International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 22; 2026
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Acknowledgements
Abbreviations
References
Author biography
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Acknowledgements
Abbreviations
References
Author biography

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2009; 5(5):489-499. doi:10.7150/ijbs.5.489 This issue Cite
Research Paper
An apoA-I mimetic peptide increases LCAT activity in mice through increasing HDL concentration
Xun Chen, Charlotte Burton, Xuelei Song, Lesley Mcnamara, Annunziata Langella, Simona Cianetti, Ching H. Chang, Jun Wang ![]()
Merck Research Laboratories, Rahway, New Jersey 07065, USA
Received 2009-5-9; Accepted 2009-7-20; Published 2009-7-28
Citation:
Chen X, Burton C, Song X, Mcnamara L, Langella A, Cianetti S, Chang CH, Wang J. An apoA-I mimetic peptide increases LCAT activity in mice through increasing HDL concentration. Int J Biol Sci 2009; 5(5):489-499. doi:10.7150/ijbs.5.489. https://www.ijbs.com/v05p0489.htm
Other stylesAbstract
Lecithin cholesterol acyltransferase (LCAT) plays a key role in the reverse cholesterol transport (RCT) process by converting cholesterol to cholesteryl ester to form mature HDL particles, which in turn deliver cholesterol back to the liver for excretion and catabolism. HDL levels in human plasma are negatively correlated with cardiovascular risk and HDL functions are believed to be more important in atheroprotection. This study investigates whether and how D-4F, an apolipoprotein A-I (apoA-I) mimetic peptide, influences LCAT activity in the completion of the RCT process. We demonstrated that the apparent rate constant value of the LCAT enzyme reaction gives a measure of LCAT activity and determined the effects of free metals and a reducing agent on LCAT activity, showing an inhibition hierarchy of Zn2+>Mg2+>Ca2+ and no inhibition with β-mercaptoethanol up to 10 mM. We reconstituted nano-disc particles using apoA-I or D-4F with phospholipids. These particles elicited good activity in vitro in the stimulation of cholesterol efflux from macrophages through the ATP-binding cassette transporter A1 (ABCA1). With these particles we studied the LCAT activity and demonstrated that D-4F did not activate LCAT in vitro. Furthermore, we have done in vivo experiments with apoE-null mice and demonstrated that D-4F (20 mg/kg body weight, once daily subcutaneously) increased LCAT activity and HDL level as well as apoA-I concentration at 72 hours post initial dosing. Finally, we have established a correlation between HDL concentration and LCAT activity in the D-4F treated mice.
Keywords: D-4F, lipoprotein, apoE, HDL, RCT, atherosclerosis, LCAT
1. Introduction
Cardiovascular disease (CVD) remains the leading cause of death in developed countries and is estimated to be the leading cause of death in developing countries by 2010 despite advanced medical therapies. Atherosclerosis, the process of accumulating cholesterol in the arteries, leads to clinical events such as myocardial infarction and stroke. It is well documented that high density lipoprotein (HDL) is inversely correlated with coronary heart diseases. HDL involves a multi-step physiological process that transports excess cholesterol from the peripheral tissues including atherogenic foam cells or macrophages in plaques to the liver for catabolism and excretion [1]. This process is called reverse cholesterol transport (RCT) and has been considered an essential function for atheroprotection by HDL. In addition, niacin and fibrates that increase HDL level have been used in clinics to prevent atherosclerotic progression. However, Torcetrapib, a cholesterol ester transfer protein (CETP) inhibitor that dramatically increases plasma HDL levels, recently failed in the Phase III clinical trial due to an increased risk of death and cardiac events. Therefore, the research focus has greatly shifted from HDL concentration to HDL functions such as its anti-inflammatory [2], anti-oxidant, and anti-thrombotic [3] functions; its stimulation of ABCG1-dependent cholesterol efflux [4]; its SR-BI-dependent delivery of cholesterol to the liver [5, 6]; and its increase of lecithin cholesterol acyltransferase (LCAT) activity. Although HDL is still believed to play an important role in atheroprotection, the lesson learned from the Torcetrapib experience is that the detailed understanding of HDL functions, metabolism and its metabolic regulators, as well as their relationship with HDL compositions, is critically important for the development of new therapies. ApoA-I, the key protein in HDL, is receiving increasing attention for developing treatments for atherosclerosis. Overexpression of apoA-I decreased atherosclerosis in animal studies [7, 8]. Infusion of the complexes of phospholipids and apoA-I or its genetic variant (R173C), apoA-IMilano, showed anti-atherosclerotic effects in animals and humans [9-13]. The benefit of apoA-I has been demonstrated to be directly involved in all steps of RCT process [14]. However, due to the high cost of production of apoA-I protein, the limitation of structural diversity, and limited route of administration, small peptides that can mimic apoA-I activities have been explored as new therapeutic agents for the treatment of atherosclerosis. Numerous mimetic peptides exhibit anti-atherosclerotic properties in different animal models and a few, such as D-4F and ETC-642 [15] are moving into human clinical trials. However, like apoA-I itself, the precise mechanism of these peptides that is responsible for atheroprotection remains unclear [16]. Most of these peptides, if not all, share amphipathic properties. Similar to apoA-I, these peptides interact with phospholipids and stimulate cholesterol efflux from microphages and foam cells, however, their effects on LCAT activity and other activities independent of RCT vary. This study investigated the biochemical characteristics of the LCAT enzyme and determined the effects of D-4F on the stimulation of LCAT activity both in vitro and in vivo, demonstrating a correlation between HDL level and LCAT activity in mice.
2. Materials and Methods
2.1. Reagents
All reagents were obtained from Sigma (St. Louis, MO) unless otherwise indicated. Cholesterol was from Steraloids Inc. (Cat. C6760; Newport, RI). [3H]cholesterol (40 Ci/mmol) was from PerkinElmer Life Sciences (Cat. NET139001MC; Waltham, MA). The LCAT activity assay kit was from EMD Biosciences (Cat. 428900; San Diego, CA). The Total Cholesterol E and HDL Cholesterol Kits were from Wako Chemical USA Inc. (Cat. 439-17501; Richmond, VA). The 4-20% Tris-Glycine native gradient gel and running buffer were from Invitrogen (Calsbad, CA) and the high molecular weight marker was from GE healthcare (Cat. 17-0445-01; Piscataway, NJ). Purified human apoA-I, rabbit anti-mouse apoA-I antibody, and purified mouse apoA-I were from Meridian Life Science Inc. (Cincinnati, OH). Goat anti-mouse apoA-I polyclonal antibody was from Rockland Immunochemicals Inc. (600-101-196; Gilbertsville, PA). DELFIA Eu-N1-labeled anti-rabbit antibody and Enhancement solution were from PerkinElmer Life Sciences (Waltham, MA). The BCA Protein Assay Kit was from Pierce Biotechnology (Cat. 23227; Rockford, IL).
2.2. In vivo study
ApoE-null mice were purchased from Jackson Laboratories (Bal Harbor, Maine). All animal experimental procedures followed the NIH guideline and were approved by the Merck Research Laboratories Instututional Animal Care and Use Committee. Mice were enrolled at 24 weeks of age after feeding with a high fat Western diet (HF; Harlan Teklad TD#88137) for 20 weeks. The D-4F peptide was prepared in PBS solution containing 50 mM mannitol and dosed at 20 mg/kg body weight subcutaneously once daily for 3 days except where indicated. Samples were collected at each designated time point and were added to tubes containing EDTA. Plasma was isolated by centrifugation and kept frozen at -80ºC for further analysis.
2.3. Determination of plasma LCAT activity
Mouse plasma LCAT activity was determined using the LCAT Activity Assay Kit according to the manufacturer's instructions with some modifications. Briefly, 5 µl aliquots of mouse plasma were incubated at 37ºC with the fluorescently labeled cholesterol in assay buffer containing 150 mM NaCl, 10 mM Tris-HCl, 4 mM β-mercaptoethanol and 1 mM EDTA at pH 7.4. The total assay volume was 200 µl. After 0, 20 and 40 min., 45 µl of the reaction mixture was added to 135 µl of the READ reagent (150 mM NaCl, 10 mM Tris-HCl, and 1 mM EDTA at pH 7.4). The conversion of cholesterol (Em. 470 nm) to cholesteryl ester (Em. 390 nm) at 340 nm excitation was determined in a fluorescence microplate reader (SPECTRAmax GEMINI, Molecular Devices Co., Sunnyvale, CA). The change of ratio of the two intensities (470/390) was calculated.
2.4. Preparation of HDL associated LCAT enzyme
Fresh human or mouse blood was drawn into tubes containing potassium EDTA (Becton Dickinson, Franklin lakes, NJ), and plasma was separated immediately by low-speed centrifugation at 2500 rpm (1430g ) for 30 min at 4ºC. Size exclusion chromatography was performed at ambient temperature using a Superose-6 10/300 column on a Bio-Rad FPLC system (Hercules, CA). A 200 µl aliquot of plasma was injected per run, and eluted in PBS with 1 mM EDTA at a flow rate 0.2 ml/min. A total of 72 fractions with 0.27 ml per fraction were directly collected into microtiter plate for further analysis. Total cholesterol in each fraction was measured using the Cholesterol E Kit, according to the manufacturer's instructions. LCAT activity was measured using the LCAT Activity Assay Kit as described above (Fig. 1). HDL fractions containing LCAT activity were pooled and stored at -80ºC. Protein concentration was determined by BCA.
Figure 1
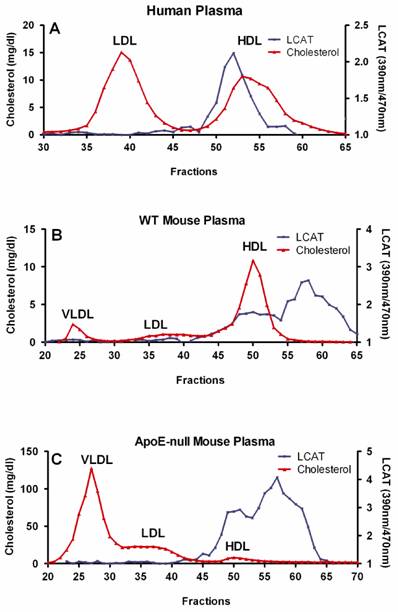
Preparation of HDL associated LCAT. Size exclusion chromatography was performed at ambient temperature using a Superose-6 10/300 column on a Bio-Rad FPLC system (Hercules, CA). A 200 µl aliquot of plasma was injected per run, and eluted in PBS with 1 mM EDTA at a flow rate of 0.2 ml/min. Total cholesterol in each fraction was measured using the Cholesterol E Kit. LCAT activity was determined as following: A. 20 µl of each fraction of human plasma were incubated at 37ºC for 5 h; B. 2.5 µl of each fraction of WT mouse plasma were incubated at 37ºC for 2 h; C. 10 µl of each fraction of apoE-null mouse plasma were incubated at 37ºC for 4 h.
2.5. LCAT substrate preparation
The substrate particles composed of L-α-Phosphatidylcholine derived from egg yolk phosphatidylcholine (EPC), cholesterol with 1% of [3H]cholesterol, and human apoA-I with a molar ratio of 100:10:1 as described [17, 18] with some modification. The molar ratio was 100:10:10 if D-4F peptide was used. A control EPC-cholesterol vesicle was also made at a molar ratio of 100:10. After lyophilizing EPC and cholesterol, apoA-I or D-4F and assay buffer (10 mM Tris, 140 mM NaCl, and 1 mM EDTA, pH 7.4) were added to the dry powder. The particles were created by sonication on ice with a microtip at power 23 for 5 min x 5 times with a 2 min pause between each cycle followed by centrifugation at 1000 rpm (228g) for 5 min at 4ºC. The supernatant was kept at 4ºC for further characterization. The particles were confirmed by native gradient gel electrophoresis and the size of particles was determined against high molecular weight markers (data not shown).
2.6. LCAT activation assay
In the LCAT activation assay, the synthetic particles were used as substrates. After a 15 min incubation of 8 µl of 10-fold diluted substrates with 0.6% bovine serum albumin (fatty acid free) in assay buffer mentioned above, β-mercaptoethanol was added to a final concentration of 2 mM. The LCAT reaction was initiated by addition of 2 µl HDL associated-human LCAT and total volume was brought to 40 µl. After 5 hours at 37 ºC the reaction was quenched by freezing the samples in a -80ºC freezer. After the samples were thawed, 20 µl aliquots were spotted on silica TLC plates. The cholesterol and cholesteryl esters were then separated by thin-layer chromatography in petroleum ether/diethyl ether/acetic acid (80:30:1, v/v/v) mixture. The cholesterol and cholesteryl ester bands were scraped and counted in scintillation cocktail. The percentage conversion of cholesterol to cholesteryl ester was quantified by their respective associated radioactivity.
2.7. Cholesterol efflux assay
Mouse macrophage cells (RAW 264.7, ATCC) were cultured in Dulbecco's Modified Eagle Medium (Invitrogen 12491) with 10% FCS and 1% Pen Strep Glutamine (GIBCO 10378) in T225 flasks to a confluency of ~80%. The cells were then seeded into 48 well plates at a concentration of 1 x 105 cells/0.2 ml /well in the same medium now supplemented with 3[H] cholesterol (Perkin-Elmer) to a final 5 µCi/ml. After 24 hours, the cells were washed once and starved overnight with serum-free medium supplemented with 0.1% lipid-free BSA. Finally, apoA1 and D-4F with or without cAMP in serum-free medium were added and incubated for another 24 hours. Subsequently, medium and cell lysate (0.5% Triton X-100 in 1 mM HEPES, pH 7.5) were collected and aliquots were mixed separately with scintillation cocktail and counted. Cholesterol efflux was calculated as the percentage of radioactivity associated with medium over the sum of radioactivity of both medium and lysate. The cAMP-dependent cholesterol efflux was determined by the difference between cholesterol measured in the presence and the absence of cAMP.
2.8. Other assays
Plasma total cholesterol and HDL were quantified by Wako cholesterol kits. Apolipoprotein A-I was determined by ELISA and Western blot using rabbit anti-mouse apoA-I and quantified against purified mouse apoA-I. In the ELISA assay, the plates were coated with goat anti-mouse apoA-I polyclonal antibody at a concentration of 0.5 µg/ml overnight at 4°C. After blocking, the purified mouse apoA-I standard was diluted to 50 ng/ml, and then 2 fold serial dilution, and mouse plasma samples were diluted 50,000 times in assay buffer, 100 µl diluted samples were added to the wells and incubated at 37°C for 2 hours. Subsequently, rabbit anti-mouse apoA-I antibody was added and incubated at 37°C for 1 hour. Finally, DELFIA Eu-N1-labeled anti-rabbit antibody was added and incubated at room temperature for 30 minutes. The signal was detected by adding the enhancement solution and measured in an EnVision multilabel plate reader.
3. Results
3.1. Determination of enzyme kinetics
In addition of apoA-I, apoE is an important protein in the lipoprotein transport system. The atheroprotective properties of apoE has been demonstrated in numerous models. ApoE-null mice on a high fat/high cholesterol diet has also been well documented as an animal model for atherosclerosis [19]. Wild type (WT) and apoE-null mice have different lipoprotein profiles, in that WT mice have high HDL and very low LDL and VLDL while apoE-null mice have high VLDL and very low HDL, with total cholesterol levels approximately 6 times higher than that of WT mice. In order to determine the role of LCAT in atherogenesis, we purified HDL-associated LCAT enzymes and measured their activities in a time course study at 37ºC. We used fluorescence-labeled cholesterol as substrate and found that cholesteryl ester is formed in a dose- and time-dependent manner in the presence of LCAT (Fig. 2A). Addition of the HDL-associated LCAT increased the rate of cholesteryl ester formation dramatically. Further, we analyzed the enzyme kinetics. LCAT reaction is a first-order reaction, and rate constants increased linearly with increasing amounts of LCAT enzyme (Fig. 2B), therefore the increase in the rate constant is a measure of LCAT activity. We define a unit of LCAT activity as an increment in the rate constant kapp of 1.0 min-1. Here, we observed that the specific LCAT activities of WT and apoE-null mice are 0.00199 ± 0.0001058 and 0.001322 ± 0.000124 min-1μg-1, respectively, indicating that the specific LCAT activity of HDL-associated LCAT enzyme from apoE-null mice is 1.5-fold lower than that from WT mice.
Figure 2
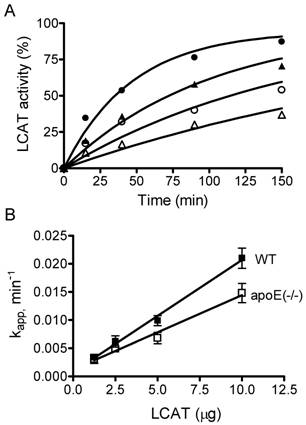
Determination of LCAT kinetics. A. The assay was performed at 37ºC as described in Materials and Methods with increasing amounts of HDL-associated LCAT purified from wild type (WT) mice (∆1.25; ○2.5; ▲5; ●10 μg). The cholesterol was converted to cholesteryl ester by LCAT and the reaction was determined by measuring the change of fluorescence intensity ratios (470 nm/390 nm) at different times shown on the abscissa. Similar experiments were performed using HDL-associated LCAT purified from apoE-null mice (data not shown). B. LCAT activity, presented as the apparent rate constant, kapp, that was obtained from each data set of A, plotted against the amount of LCAT added. The goodness-of-fit of linear regression (r2) is 0.98 for both WT (■) and apoE-null (□) mouse HDL-associated LCAT enzymes. Their specific LCAT activities are 0.00199 ± 0.0001058 and 0.001322 ± 0.000124 min-1μg-1 for WT and apoE-null mice, respectively. Similar results were obtained in two other experiments.
3.2. Effects of metals and reducing agent on LCAT activity
To optimize the assay condition and understand physiological regulation by metals, the effects of divalent cations and reducing agent β-mecaptoethanol on LCAT activity were examined. We used EPC-cholesterol vesicles as substrate and added human apoA-I at a final concentration of 0.5 µM which mimics the condition of the pre-formed particles of EPC/Ch/apoA-I (100:10:1, mol/mol/mol). The reaction was initiated by adding HDL-associated human LCAT enzyme as described in Materials and Methods. With the addition of 1 mM EDTA, an inhibition hierarchy of Zn2+>Mg2+>Ca2+ was observed, providing IC50 values of 1.1, 7.3, and 21.7 mM for Zn2+, Mg2+ and Ca2+, respectively (Fig. 3A, 3B, 3C). The IC50 observed for Zn2+ is close to the EDTA concentration added, therefore the IC50 for free (unchelated) Zn2+ might be very low and would need to be carefully determined as described for greater accuracy [20]. No inhibition was observed with β-mercaptoethanol at concentrations up to 10 mM (Fig. 3D).
Figure 3
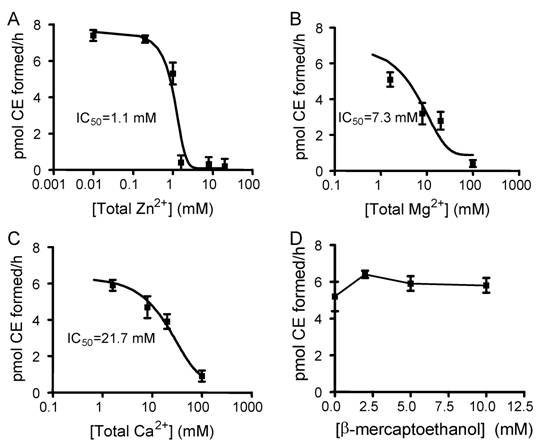
The effects of divalent cations and β-mecaptoethanol on LCAT activity. The assay was performed as described in Materials and Methods with a serial dilution of metals and β-mecaptoethanol. With EPC-cholesterol as substrate, 0.5 µM of apoA-1 and 2 μg of HDL-associated human LCAT were added. Fig. A, B, and C show an inhibition hierarchy of Zn2+>Mg2+>Ca2+, giving the IC50 values of 1.1 mM , 7.3 mM, and 21.7 mM for Zn2+, Mg2+ and Ca2+, respectively. Fig. D shows no inhibition with β-mercaptoethanol up to 10 mM. Error bars represent standard deviations of duplication in two independent experiments.
3.3. In vitro LCAT activation properties of apoA-I and D-4F
To investigate the ability of human apoA-I and the mimetic peptide D-4F to activate LCAT, apoA-I and D-4F particles were synthesized with EPC and cholesterol as described in Materials and Methods. The particle sizes showed diameters of 8.6-12.2 nm on a native gel separated by electrophoresis (data not shown). The bands on the native gel were broader than particles made with Dipalmitoylphosphatidylcholine (DPPC) which had both size and discoidal shape confirmed by electron microscope (EM) (Fig. 4F).
ABCA1-dependent cholesterol efflux from peripheral tissues is the first step in the RCT process. In the mouse macrophage cell line, RAW 264.7, the basal expression level of ABCA1 is very low, but can be significantly increased by adding cAMP (Fig. 4A). This characteristic is useful for determining the ABCA1-dependent cholesterol efflux in vitro. ApoA-I added to RAW cells without cAMP produced almost no efflux activity, but in the presence of cAMP efflux levels increased dramatically (Fig. 4B). The activities of free apoA-I, apoA-I particles, free D-4F and D-4F particles in the stimulation of ABCA1-dependent cholesterol efflux from macrophages were assayed, resulting in EC50 values of 101 nM, 103 nM, 2.2 μM and 2.8 μM, respectively (Fig. 4C, 4D). ApoA-I or D-4F particles or EPC-cholesterol vesicles were used as substrates in the LCAT activation assay. As shown in Fig. 5A and 5B, human HDL-associated LCAT had a basal LCAT activity converting 4.6% to cholesteryl ester; proteoliposome containing ApoA-I increased LCAT activity by 5.9 fold while peptidosome containing D-4F showed no LCAT activation in the assay.
Figure 4
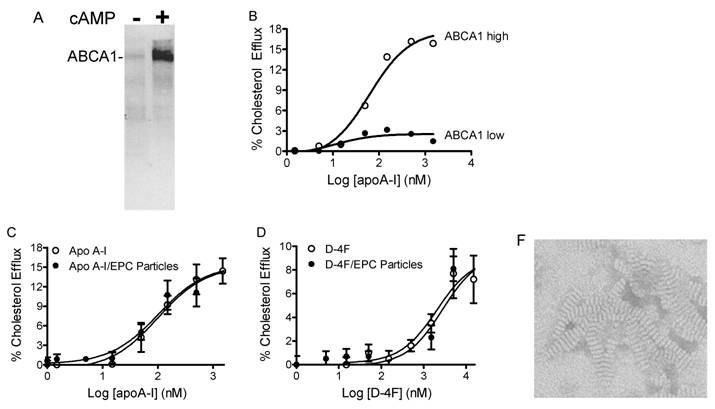
ABCA1 dependent cholesterol efflux assay. The assay details are described in Materials and Methods. A, ABCA1 expression levels in RAW cells were determined by western blot with anti-mouse ABCA1 antibody. Very low basal expression levels of ABCA1 are dramatically increased by the addition of cAMP. B, apoA-I-stimulated cholesterol efflux from macrophage is ABCA1 level dependent. C, apoA-I and its formulated particles show activity in stimulating cholesterol efflux through the ABCA1 transporter, providing EC50 values of 101 nM and 103 nM, respectively. D, D-4F and its formulated particles show activity in stimulating cholesterol efflux through the ABCA1 transporter, providing EC50 values of 2.2 μM and 2.8 μM, respectively. F, both size and discoidal shape of D-4F particles made with Dipalmitoylphosphatidylcholine (DPPC) (D4F:DPPC = 1:10) were confirmed by electron microscope (Magnification 45000, disk size; 6 x 12 nm).
The ability of apoA-I and D-4F to activate LCAT activity was also determined using the method that Datta et al. used to demonstrate LCAT activation by the 5F and 6F, analogs of D-4F [21]. Following that method, our EPC-cholesterol vesicles were used as substrates and apoA-I and D-4F were titrated (Fig. 5C). Again, apoA-I showed a dose-dependent activation of LCAT with an EC50 of 72.8 nM, while D-4F was unable to activate LCAT up to 28 µg/ml (12.1 μM).
Figure 5
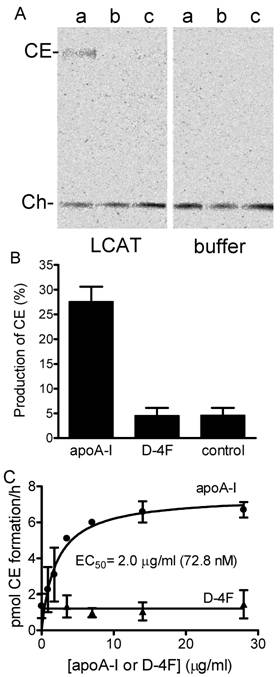
LCAT activation with apoA-1 and D4F peptide. A, phosphorimaging results of LCAT assay. The assay was performed as described in Materials and Methods. Pre-formed apoA1/EPC/cholesterol (a), D-4F/EPC/cholesterol (b) and EPC-cholesterol particles (c) as substrates, incubated with 2 μg of HDL- associated human LCAT (left panel) or assay buffer (right panel) at 37ºC. After 5 hours the reaction was stopped by freezing the samples. The cholesteryl ester formed was separated from free cholesterol by TLC and visualized by phosphoimaging. B, production of cholesteryl ester from the same assay. The cholesterol and cholesteryl ester bands on TLC plate were also visualized by exposing the plate to iodide vapour and the visible bands were scraped from the plate and counted for [3H] signal in scintillation fluid. The conversion of cholesterol to cholesteryl ester was calculated based on their associated radioactivity. Experiments were done at least three times in duplicates. C, EPC/cholesterol vesicle as substrate. Increasing amounts of apoA-1 (●) and D-4F (▲) were incubated with 2 μg partially purified HDL- associated human LCAT. Error bars represent two independent experiments with duplication.
3.4. Dosing D-4F in mice
In light of the effect of D-4F on LCAT activity in vitro, we performed additional experiments to measure plasma LCAT activity after treatment with D-4F in vivo. 20 mg/kg of D-4F peptide or 50 mM mannitol were injected into apoE-null mice as a single dose or once daily for 3 days. Twenty-four hours after a single dose (24 h) or after last injection of three doses (72 h), mice were sacrificed and plasma samples were analyzed for different parameters. A significant increase of LCAT activity was observed in the 72 h D-4F treated group but not in the 24 h group (Fig. 6A). The plasma samples were analyzed by FPLC and each fraction was assayed for total cholesterol levels. Total cholesterol levels increased slightly in the 24 h group (1,512 ± 54 mg/dl) and slightly decreased in 72 h group (1,218 ± 79 mg/dl) compared to the control group (1,301 ± 124 mg/dl), but none reached statistical significance. Surprisingly, HDL levels did not change 24 h after first dose but increased significantly 24 h after the third dose (from 14.3 ± 3.8 to 28.1 ± 3.5 mg/dl). When HDL cholesterol concentration was plotted against LCAT activity, the treated group (72h) had a parallel increase in both LCAT activity and HDL level, which demonstrated that LCAT activity is positively correlated with plasma HDL cholesterol concentration (Fig. 6B). Plasma taken 1 h and 4 h after the first dosing of D-4F showed no differences from the vehicle controls in LCAT activity or HDL levels, even though D-4F reached its highest concentration in plasma at 4 h. (data not shown). To understand whether the HDL increase is accompanied by an increase in apoA-I, apoA-I ELISAs with rabbit anti-mouse apoA-I antibody were done and confirmed by western blots. The apoA-I protein in the 72 h D-4F group significantly increased 1.27-fold compared to the vehicle group (Fig. 6C).
Figure 6
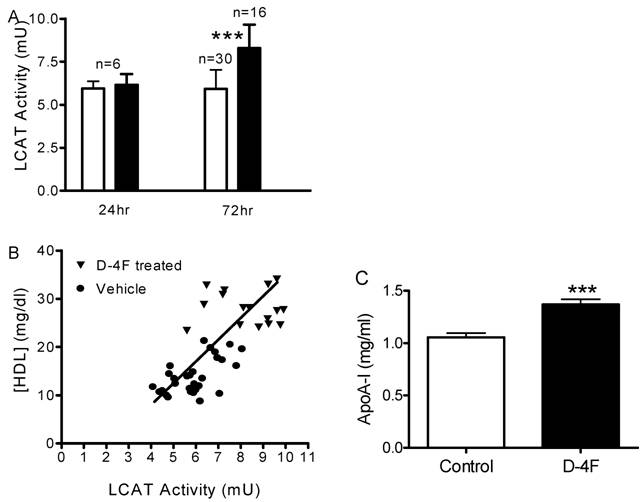
D-4F increases plasma LCAT activity, HDL, and apoA-I in vivo. ApoE-null mice were injected subcutaneously with D-4F peptide at 20 mg/kg once daily for 3 days, and plasma samples were collected at different time points. LCAT activity and HDL concentrations were measured as described in Materials and Methods. A, plasma LCAT activity of apoE-null mouse after D-4F treatment (□ Vehicle, ■ D-4F treated). The plasma samples were collected 24 hours after first (24 h) and third (72 h) dosing. No change of LCAT activity was seen at 24 hours after single dosing while a significant increase of LCAT activity was seen 24 hours after the third dosing (p = 2x10-6). Results were from two independent experiments. B, correlation between LCAT activity and HDL concentration. LCAT activities of control plasma (●) and 24 hr after third dose with D-4F (▼) were plotted against their HDL concentrations. The goodness-of-fit of linear regression (r2) is 0.64. C, apoA-I ELISA of apoE-null mouse plasma collected from control group (open bar) and 24 hours after third dose with D-4F (closed bar). Error bars represented the standard division (*: P<0.05, ***: P<0.001).
4. Discussion
Lecithin cholesterol acyltransferase (LCAT) is an enzyme responsible for free cholesterol (FC) esterification, which is critical for HDL maturation and the completion of the RCT process. It circulates in plasma, primarily associates with HDL, and is activated by apoA-I [22, 23]. Genetic mutations in human LCAT genes cause familial LCAT deficiency [24] and fish eye disease [25], which result in a decrease of plasma HDL and accumulation of cholesterol in organs. LCAT activity was found significantly decreased in atherosclerotic patients [26] and this decrease was accompanied by lower HDL cholesterol levels as well as lower apoA-I mass due to its rapid catabolism [27, 28]. A number of animal models have also been generated to investigate the role of LCAT activity in atherosclerosis [29-32]. However, reports showing the effects of LCAT on atherosclerosis are controversial. ApoA-I mimetic peptides with 18-22 amino acids are designed to mimic apoA-I's ability to form class A amphipathic helices without sharing the sequence similarity of apoA-I protein. Their sequence diversity offers them opportunity to surpass apoA-I's atheroprotection. However, mimicking all of the atheroprotective properties of apoA-I protein (243 amino acids) that contains 10 different amphipathic helices with one helix could be very challenging. Both mutagenesis studies and human genetic data suggest that different helices of apoA-I are important for different functions. For example, the mutations at the N-terminal (helices 2-7) are associated with familial amyloidosis while the mutations at the C-terminal (helices 4-10) are associated with LCAT activation, despite the fact that these mutations are associated with low HDL levels in plasma [33]. RCT is a process that requires all proteins in the pathway to work in concert. Then, how can a mimetic peptide enhance the RCT process if it only increases cholesterol efflux from peripheral cells without increasing other enzyme and receptor activities such as LCAT and SR-BI? To answer this question we purified HDL-associated LCAT from wild type and apoE-null mice and studied LCAT activity and its kinetics (Fig. 2). Despite the fact that plasma HDL cholesterol concentration is 6-fold lower, apoA-I concentration is 1.5-fold lower in apoE-null mice than in wild type mice (Fig. 1, 6B, and 6C, data not shown). Correspondingly, the specific activity of purified HDL-associated LCAT from apoE-null mice is 1.5-fold lower than that from wild type mice (Fig. 2), suggesting that 1) the HDL composition alters LCAT activity, and 2) poor LCAT activity might play a role in apoE-null mice for atherogenesis. In addition, in atherogenesis, poor LCAT activity may result as a consequence of enhanced haptoglobin level on apoA-I function [16, 34-36]. Since a single amino acid mutation at any place in helices 4-10 of the apoA-I protein is associated with LCAT activation, a subtle change in a mimetic peptide might alter its capability to activate LCAT [21]. To determine the LCAT activation by D-4F, we performed two types of assays. One was using reconstituted pre-beta nano-particles (peptide/ or apoA-I/EPC/cholesterol) as substrates, and initiating the LCAT reaction by the addition of LCAT enzyme (Fig. 5A, 5B); another was using EPC-cholesterol vesicles as substrate with addition of a serial diluted peptide or apoA-I followed by adding LCAT enzyme (Fig. 5C). ApoA-I showed good LCAT activation but D-4F did not exhibit any activation in either assay. This conclusive finding raised interesting questions on how D-4F can complete the RCT process and increase cholesterol excretion in vivo [37]. Also, does D-4F, as a dominant negative inhibitor, block endogenous apoA-I-induced LCAT activity, which in turn increases pre-β HDL and decreases mature HDL whose function is considered more important for atheroprotection? To address these issues we performed in vivo studies with apoE-null mice. Our study with multiple dosing of D-4F in apoE-null mice showed that total cholesterol but not HDL cholesterol increased at 24 hours after the first subcutaneous injection. However, HDL cholesterol level and apoA-I concentration increased 24 hours after the third injection. Notably, it has been reported in literature that D-4F had no effect on total cholesterol or HDL-cholesterol concentrations [38]. This discrepancy might be due to different animal models, dosing regimens, routes of administration, and the time that samples were collected. Our study showed that D-4F increases LCAT activity in vivo through an increase of apoA-I as well as HDL concentration. The correlation demonstrated between LCAT and HDL concentrations indicates that the D-4F-induced HDL DOES have a good function in stimulating LCAT activity. Recently Song et al. (manuscript in preparation) have demonstrated that D-4F is capable of forming HDL-like particles and delivering cholesterol to the liver cells selectively through SR-BI and enhances the cholesterol delivery by native HDL. Our results along with other in vivo studies [37, 39] suggest that the RCT pathway is an important therapeutic target for new drug discovery and development for the treatment of atherosclerosis.
Notably, the concentration of total apoA-I protein in human plasma is in the range of 35-55 μM (1-1.5 mg/ml), however the EC50 of LCAT activation by free apoA-I is only 72.8 nM (2 μg/ml) (Fig. 5C) which is similar to the EC50 values of free or discoidal apoA-I in stimulating ABCA1-dependent cholesterol efflux (101 or 103 nM, Fig. 4C). These similarities suggest that free and discoidal apoA-I are the most active forms in reverse cholesterol transport and that slight changes of their concentrations in circulation might alter RCT process dramatically. These also suggest that apoA-I infusion at a very low dose (such as 1 mg/kg body weight), reaching a plasma concentration above 100 nM (>EC50) might be sufficient for the treatment of atherosclerosis.
There are many challenges in drug discovery [40] and development by targeting HDL. The mechanism of HDL functions and the effects of the complex remodeling of the particles' composition are poorly understood. There is no ideal mouse model with lipoprotein profiles and atherosclerotic lesion biology similar to that of humans, partly due to the mouse's inherent lack of CETP. In addition, large population outcome trials (cost and time consuming) are required because high HDL levels alone do not always translate to a clinical benefit. No endpoint surrogates other than medical events and death have been validated although magnetic resonance imaging (MRI) and intravascular ultrasound (IVUS) have shown a great potential for application [13]. Therefore, the detailed understanding of each agent that targets HDL functions, modulations and its regulations may lead to the discovery of good biomarkers and provide valuable guidance for clinical trials and applications.
Acknowledgements
We thank My-Hanh Lam, Ying Chen and Denise Milot for technical assistance. We are grateful to Dr. Carlo Tacchetti and Dr. Cristina Gagliani for electron microscope (EM) analysis.
Abbreviations
CVD: Cardiovascular disease; ABCA1: ATP binding cassette transporter A1; apoA-I: apolipoprotein A-I; LCAT: lecithin cholesterol acyltransferase; RCT: reverse cholesterol transport; CETP: cholesteryl ester transfer protein; EPC: egg yolk phosphatidylcholine; DPPC: Dipalmitoylphosphatidylcholine; SR-BI: scavenger receptor B1.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. J Lipid Res. 1995;36:211-28
2. Barter PJ, Nicholls S, Rye KA, Anantharamaiah GM, Navab M, Fogelman AM. Antiinflammatory properties of HDL. Circ Res. 2004;95:764-72
3. Mineo C, Deguchi H, Griffin JH, Shaul PW. Endothelial and antithrombotic actions of HDL. Circ Res. 2006;98:1352-64
4. Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci U S A. 2004;101:9774-9
5. Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518-20
6. Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34-47
7. Rubin EM, Ishida BY, Clift SM, Krauss RM. Expression of human apolipoprotein A-I in transgenic mice results in reduced plasma levels of murine apolipoprotein A-I and the appearance of two new high density lipoprotein size subclasses. Proc Natl Acad Sci U S A. 1991;88:434-8
8. Paszty C, Maeda N, Verstuyft J, Rubin EM. Apolipoprotein AI transgene corrects apolipoprotein E deficiency-induced atherosclerosis in mice. J Clin Invest. 1994;94:899-903
9. Shah PK, Yano J, Reyes O, Chyu KY, Kaul S, Bisgaier CL. et al. High-dose recombinant apolipoprotein A-I(milano) mobilizes tissue cholesterol and rapidly reduces plaque lipid and macrophage content in apolipoprotein e-deficient mice. Potential implications for acute plaque stabilization. Circulation. 2001;103:3047-50
10. Tangirala RK, Tsukamoto K, Chun SH, Usher D, Pure E, Rader DJ. Regression of atherosclerosis induced by liver-directed gene transfer of apolipoprotein A-I in mice. Circulation. 1999;100:1816-22
11. Chiesa G, Sirtori CR. Use of recombinant apolipoproteins in vascular diseases: the case of apoA-I. Curr Opin Investig Drugs. 2002;3:420-6
12. Ameli S, Hultgardh-Nilsson A, Cercek B, Shah PK, Forrester JS, Ageland H. et al. Recombinant apolipoprotein A-I Milano reduces intimal thickening after balloon injury in hypercholesterolemic rabbits. Circulation. 1994;90:1935-41
13. Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M. et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. Jama. 2003;290:2292-300
14. Eisenberg S. High density lipoprotein metabolism. J Lipid Res. 1984;25:1017-58
15. Sethi AA, Amar M, Shamburek RD, Remaley AT. Apolipoprotein AI mimetic peptides: possible new agents for the treatment of atherosclerosis. Curr Opin Investig Drugs. 2007;8:201-12
16. Spagnuolo MS, Cigliano L, D'Andrea LD, Pedone C, Abrescia P. Assignment of the binding site for haptoglobin on Apolipoprotein A-I. J Biol Chem. 2005;280:1193-98
17. Sviridov D, Hoang A, Sawyer WH, Fidge NH. Identification of a sequence of apolipoprotein A-I associated with the activation of Lecithin:Cholesterol acyltransferase. J Biol Chem. 2000;275:19707-12
18. Kypreos KE, Zannis VI. Pathway of biogenesis of apolipoprotein E-containing HDL in vivo with the participation of ABCA1 and LCAT. Biochem J. 2007;403:359-67
19. Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG. et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343-53
20. Kodali S, Galgoci A, Young K, Painter R, Silver LL, Herath KB. et al. Determination of selectivity and efficacy of fatty acid synthesis inhibitors. J Biol Chem. 2005;280:1669-77
21. Datta G, Chaddha M, Hama S, Navab M, Fogelman AM, Garber DW. et al. Effects of increasing hydrophobicity on the physical-chemical and biological properties of a class A amphipathic helical peptide. J Lipid Res. 2001;42:1096-104
22. Fielding CJ, Shore VG, Fielding PE. Lecithin: cholesterol acyltransferase: effects of substrate composition upon enzyme activity. Biochim Biophys Acta. 1972;270:513-8
23. Jonas A. Lecithin cholesterol acyltransferase. Biochim Biophys Acta. 2000;1529:245-56
24. Guerin M, Dachet C, Goulinet S, Chevet D, Dolphin PJ, Chapman MJ. et al. Familial lecithin:cholesterol acyltransferase deficiency: molecular analysis of a compound heterozygote: LCAT (Arg147 --> Trp) and LCAT (Tyr171 --> Stop). Atherosclerosis. 1997;131:85-95
25. Funke H, von Eckardstein A, Pritchard PH, Albers JJ, Kastelein JJ, Droste C. et al. A molecular defect causing fish eye disease: an amino acid exchange in lecithin-cholesterol acyltransferase (LCAT) leads to the selective loss of alpha-LCAT activity. Proc Natl Acad Sci U S A. 1991;88:4855-9
26. Solajic-Bozicevic N, Stavljenic A, Sesto M. Lecithin:cholesterol acyltransferase activity in patients with acute myocardial infarction and coronary heart disease. Artery. 1991;18:326-40
27. Rader DJ, Ikewaki K, Duverger N, Schmidt H, Pritchard H, Frohlich J. et al. Markedly accelerated catabolism of apolipoprotein A-II (ApoA-II) and high density lipoproteins containing ApoA-II in classic lecithin: cholesterol acyltransferase deficiency and fish-eye disease. J Clin Invest. 1994;93:321-30
28. Solajic-Bozicevic N, Stavljenic-Rukavina A, Sesto M. Lecithin-cholesterol acryltransferase activity in patients with coronary artery disease examined by coronary angiography. Clin Investig. 1994;72:951-6
29. Berard AM, Foger B, Remaley A, Shamburek R, Vaisman BL, Talley G. et al. High plasma HDL concentrations associated with enhanced atherosclerosis in transgenic mice overexpressing lecithin-cholesteryl acyltransferase. Nat Med. 1997;3:744-9
30. Hoeg JM, Santamarina-Fojo S, Berard AM, Cornhill JF, Herderick EE, Feldman SH. et al. Overexpression of lecithin:cholesterol acyltransferase in transgenic rabbits prevents diet-induced atherosclerosis. Proc Natl Acad Sci U S A. 1996;93:11448-53
31. Brousseau ME, Kauffman RD, Herderick EE, Demosky SJJr, Evans W, Marcovina S. et al. LCAT modulates atherogenic plasma lipoproteins and the extent of atherosclerosis only in the presence of normal LDL receptors in transgenic rabbits. Arterioscler Thromb Vasc Biol. 2000;20:450-8
32. Ng DS. Insight into the role of LCAT from mouse models. Rev Endocr Metab Disord. 2004;5:311-8
33. Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med. 2006;84:276-94
34. Salvatore A, Cigliano L, Bucci EM, Corpillo D, Velasco S, Carlucci A, Pedone C, Abrescia P. Haptoglobin binding to apolipoprotein A-I prevents damage from hydroxyl radicals on its stimulatory activity of the enzyme lecithin-cholesterol acyl-transferase. Biochemistry. 2007;46:11158-68
35. Davit-Spraul A, Therond P, Leroy A, Palmade-Rieunier F, Rousset C, Moatti N, Legrand A. Inhibition of lecithin cholesterol acyltransferase by phosphatidylcholine hydroperoxides. FEBS Lett. 1999;447:106-10
36. Subbaiah PV, Liu M. Disparate effects of oxidation on plasma acyltransferase activities: inhibition of cholesterol esterification but stimulation of transesterification of oxidized phospholipids. Biochim Biophys Acta. 1996;1301:115-26
37. Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR. et al. Oral D-4F causes formation of pre-beta high-density lipoprotein and improves high-density lipoprotein-mediated cholesterol efflux and reverse cholesterol transport from macrophages in apolipoprotein E-null mice. Circulation. 2004;109:3215-20
38. Navab M, Anantharamaiah GM, Reddy ST, Van Lenten BJ, Ansell BJ, Fogelman AM. Mechanisms of disease: proatherogenic HDL--an evolving field. Nat Clin Pract Endocrinol Metab. 2006;2:504-11
39. Navab M, Anantharamaiah GM, Hama S, Garber DW, Chaddha M, Hough G. et al. Oral administration of an Apo A-I mimetic Peptide synthesized from D-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation. 2002;105:290-2
40. Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A. et al. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature. 2006;441:358-61
Author biography
Dr. Jun Wang, VP and Head of Biology at Sundia MediTech. Before joining Sundia MediTech, Dr. Wang served at Merck & Co., Inc. for 9 years leading multiple drug-discovery programs on principles of food-animal growth promotion, infectious diseases, diabetics and cardiovascular diseases. Prior to Merck, Dr. Wang worked at a biotech company in Seattle as the head of biology section and served as an assistant professor in University of Texas Southwestern Medical Center and University of Washington for 5 years. Dr. Wang has published numerous papers as the corresponding author in leading scientific journals. Lately, Dr. Wang has been invited by many organizations including University of Cambridge, Royal Society of Chemistry, Naito Foundation etc. for presentations on his discovery of novel candidates of antibiotics (Platensimycin and Platencin). Dr. Wang got his BS (1982) in biophysics from Fudan University and received his Ph.D. in molecular biology and biochemistry from Kobe University (Japan). Post-doc training was followed in the Department of Pharmacology at UT Southwestern Medical Center (Dallas).
![]() Correspondence to: Jun Wang, Ph.D., Department of Biology, Sundia MediTech Company, Ltd. Building 8, 388 Jialilue Road, Zhangjiang Hightech Park, Shanghai 201203, China Tel: 86-21-51098642 x 128; Fax: 86-21-51903505 E-mail: jwangcom
Correspondence to: Jun Wang, Ph.D., Department of Biology, Sundia MediTech Company, Ltd. Building 8, 388 Jialilue Road, Zhangjiang Hightech Park, Shanghai 201203, China Tel: 86-21-51098642 x 128; Fax: 86-21-51903505 E-mail: jwangcom
Citation styles
APA
Chen, X., Burton, C., Song, X., Mcnamara, L., Langella, A., Cianetti, S., Chang, C.H., Wang, J. (2009). An apoA-I mimetic peptide increases LCAT activity in mice through increasing HDL concentration. International Journal of Biological Sciences, 5(5), 489-499. https://doi.org/10.7150/ijbs.5.489.
ACS
Chen, X.; Burton, C.; Song, X.; Mcnamara, L.; Langella, A.; Cianetti, S.; Chang, C.H.; Wang, J. An apoA-I mimetic peptide increases LCAT activity in mice through increasing HDL concentration. Int. J. Biol. Sci. 2009, 5 (5), 489-499. DOI: 10.7150/ijbs.5.489.
NLM
Chen X, Burton C, Song X, Mcnamara L, Langella A, Cianetti S, Chang CH, Wang J. An apoA-I mimetic peptide increases LCAT activity in mice through increasing HDL concentration. Int J Biol Sci 2009; 5(5):489-499. doi:10.7150/ijbs.5.489. https://www.ijbs.com/v05p0489.htm
CSE
Chen X, Burton C, Song X, Mcnamara L, Langella A, Cianetti S, Chang CH, Wang J. 2009. An apoA-I mimetic peptide increases LCAT activity in mice through increasing HDL concentration. Int J Biol Sci. 5(5):489-499.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.