway by transiently or stably expressing Aurora-A in other cell lines.
It was found that levels of p53 is induced in cells of early passage numbers p53, which returned to the control levels after prolonged cell culture. Mammary tumor development induced by MMTV-Aurora-A was accelerated on p53(+/-) background (11). These results support the notion that p53 plays a role of a gate-keeper in Aurora-A-induced tumorigenesis.
We found that there is a correlation between sensitivities to both UV and Aurora-A inhibitor VX-680 and the phosphorylation status of Akt Ser473. Three of Aurora-A stable clones isolated independently demonstrated similar response to these stimuli. Although we recognize that other cell types need to be studied, we consider that collaboration of Aurora-A and Akt pathway is crucial for cell transformation. It has been demonstrated that Aurora-A inhibitor, VX-680, can cause cell death of series of human cancer cell lines (12). In our cell culture model, cells showed apoptotic outcome in response to VX-680 treatment only in early time period of Aurora-A expression. It remains unclear why these cells not only stop the cell growth but also proceed to apoptosis after VX-680 treatment. It is suggested that overexpression of Aurora-A can cause activation of pro-apoptotic pathway in early stages of cell culture, and that this apoptotic pathway is suppressed after long duration of Aurora-A overexpression. Inhibition of mTOR with rapamycin resulted in apoptosis, suggesting that mTOR is anti-apoptotic in Aurora-A cells. In that sense, inhibition of Aurora-A kinase by compounds in late stage of Aurora-A cancer may not suppress tumor phenotype effectively. In our previous MMTV-Aurora-A mice model, latency of tumor development is quite long, so it is possible that Aurora-A triggers either activation of unidentified oncogenic pathways or inactivation of tumor suppressive pathways happened in those time period. Identification of these 'factors' is quite important for detection and treatment of Aurora-A tumors.
Acknowledgements
We thank the members of the Ouchi laboratory for helpful discussion. We dedicate this work to our great mentor, Dr. Hidesaburo Hanafusa, Professor Emeritus at The Rockefeller University who passed March 15, 2009. He devoted his life to science and teaching of young scientists. This work was supported by RO1CA79892 (NCI), RO1CA90631 (NCI), Susan G. Komen Investigator Initiated Grant and AVON Breast Cancer Pilot Project (all for T.O.).
Abbreviations
siRNA: small interference RNA; BRCA1: breast cancer gene 1; DMEM: Dulbecco modified eagle media; PAGE: poly acrylamide gel electrophoresis; ATM: ataxia telangiectasia mutated; ATR: ATM related.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Fu J, Bian M, Jiang Q. et al. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res. 2007;5:1-10
2. Ducat D, Zheng Y. Aurora kinases in spindle assembly and chromosome segregation. Exp Cell Res. 2004;301:60-67
3. Hirota T, Kunitoku N, Sasayama T. et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114:585-598
4. Tanaka T, Kimura M, Matsunaga K. et al. Centrosomal kinase AIK1 is overexpressed in invasive ductal carcinoma of the breast. Cancer Res. 1999;59:2041-2044
5. Nishida N, Nagasaka T, Kashiwagi K. et al. High copy amplification of the Aurora-a gene is associated with chromosomal instability phenotype in human colorectal cancers. Cancer Biol. Ther. 2007;6:525-533
6. Compérat E, Camparo P, Haus R. et al. Aurora-A/STK-15 is a predictive factor for recurrent behaviour in non-invasive bladder carcinoma: a study of 128 cases of non-invasive neoplasms. Virchows Arch. 2007;450:419-424
7. Li D, Zhu J, Firozi PF. et al. Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin Cancer Res. 2003;9:991-997
8. Lassmann S, Shen Y, Jütting U. et al. Predictive value of Aurora-A/STK15 expression for late stage epithelial ovarian cancer patients treated by adjuvant chemotherapy. Clin Cancer Res. 2007;13:4083-4091
9. Tong T, Zhong Y, Kong J. et al. Overexpression of Aurora-A contributes to malignant development of human esophageal squamous cell carcinoma. Clin Cancer Res. 2004;10:7304-7310
10. Ouchi M, Fujiuchi N, Sasai K. et al. BRCA1 phosphorylation by Aurora-a in the regulation of G2 to M transition. J Biol Chem. 2004;279:19643-19648
11. Wang X, Zhou YX, Qiao W. et al. Overexpression of aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene. 2006;25:7148-7158
12. Harrington EA, Bebbington D, Moore J. et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;101:262-267
13. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261-1274
14. Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403-414
15. Stambolic V, MacPherson D, Sas D. et al. Regulation of PTEN transcription by p53. Mol Cell. 2001;8:317-325
16. Lee S, Comer FI, Sasaki A. et al. TOR complex 2 integrates cell movement during chemotaxis and signal relay in Dictyostelium. Mol Biol Cell. 2005;16:4572-4583
17. Sarbassov DD, Ali SM, Sengupta S. et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159-168
18. Jacinto E, Facchinetti V, Liu D. et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125-137
19. Rowse GJ, Ritland SR, Gendler SJ. Genetic modulation of neu proto-oncogene-induced mammary tumorigenesis. Cancer Res. 1998;58:2675-2679
20. Lin S, Du P, Jafari N. et al. Using Free and Open-Source Bioconductor Packages to Analyze Array Comparative Genomics Hybridization (aCGH) Data. Curr Genomics. 2009;10:60-63
Author contact
![]() Correspondence to: Toru Ouchi, ENH, Department of Medicine, Feinberg School of Medicine, Northwestern University, 1001 University Place, Evanston, IL 60201, USA. Tel: 224-364-7687, Fax: 224-364-7402, Email: t-ouchiedu
Correspondence to: Toru Ouchi, ENH, Department of Medicine, Feinberg School of Medicine, Northwestern University, 1001 University Place, Evanston, IL 60201, USA. Tel: 224-364-7687, Fax: 224-364-7402, Email: t-ouchiedu