Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(5):451-457. doi:10.7150/ijbs.5.451 This issue Cite
Short Research Communication
Construction of a full-length cDNA Library from Chinese oak silkworm pupa and identification of a KK-42-binding protein gene in relation to pupa-diapause termination
Yu-Ping Li1, Run-Xi Xia1, Huan Wang1, Xi-Sheng Li2, Yan-Qun Liu1, 3 ![]() , Zhao-Jun Wei4
, Zhao-Jun Wei4 ![]() , Cheng Lu3, Zhong-Huai Xiang3
, Cheng Lu3, Zhong-Huai Xiang3
1. College of Bioscience and Biotechnology, Shenyang Agricultural University, Shenyang 110161, China
2. Sericultural Institute of Liaoning Province, Fengcheng 118100, China
3. The Key Sericultural Laboratory of Agricultural Ministry, Southwest University, Chongqing 400716, China
4. School of Biotechnology and Food Engineering, Hefei University of Technology, Hefei 230009, China
Received 2009-3-25; Accepted 2009-6-7; Published 2009-6-24
Abstract
In this study we successfully constructed a full-length cDNA library from Chinese oak silkworm, Antheraea pernyi, the most well-known wild silkworm used for silk production and insect food. Total RNA was extracted from a single fresh female pupa at the diapause stage. The titer of the library was 5 × 105 cfu/ml and the proportion of recombinant clones was approximately 95%. Expressed sequence tag (EST) analysis was used to characterize the library. A total of 175 clustered ESTs consisting of 24 contigs and 151 singlets were generated from 250 effective sequences. Of the 175 unigenes, 97 (55.4%) were known genes but only five from A. pernyi, 37 (21.2%) were known ESTs without function annotation, and 41 (23.4%) were novel ESTs. By EST sequencing, a gene coding KK-42-binding protein in A. pernyi (named as ApKK42-BP; GenBank accession no. FJ744151) was identified and characterized. Protein sequence analysis showed that ApKK42-BP was not a membrane protein but an extracellular protein with a signal peptide at position 1-18, and contained two putative conserved domains, abhydro_lipase and abhydrolase_1, suggesting it may be a member of lipase superfamily. Expression analysis based on number of ESTs showed that ApKK42-BP was an abundant gene in the period of diapause stage, suggesting it may also be involved in pupa-diapause termination.
Keywords: Chinese oak silkworm, Antheraea pernyi, cDNA library, Expressed sequence tag, KK-42-binding protein, diapause termination
1. Introduction
Economically important silk-producing insects mainly belong to two families, Bombycidae and Saturniidae, of order Lepidoptera. The domesticated silkworm, Bombyx mori, a member of family Bombycidae, is the most well-studied lepidopteran model system [1]. The framemap drafts of B. mori genome have been reported [2, 3]. Recently, great advancements have been achieved in the research of B. mori cDNA [4, 5]. These works contributed greatly to rapidly clone and identify the functional genes of B. mori.
Chinese oak silkmoth, Antheraea pernyi, is the most well-known species among wild silkmoths of family Saturniidae, and is commercially cultivated mainly in China, India, Korea and Japan for silk production. At present, it is mostly used as a source of insect food (larva, pupa, moth) and for cosmetics. It undergoes a winter diapause as a pupa. According to the historic records, Chinese oak silkmoth originated in Shandong province of China, and began being used during the Han dynasty (40 B.C.) [6, 7]. There are currently more than one hundred varieties in China which are divided into four lines based on the larva skin color: yellow, blue, white, and yellow-cyan.
However, only about 40 functional genes of A. pernyi were cloned and partially studied to data [8] and information with reference to cDNA library of A. pernyi is scarce. To identify more genes of Chinese oak silkworm, including the characterization of specific expressed, new or unknown genes and further study their functions, construction of full-length cDNA libraries of Chinese oak silkworm is an efficient method [9]. EST analysis is an effective approach for novel gene identification, homologous gene comparison, and transcription profiling [10]. In this paper, we constructed a full-length cDNA library from Chinese oak silkworm pupa at the diapause stage by the SMART technique. Partially sequenced ESTs allowed us to identify a new gene coding KK-42-binding protein of A. pernyi that may be involved in pupa-diapause termination.
2. Materials and Methods
Sample
Chinese oak silkworm variety, Shenhuang No. 1, was selected to construct the cDNA library in this study. We bred the new variety over six years for 12 generations by cross-breeding Qing No. 6 (yellow-cyan line) and Fangshanhuang (yellow line). This variety was the first yellow line variety of Chinese oak silkworm adapted to be reared in Northeast China because of a lot of excellent economic characters. The cocoons (pupae) were kept naturally at room temperature until total RNA isolation when they were in the period of diapause stage.
Total RNA extraction and full-length cDNA library construction
Total RNA was extracted with Trizol reagent (Invitrogen, USA) according to the manufacturer's instructions. The RNA integrity was evaluated by gel electrophoresis on denaturing formaldehyde agarose. The quantity of the RNA was quantified by ultraviolet spectroscopy.
First and double-strand cDNAs were synthesized according to the protocol of the Creator SMART cDNA Library Construction kit (Clontech, USA). Double-strand cDNA synthesis was analyzed by visualization on 1% agorase gel. The ds-cDNA was digested with Sfi I restriction enzyme and size fractionated from a low-melt agarose gel to recover cDNA fractions longer than 800 bp. The cDNA fragments were directionally ligated into Sfi I degisted pDNR-LIB vector. The ligation mixture was transferred into the competent cells of E. coli DH10B by electroperation.
The unamplified cDNA library was titered by calculating of clone numbers on plates, the percentage of recombinant clones was calculated by sequencing 3 × 96 clones selected randomly. Colony PCR was used to confirm the size of inserted fragments in the library. Amplified product (5 μl) was analyzed by 1.2% agarose gel electrophoresis.
Expressed sequence tags sequencing and data analysis
cDNA clones were selected randomly from the cDNA library and sequenced. Plasmid DNAs were single-pass sequenced at the 5' end on an ABI 3730 Genetic Analyzer (Applied Biosystems) using the T7 promoter primer. Sequence files with quality values were produced and processed locally. Raw sequences were first trimmed to remove vector sequence and low-quality sequences using “Crossmatch” program. ESTs with length less than 100 bp were also discarded. The high-quality sequences were assembled and clustered using CAP3 program with the default options (http://deepc2.psi.iastate.edu/aat/cap/). An wild silkmoth cDNA database, WildSilkbase, has been developed [11], so the EST sequences of wild silkmoths and other insects including silkworm B. mori, are available to identify the cloned cDNA sequences at the BLAST search web site (http://www.cdfd.org.in/wildsilkbase/). The processed cDNA sequences were used to compare with the cDNA database of wild silkmoths with an E-value criterion of e-10 or a score of 100. If the cloned cDNA sequence was judged as not significantly matching with wild silkmoths cDNAs, we performed further the BLAST search at the GenBank database to compare all available ESTs and genes to date (http://www.ncbi.nlm.nih.gov/blast).
3. Results and Discussions
Construction of Chinese oak silkworm pupal cDNA library
A single fresh female pupa was used to extract total RNA with Trizol reagent. Agarose gel (1%) electrophoresis of RNA showed that high quality of total RNA was isolated from the pupa of Chinese oak silkworm (Fig. 1A), as shown by clear presence of bands of 28S and 18S. The concentration of the total RNA was 1.86 μg/μl. The ratio of OD260/OD280 to the total RNA was 2.03, well within the range between 2.0 and 2.2, indicating the total RNA isolated was suitable for a cDNA library construction.
Agarose gel electrophoresis of total RNA extracted from Chinese oak silkworm pupa, the synthesized double-strand cDNA (A), and the PCR products of insertion fragments from the clones selected randomly (B). Lane 1: total RNA. Lane 2: ds-cDNA. Lanes 3-14 show the PCR products of different clones. Lane M: marker (5 000, 3 000, 2 000, 1 000, 750, 500, 200, and 100 bp).
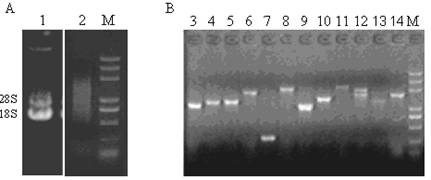
Two microgram of total RNA was subjected to reverse transcription for synthesis of the first and double-strand cDNAs. Double-strand cDNA after second strand synthesis was concentrated on range of 2 000 bp to 500 bp (Fig. 1A), suggesting that double-strand cDNAs were successfully synthesized. After ligation and transformation, we picked randomly 12 clones to perform the colony PCR with M13 primers for confirming the size of inserted fragments within recombination plasmids. The amplified cDNA fragments ranged from 800 bp to 2 500 bp (Fig. 1B), and 90% of insertion fragments was more than 1 kb in size, suggesting that the insertion fragments harbored most of the mRNAs and reached the requirement for further studies on gene structure, translation, and expression[12]. Theoretically, a cDNA library should contain at least 3.3 × 105 independent clones so that a clone derived from a low abundance mRNA would be screened out with probability of 99% from the library [12]. The capacity of the unamplified constructed cDNA library was 5 × 105 cfu/ml after calculation of clone numbers, which should meet almost all requirements to find a cDNA derived from a low-abundance mRNA. The recombination rate of the unamplified cDNA library was 95% by EST sequencing of 288 selected randomly clones. Thus, we successfully constructed a full-length cDNA library from Chinese oak silkworm pupa with high quality, providing a useful resource for the functional genomic research of Chinese oak silkworm.
Characterization of Chinese oak silkworm pupal cDNA library
EST sequencing for selected independent clones from the cDNA library has been proved to be a quick and efficient approach to assess library quality [13]. Two hundred and eighty-eight white clones were picked randomly for EST sequencing. The average meaningful readable sequence size was approximately 490 bp. After removal of the vector sequences and low-quality sequences, 250 effective sequences from the total cDNA sequences were obtained, with the total reading valid length of 75 624 bp. A total of 175 EST clusters consisting of 24 contigs and 151 singlets, also known as unigenes, were generated using online CAP3 program with the default options. The length of the unigenes obtained from EST sequencing was between 111 bp and 765 bp, with the average size of 430 bp. ESTs longer than 100 bp were retained for later analysis as the effective sequences [14]. The EST sequences were deposited in the GenBank under accession no. from GH334838 to GH335061. Twelve contigs had three or more ESTs, with the largest one containing 21 ESTs (8.4% of 250 effective ESTs) which codes mitochondrion 16S ribosomal RNA of A. pernyi (AY242996) [15], the second largest one containing 10 ESTs which has 89% sequence identity with a known EST of A. assama without function annotation (FG224715) [11], and the third largest one containing eight ESTs coding a KK-42-binding protein (see below for details).
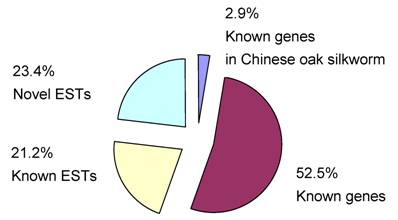
We divided all of the ESTs into four groups, which were named known genes in Chinese oak silkworm, known genes (significantly homologous to the known genes with function annotation), known ESTs (significantly homologous to the known ESTs without function annotation), and novel ESTs (no matching sequences in GenBank). The results of homology comparisons showed that five clustered EST sequences (2.9% of the 175 unigenes) are known genes in Chinese oak silkworm, 92 (52.5%) are known genes in insects, 37 (21.2%) are known ESTs in insects, and 41 (23.4%) are novel ESTs (Fig. 2). By analysis of the 97 known genes, the integrity of the full-length sequences in the cDNA library reached 91%.
Classification of all ESTs determined in this study.
By comparing the 170 clustered ESTs except for five known A. pernyi genes with the known cDNA sequences of other wild silkmoths including A. assma, A. mylitta, A. yamamai and A. polyphemus, and Samia cynthia ricini determined to date, we identified 60 (34.3%) ESTs of A. pernyi that had not found match sequences and 110 (65.7%) had homologous sequences including 30 known ESTs and 80 known genes. Of the 60 ESTs without match sequences in other wild silkmoths, only 19 ESTs were the known ESTs and functional genes in insects (7 and12, respectively).
The five known genes of A. pernyi observed in this analysis included mitochondrion 16S ribosomal RNA gene with 21 ESTs, 18S ribosomal RNA gene with five ESTs, elongation factor 1 alpha gene with four ESTs, ribosomal protein L8 gene with one ESTs, and vitellogenin gene with one EST. Expression analysis based on number of ESTs showed that mitochondrion 16S ribosomal RNA gene was over-expressed in the constructed cDNA library. This phenomenon might be due to some external stimulation, as described previously [16]. Mitochondrial RNA has fewer chemical tags, and the absence of modifications causes mitochondria RNA to activate the immune response. When a tissue was damaged by injury, infection, or inflammation, the mitochondrial RNA was released by cells, acting as a signal to the immune system to recognize the damage and help defend and repair the tissue [16]. Although there were two ribosomal RNA genes in mitochondria, no EST coding 12S rRNA gene was observed in this study. We could not explain the reason of no expression of EST coding 12S rRNA gene in this cDNA library.
In this study, 16 ESTs coding some ribosomal proteins in A. pernyi were identified including cytoplasmic L3, L5, L7, L7A, L8, L10, L10Ab, L15, S4, S5, S6, S9, S17, S3Ae, P0, and mitochondrial L2. The fifteen ESTs coding cytoplasmic ribosomal protein in A. pernyi shared 92% ~ 99% identities with those in B. mori at amino acid sequence level; whereas the one coding the mitochondrial ribosomal protein L2 showed 70% identity with that in B. mori. We also identified 18 ESTs coding various enzyme proteins of A. pernyi, which showed 79% ~ 98% identities with those of B. mori at amino acid sequence level. However, two enzymes identified in A. pernyi including aspartyl-tRNA synthetase and protein phosphatase 1 catalytic subunit beta had no match sequences in B. mori available to date. The other ESTs coded a number of proteins, such as cell division protein, GTP-binding protein, heat shock protein, KK-42-binding protein, and some chorions. The roles of these genes mentioned above have never been characterized in Chinese oak silkworm. We describe below in more detail the KK-42-binding protein (KK42-BP) that is a diapause-related protein.
Identification and characterization of a KK-42-binding protein related to pupa-diapause termination
The cDNA clone (clone name Appu0240) in the cDNA library was used to complete the full-length cDNA sequence. The full-length cDNA in this clone was 1795 bp in length, having a 5' untranslated region (UTR) of 27 bp, a 3' UTR of 259 bp with a canonical polyadenylation signal sequence AATAAA and a poly (A) tail, and an open reading frame (ORF) of 1509 bp encoding a polypeptide of 502 amino acids (Fig. 3). The predicted molecular weight of this protein was 57192 Da and isolectric point was 6.4. Predicted protein sequences of this cDNA shared 95% identity with KK42-BP of Antheraea yamamai with GenBank accession no. AB081090 (Fig. 4). No other homologous proteins were found in GenBank database by BLAST search. We therefore referred to the protein as ApKK42-BP (FJ744151).
The complete nucleotide and deduced amino acid sequences of A. pernyi KK-42-binding protein. The amino acid residues are represented by one-letter symbols. The initiation codon (ATG) and termination codon (TGA) are underlined. The abhydro_lipase domain is boxed; the abhydrolase_1 domain is boxed and shaded. The putative polyadenylation signals are double-underlined.

Sequence alignment of A. pernyi KK-42-binding protein (FJ744151) with homologue of A. yamamai (AB081090). Grey shades indicate the different amino acid residues.

KK-42 is an imidazole insect growth regulator and can terminate diapause of the pharate larvae of A. yamamai [17]. The KK42-BP was first isolated by chromatography and SDS-PAGE methods from A. yamamai in which this protein appeared throughout the periods of pre-diapause and diapause, and disappeared after the KK-42 application and long period of chilling [18], thus it was the protein associated with diapause termination with imidazole [18, 19]. Unlike Japanese oak silkworm that undergoes a winter diapause as a pharate first instar larva resting within the eggshell, Chinese oak silkmoth undergoes a winter diapause as a pupa. We have reported that an imidazole derivate, Jinlu, could induce tetramolter into trimolter in A. pernyi treated in early period of 3rd instar [20], as observed in A. yamamai [21]. The ApKK42-BP was observed to be an abundant gene with eight ESTs in 250 effective ESTs sequenced in this study in the period of diapause stage. These results suggested the ApKK42-BP may also be involved in pupa-diapause termination. To address this question, the expression pattern at different development stages will be further investigated.
In this study, we characterized the ApKK42-BP in details. The KK42-BP cDNA sequence of A. yamamai was previously isolated and deposited in GenBank database, however no other information could be available on its molecular characterization of this gene. Subcellular localization prediction showed ApKK42-BP was an extracellular protein (Reliability Index = 2; Expected Accurcy = 74) by SubLoc v1.0 server (http://www.bioinfo.tsinghua.edu.cn/SubLoc/). Prediction of transmembrane helices indicated the protein was not a membrane protein by TMHMM Server v2.0 and the predicted signal peptide was located at position 1-18 by SignalP 3.0 server (http://www.cbs.dtu.dk/services). Detection of the conserved domains in CDD database (http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) showed that ApKK42-BP had two putative conserved domains: abhydro_lipase and abhydrolase_1. The former located at position 179 - 243 was a member of the abhydro_lipase superfamily; the latter located at position 257 - 375 was a member of the esterase_lipase superfamily (Fig. 3). These results suggested ApKK42-BP may be a lipase. Lipase is an enzyme that catalyzes the hydrolysis of ester bonds in water-insoluble, lipid substrates and plays essential roles in the digestion, transport and processing of lipids (e.g. triglycerides, fats). In A. pernyi, it has been known that lipid substrates, especially triglyceride, were reserved in fat body as the high energy substance to maintain the metabolism consumption at the pupa-diapause stage. It has been speculated that KK42-BP may be involved in diapause termination as a receptor of an endogenous signaling compound [19]. Therefore, the exact roles of ApKK42-BP needs to be investigated, especially the lipase activities should be further affirmed.
In conclusion, we have first constructed a full-length cDNA library from Chinese oak silkworm pupa using the SMART method. The full-length cDNA of the KK-42-binding protein, a candidate diapause-related protein, was cloned from Chinese oak silkworm pupa and first characterized by bioinformatics analysis. Although we represented the first comprehensive set of gene sequences for A. pernyi that were not reported previously, the clones in the full-length cDNA library will be further sequenced to gain insight into the functional study of Chinese oak silkworm.
Acknowledgements
This work was supported in part by grants from the National Natural Science Foundation of China (30800803), the Program for New Century Excellent Talents in University (NCET-07-0251), the National Modern Agriculture Industry Technology System Construction Project (Silkworm and Mulberry), the Scientific Research Project for Commonweal Industry of Agricultural Ministry (nyhyzx07-020-17), the Science and Technology Fund of Anhui Province for Outstanding Youth (08040106803), the Scientific Research Project for High School of the Educational Department of Liaoning Province (2008643), respectively.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Nagaraju J, Goldsmith MR. Silkworm genomics - progress and prospects. Current Science. 2002;83:415-425
2. Mita K, Kasahara M, Sasaki S. et al. The genome sequence of silkworm, Bombyx mori. DNA Res. 2004;11:27-35
3. Xia Q, Zhou Z, Lu C. et al. A draft sequence for the genome of the domesticated silkworm (Bombyx mori). Science. 2004;306:1937-1940
4. Wang J, Xia Q, He X. et al. SilkDB: a knowledgebase for silkworm biology and genomics. Nucleic Acids Res. 2005;33:D399-402
5. Zhang YZ, Chen J, Nie ZM. et al. Expression of open reading frames in silkworm pupal cDNA library. Appl Bioch Biotech. 2007;36:327-343
6. Zhang K. Origin and radiation of oak silkworm in China. Acta Sericologia Sinica. 1982;18:112-116
7. Gu KB. Review of phylogeny on tussah sericulture. Agri Archaeol. 1995;3:206-214
8. Liu YQ, Jiang DF. Research progress on functional genes of the Tussah, Antheraea pernyi. Acta Sericologia Sinica. 2008;34:568-574
9. Gao PF, Cao GQ, Zhao HT. et al. Molecular cloning and characterization of pigeon (Columba liva) ubiquitin and ubiquitin-conjugating enzyme genes from pituitary gland library. Int J Biol Sci. 2009;5:34-43
10. Li N, Zhao ZH, Liu ZL. et al. Analysis of expressed seuquence tags from porcine liver organ. Scientia Agricultural Sinica. 2002;35:1525-1528
11. Arunkumar KP, Tomar A, Daimon T. et al. WildSilkbase: An EST database of wild silkmoths. BMC Genomics. 2008;9:338
12. Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd ed. New York: Cold Spring Harbor Laboratory Press. 2001
13. Peterson LA, Brown MR, Carlisle AJ. et al. An improved method for construction of directionally cloned cDNA libraries from microdissected cells. Cancer Res. 1998;58:5326-5328
14. Nie RE, Yang XK, Liu ZQ. cDNA library construction and analysis of some ESTs of Chrysoperla nipponensis (Okamoto) (Neuroptera: Chrysopidae). Acta Entomogical Sinica. 2008;51:792-797
15. Liu YQ, Li YP, Pan MH. et al. The Complete Mitochondrial Genome of the Chinese Oak Silkmoth, Antheraea pernyi (Lepidoptera: Saturniidae). Acta Bioch Bioph Sin. 2008;40:693-703
16. Karik K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165-175
17. Kuwano E, Fujisawa T, Suzuki K. et al. Termination of egg diapause by imidazoles in the silkmoth, Antheraea yamami. Agric Biol Chem. 1991;55:1185-1186
18. Shimizu T, Shiotsuki T, Seino A. et al. Identification of an imidazole compound-binding protein from diapausing pharate first instar larvae of the wild silkmoth Antheraea yamamai. J Insect Biotechnol Sericology. 2002;71:35-42
19. Yang P, Tanaka H, Kuwano E. et al. A novel cytochrome P450 gene (CYP4G25) of the silkmoth Antheraea yamamai: Cloning and expression pattern in pharate first instar larvae in relation to diapause. J Insect Physiol. 2008;54:636-643
20. Qin L, Liu YQ, Zhang T. et al. Study on application of imidazole derivative on Chinese Tusser (Antheraea pernyi). J Shenyang Agricultural Univ. 1999;30:31-34
21. Hong J, Ye GY, Ling YL. et al. Effect of Jinlu, an imidazole derivate on ultrastructure of larval silk gland cells of Antheraea yamamai (Lep.: Saturniidae). J Chinese Electron Microsc Soc. 1999;18:583-594
Author contact
![]() Correspondence to: Y. Q. Liu, College of Bioscience and Biotechnology, Shenyang Agricultural University, Shenyang 110161, People's Republic of China; Tel: +86-24-8848-7163; Fax: +86-24-8841-1127; E-mail: liuyqlypcom.cn. Or to: Z. J. Wei, Department of Biotechnology, Hefei University of Technology, Hefei 230009; People's Republic of China; Tel: 86-551-2901505-8412; Fax: 86-551-2901507; E-mail: zjweiedu.cn
Correspondence to: Y. Q. Liu, College of Bioscience and Biotechnology, Shenyang Agricultural University, Shenyang 110161, People's Republic of China; Tel: +86-24-8848-7163; Fax: +86-24-8841-1127; E-mail: liuyqlypcom.cn. Or to: Z. J. Wei, Department of Biotechnology, Hefei University of Technology, Hefei 230009; People's Republic of China; Tel: 86-551-2901505-8412; Fax: 86-551-2901507; E-mail: zjweiedu.cn