International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 22; 2026
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
The Insulin/ IGF Response...
Transforming Growth Factor...
Target of Rapamycin Kinases:...
The STAT Family: Regulation of...
Programs of Cell Death or...
Tumor Suppressor p53: Regulation...
The Cell Cycle: Regulation of...
Conclusion
References
Introduction
The Insulin/ IGF Response...
Transforming Growth Factor...
Target of Rapamycin Kinases:...
The STAT Family: Regulation of...
Programs of Cell Death or...
Tumor Suppressor p53: Regulation...
The Cell Cycle: Regulation of...
Conclusion
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(1):9-50. doi:10.7150/ijbs.6.9 This issue Cite
Review
An Overview of Stress Response and Hypometabolic Strategies in Caenorhabditis elegans: Conserved and Contrasting Signals with the Mammalian System
Benjamin Lant, Kenneth B. Storey ![]()
Institute of Biochemistry, Carleton University, Ottawa, Ont., Canada
Received 2009-9-11; Accepted 2009-11-25; Published 2010-1-7
Citation:
Lant B, Storey KB. An Overview of Stress Response and Hypometabolic Strategies in Caenorhabditis elegans: Conserved and Contrasting Signals with the Mammalian System. Int J Biol Sci 2010; 6(1):9-50. doi:10.7150/ijbs.6.9. https://www.ijbs.com/v06p0009.htm
Other stylesAbstract
Studies of the molecular mechanisms that are involved in stress responses (environmental or physiological) have long been used to make links to disease states in humans. The nematode model organism, Caenorhabditis elegans, undergoes a state of hypometabolism called the 'dauer' stage. This period of developmental arrest is characterized by a significant reduction in metabolic rate, triggered by ambient temperature increase and restricted oxygen/ nutrients. C. elegans employs a number of signal transduction cascades in order to adapt to these unfavourable conditions and survive for long times with severely reduced energy production. The suppression of cellular metabolism, providing energetic homeostasis, is critical to the survival of nematodes through the dauer period. This transition displays molecular mechanisms that are fundamental to control of hypometabolism across the animal kingdom. In general, mammalian systems are highly inelastic to environmental stresses (such as extreme temperatures and low oxygen), however, there is a great deal of conservation between the signal transduction pathways of nematodes and mammals. Along with conserving many of the protein targets in the stress response, many of the critical regulatory mechanisms are maintained, and often differ only in their level of expression. Hence, the C. elegans model outlines a framework of critical molecular mechanisms that may be employed in the future as therapeutic targets for addressing disease states.
Keywords: Hypometabolism, Lifespan extension, Diapause, Post-translational modification, Transcriptional regulation, Apoptosis
Introduction
Metabolism is dynamic, and all living organisms must to respond to a wide variety of needs, signals and stresses in order to remain alive. Most often these responses adjust metabolism to meet new demands or to cope with internal or external stressors, but sometimes death responses are triggered to eliminate damaged or unneeded cells for the betterment of the whole organism. All organisms respond to many types of environmental stresses (e.g. heat/cold, oxygen lack, desiccation, food limitation, etc.) with coordinated adjustments to metabolism that deal with the consequences and/or injuries caused by the stress. In some cases, behavioural, physiological or biochemical adjustments are sufficient to adapt the organism to new conditions, and normal life can continue. However, when environmental stress reaches a critical level, organisms need to make choices. Death is usually not the preferred option, but sometimes one generation will take steps to ensure that the next generation has a better chance of survival. For example, short photoperiod and cooling temperatures cue females of various insect species such as silkworms to produce diapause eggs that can overwinter [1]. Embryos develop for just a brief time, and then development is arrested (at the G2 phase of the cell cycle in silkworms) and the eggs transition into a dormant state called diapause where they can survive for many months until conditions are again favorable for the resumption of embryogensis [2]. Many other organisms have comparable procedures; for example, brine shrimp shift from birthing nauplius larvae under favourable conditions to producing encysted gastrulae when environmental conditions are tough [3]. Embryonic diapause (also called delayed implantation) also occurs in many other species including a variety of mammals where it allows mating to occur but development to be delayed by arresting embryogensis until environmental conditions improve or until lactation stops when a previous litter of offspring are weaned [4, 5].
Entry into a hypometabolic state, where metabolic rate is strongly suppressed and normal activities are suspended, is actually a widespread response to stress across phylogeny providing organisms with a greater chance to survive through stressful times. As an example, entry into a dormant state where metabolic rate falls to just 10% of normal resting rate provides an automatic 10-fold extension of the time that organisms can survive using only stored endogenous fuel reserves to support their energy needs. Such a time extension is typically more than enough to ensure survival through predictable seasonal periods of environmental stress particularly when paired with a strong enhancement of body fuel reserves in the weeks leading up to the entry into hypometabolism. Although only a minor part of the human experience [6], hypometabolism appears in many formats across the animal kingdom, as well as in plants and microbes. A hypometabolic state where metabolic rate is typically suppressed by at least 70% and in many cases to >95% or greater is a critical part of phenomena including winter hibernation by mammals, nightly torpor by small birds and mammals, estivation and anhydrobiosis under summer drought conditions, anaerobiosis in response to oxygen depletion, cold-hardiness and many other forms of dormancy or diapause (Selected reviews: [7 - 13]).
Known principles of hypometabolism are proving to be highly conserved across the animal kingdom and include (a) an overall strong suppression of metabolic rate, (b) differential control over the rates of individual metabolic processes so that energy use is reprioritized to favor core vital cell functions (e.g. maintenance of membrane potential difference) while largely shutting off “optional” activities (e.g. protein synthesis, cell division, growth and development), and (c) implementation of actions that protect cells and preserve viability over what could be many months of dormancy [12]. Actions that are important to life extension during dormancy can include changing to optimal patterns of fuel use (e.g. states of aestivation or hibernation rely heavily on lipid reserves whereas anaerobiosis requires a switch to carbohydrate fuels), protecting and stabilizing cellular macromolecules by enhanced production of chaperone proteins or antioxidant defenses, and implementing stress-specific actions such as colligative protectants that defend against cell water loss during freezing or drying. Furthermore, it is now clear from studies on a wide variety of animal systems of hypometabolism, that most of the molecular mechanisms of metabolic rate depression are highly conserved across phylogeny so that the principles of hypometabolism are becoming well-defined [12].
Considerable research in recent years has defined a number of critical molecular mechanisms that regulate transitions to/from hypometabolic states. Central among these is posttranslational modification of cellular enzymes and other proteins to provide coordinated transitions between high and low activity states. Reversible protein phosphorylation by the action of protein kinases and protein phosphatases is by far the most widespread and potent mechanism controlling hypometabolism, but in recent years, the important contributions of a number of other posttranslational mechanisms are becoming increasingly recognized (e.g. methylation, acetylation, SUMOylation) (Selected reviews: [14, 15]). Research in recent years has also started to define mechanisms that regulate strong global suppression of transcription and translation while at the same time enhancing the expression of selected proteins with cytoprotective actions. Much research to date on hypometabolism has been concerned with energetics, fuel catabolism, and controls on energy-expensive cell functions such as controls on ion-motive ATPases, gene transcription and protein translation. Chaperones and antioxidant defenses are also frequent topics. Research progresses at a different pace and with different emphasis in model species of hibernation, estivation, anaerobiosis, etc. Overall, however, there is a growing interest and a move towards topics that include stress signaling (how are external stresses transduced and transmitted to regulate and coordinate thousands of cell functions), reversible regulation of cell cycle, growth and development processes, the potential involvement of selective degradative processes (e.g. proteasome control, apoptosis and autophagy), and the mechanisms that stabilize/preserve cell viability to achieve long-term life extension in the hypometabolic state. Whereas these ideas are being analyzed at different levels and with different intensities in many different models of hypometabolism and are contributing to a unified theory of the principles of metabolic rate depression, the question becomes - is there a model animal that we can use to provide insights into these new topics and help us lay the framework for expanded studies of the principles of hypometabolism across phylogeny?
The answer is yes, and although the majority of scientists working on hypometabolism are interested in megafauna (e,g. hibernating mammals, estivating frogs, anoxia tolerant turtles, diapause in insects, etc.), it is to mesofauna that we need to turn for guidance on the new advances in understanding the regulation of hypometabolism. Specifically, the model is the nematode, Caenorhabditis elegans. The remainder of this review will survey current information and ideas about the regulation of a form of stress-induced larval diapause in nematodes, called the dauer state. In conjunction with this, a focus will be placed on the mechanisms of the relatively inelastic (non-hibernating) mammalian system. This comparative study will highlight conserved and contrasting molecular mechanisms that deal with stress states - both those that lead to tolerance of stress (C. elegans model system), and those that can result in disease/injury states (mammalian systems).
The Insulin/ IGF Response Pathway: Stress Response and Dauer Regulation
In the field of metabolic rate depression (MRD), one model that can experience numerous stress conditions at once, in a period of stasis natural to its development, is the nematode, C. elegans. As a model animal, C. elegans, has become widely used due to several features including their genetic tractability, short generation time, ease of culturing, and a fully sequenced genome. This allows for gene disruption with relative ease, via knockouts or knockdowns (RNAi), providing for extensive studies on gene expression [16]. Under both natural environmental or lab culture conditions, C. elegans can be faced with stresses including overcrowding, limited food, elevated temperature, increased desiccation and low levels of oxygen. Given that populations of these tiny organisms have little chance of dispersing themselves to a more hospitable environment, a primary response to stress is to enter a phase of arrested development, thereby postponing their entry into the reproductively mature stage until such time as conditions are more favourable [16]. This most widely characterized period of arrest occurs between the second and third stage of larval development, with the resulting nematodes becoming 'dauer' larvae. During this period, the larvae do not eat and are extremely resistant to external conditions. As in all models of MRD, physiological changes reduce movement, respiration rate and digestion, while metabolic changes include a drastic reduction in ATP expenditure by altering critical metabolic processes. The oxidative stress imposed during dauer signaling conditions is one of the most critical to manage, and maintaining antioxidant defenses has been shown to significantly increase the lifespan of the nematodes [17]. In the dauer stage, larvae can survive up to ten times as long as 'non-dauer' mature adults and are essentially 'ageless' in this period as it does not affect their post-dauer lifespan [16]. Under natural conditions, changes in environmental temperature (typically a rise) is perhaps the most common signal for entry into dauer, with population factors (e.g. overcrowding, reduced sustenance) subsequently triggering the release of 'dauer promoting' pheromones that lead to the formation of dauer larvae. At the metabolic level, neuronal signals are mediated through signaling pathways of 'abnormal dauer formation' (daf) genes; predominantly via the insulin response pathway [18].
The insulin response pathway is initiated through insulin and insulin-like growth factor (IGF) signaling at the cell membrane through the insulin receptor and the insulin-like growth factor 1 receptor (IGF-IR). Mutations/deficiencies in these receptors are known to provoke lifespan extension in a number of animal models [19]. Downstream of the receptor, insulin receptor substrates (IRSs) and the phosphatidylinositol-3 kinase (PI3K) are also known to have conserved roles in lifespan determinacy [19]. The primary downstream target of these receptor targets is Akt (also known as protein kinase B). Akt has numerous important downstream targets that are conserved from C. elegans to humans, which can initiate survival responses throughout prolonged periods of environmental stress and can determine lifespan of the cell through regulating critical cellular processes. The Akt pathway has been particularly well-studied in the case of diabetes because it is responsive to insulin, and is known to have downstream effects on glucose transport, protein synthesis, glycogen synthesis, apoptosis and anti-apoptosis response pathways [20]. The exact mechanism of Akt activation initially involves the rapid conversion of the plasma membrane lipid, PIP2, to phophatidylinositol-3, 4, 5-trisphosphate (PIP3), by PI3K, which causes PIP3 to become an active secondary messenger. PIP3 subsequently activates the 3'-phosphoinositide dependent kinase-1 (PDK1) (at the PH domain), which in turn phosphorylates and activates Akt, targeting the Pleckstrin homology in the Akt N-terminal region (Fig. 1) [21]. The key feature of this signal transduction is the phosphorylation of subsequent substrates, finally allowing active Akt to phosphorylate its numerous downstream targets. The phosphorylation state of Akt, intuitively, is also the key point of regulation, determining PI3K/Akt pathway activity or inactivity. Protein phosphatases, primarily the 'phosphatase and tensin homologue deleted on chromosome 10' (PTEN) and protein phosphatase 2a (PP2A), act to suppress this signaling by removing phosphate groups from members of this pathway (Fig. 1). PTEN, more accurately a lipid phosphatase, targets the 3' phosphate on PIP3, which was phosphorylated by PI3K, returning the messenger to its PIP2 state. Consequently, this prevents downstream activation of PDK1 and Akt. In conjunction with the role of Akt in cellular survival, development and proliferation, deletions in PTEN have been shown to induce tumors in mice, and in many mammalian cancers, PTEN is lost or mutated [22].
Figure 1
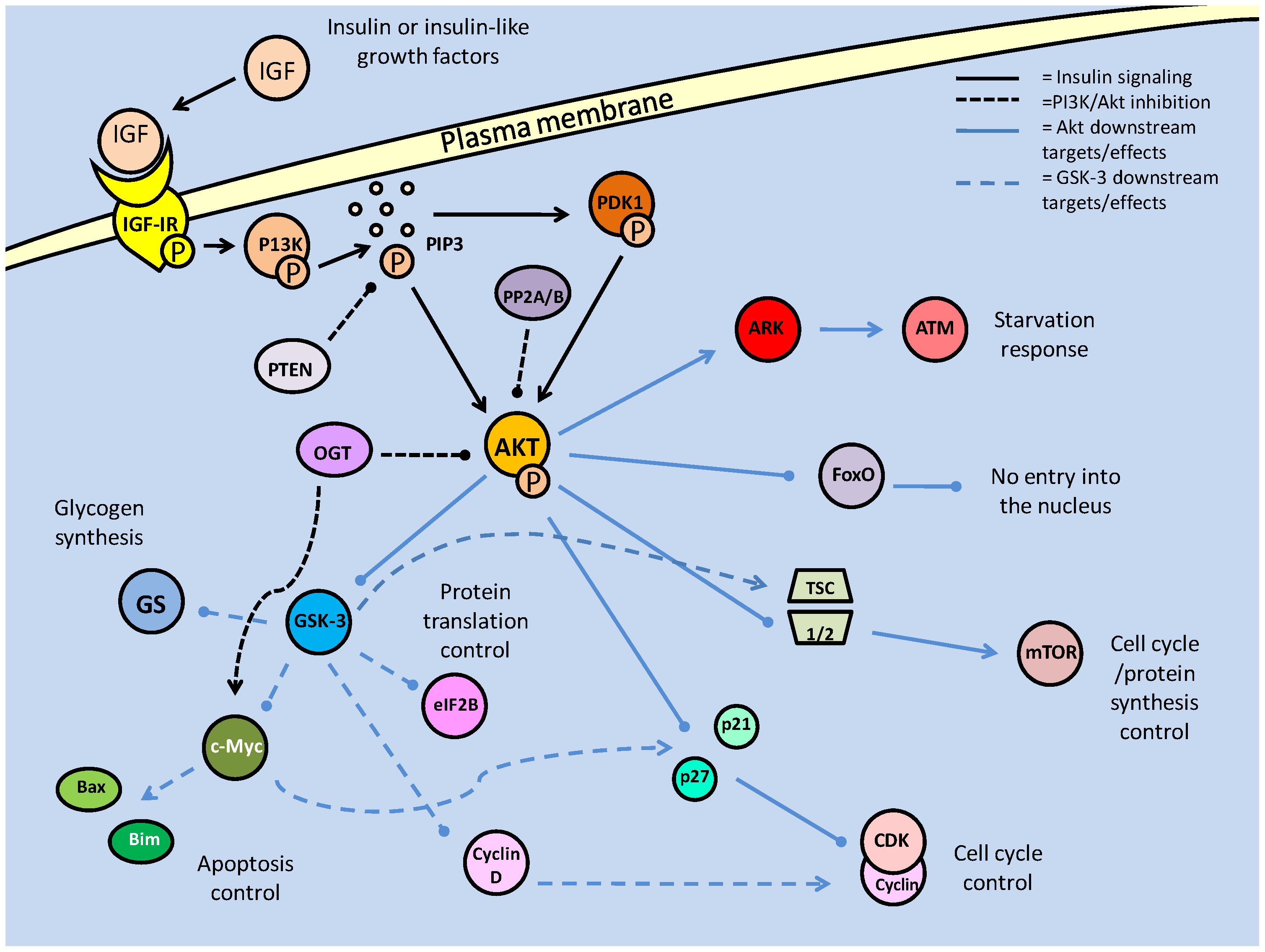
Summary of the insulin/IGF signaling pathway, using the terminology for mammalian systems. Binding of the ligand to the insulin like growth factor-I Receptor initiates the signaling cascade, which is propagated by PI3K. Akt acts as the signaling hub for the majority of the downstream effects such as cell cycle control, glycogen synthesis and apoptotic suppression. The phosphorylation state of Akt is critically regulated by protein phosphatases (PTEN/PP2) and protein kinases (PDK-1).
Also acting to reduce the activity of Akt are members of the PP2 family of phosphatases. PP2A has long been known to regulate the phosphorylation state of Akt, but only recently has PP2B (also called calcineurin) been implicated in the same role [23]. Both phosphatases directly dephosphorylate active Akt, and have been thought to do so primarily in order to promote the activity forkhead box transcription factors. Interestingly, in myocytes, FoxO activity contains a feedback inhibition loop through the production of the ubiquitin ligase, Atrogin-1, which targets calcineurin (PP2B) [23]. An alternative post-translational modification, the addition of O-linked β-N-acetylglucosamine (O-GlcNAc), is also known to inactivate PI3K downstream activity, likely by displacing phosphate groups on Akt.
The relatively novel discovery that additions of O-GlcNAc to proteins is both ubiquitous and functionally significant, has indicated that a number of signaling pathways may be modified, unbeknownst to researchers, by this mechanism [45]. Because of the small size, and uncharged nature of the molecule, detection of its addition to proteins is incredibly difficult, but its effects on phosphorylation state have made its responses discernable. This glycosylation consequently affects many critical procedures such as apoptosis regulation and cytoskeletal formation [45]. Addition of the molecule or 'O-GlcNAcylation', is mediated by the 'O-GlcNAc-transferase' (ogt), and reversed by O-GlcNAcase (oga). The mechanism whereby O-GlcNAc affects phosphorylation can be either direct, through competitive binding of phosphorylation sites, or indirect by binding nonspecific sites which affect the physical conformation of phosphorylation sites, reducing their accessibility. In terms of the Akt pathway, it was seen that O-GlcNAcylation, ogt targeting Ser473, prevented Akt phosphorylation and initiated apoptosis [46]; a process which typical Akt activity suppresses. This Akt inhibitory activity could also extend into the dauer model, given the importance of its regulation in pre-dauer conditions. Hence, the regulation of Akt phosphorylation state is critical to the success of the majority of PI3K signals (Fig. 1).
Once active and released into the cytosol, Akt is able to commence signal transduction has targets affecting numerous cellular processes and pathways, but typically promoting cellular survival and proliferation. Downstream targets include glycogen synthase kinase 3 (GSK-3), cell cycle regulators (p21, p27), protein synthesis (mTOR and TSC2, and subsequently P70S6K/ 4EBP) and cell survival factors (Bad, Forkhead box transcription factors) (Fig. 1) [21]. Several of these targets are discussed next although discussion of mTOR is deferred to a later section.
Regulation of the Forkhead Box transcription factors
Among the most important Akt targets are the members of the forkhead box family of transcription factors. In C. elegans, these are critical for entry into the dauer stage [24], and in stress-adapted animals, they have multiple and often conflicting responses when active. The Forkhead Box family of transcription factors, also referred to as the Forkhead related (FKHR) transcription factors, are a highly conserved group across phylogeny with a number of subfamilies. Indeed, with in the subtype 'other' or FoxO, the mammalian proteins (often called FKHRs) and nematode proteins (called DAF-16) can substitute for each other; for example, mammalian FKHR (DAF-16a or FoxO1) and FKHRL1 (DAF-16b or FoxO3) have been used as transgenic substitutes for the DAF proteins in C. elegans in order to initiate dauer formation [24]. FoxOs are increasingly recognized as an important group of proteins in the regulation of cellular metabolism. Their activity is linked to oxidative stress responses, cellular proliferation and apoptosis pathways and they are one of the major known targets of the Akt pathway [25]. Through the PI3K/Akt pathway, FoxO transcription factors are phosphorylated, which prevents their entrance into the nucleus, nullifying their transcriptional activity. They may be phosphorylated on up to three sites by Akt, and in certain cases by serum-glucocorticoid kinase (SGK) (also activated by the PI3K pathway), which directly phosphorylates the nuclear localization signal (NLS) causing the recruitment of 14-3-3 binding proteins (Fig. 2) [17]. Signaled by the phosphorylated residues (particularly serine), the 14-3-3 protein binds the FoxO transcription factor, obstructing its NLS, preventing nuclear (re)entry and changing FoxO conformation to expose its NES, which if the transcription factor is in the nucleus, causes its expulsion [26]. Regardless of the original nuclear/cytoplasmic location, prior to phosphorylation, binding of the 14-3-3 protein anchors the transcription factor in the cytoplasm, and is also suspected of inhibiting any DNA binding activity, due to the location of 1 4-3-3 binding which is proximal to the FoxO DNA binding domain (Fig. 2) [26]. Hence phosphorylation of FoxOs is the primary means of their transcriptional regulation.
Figure 2
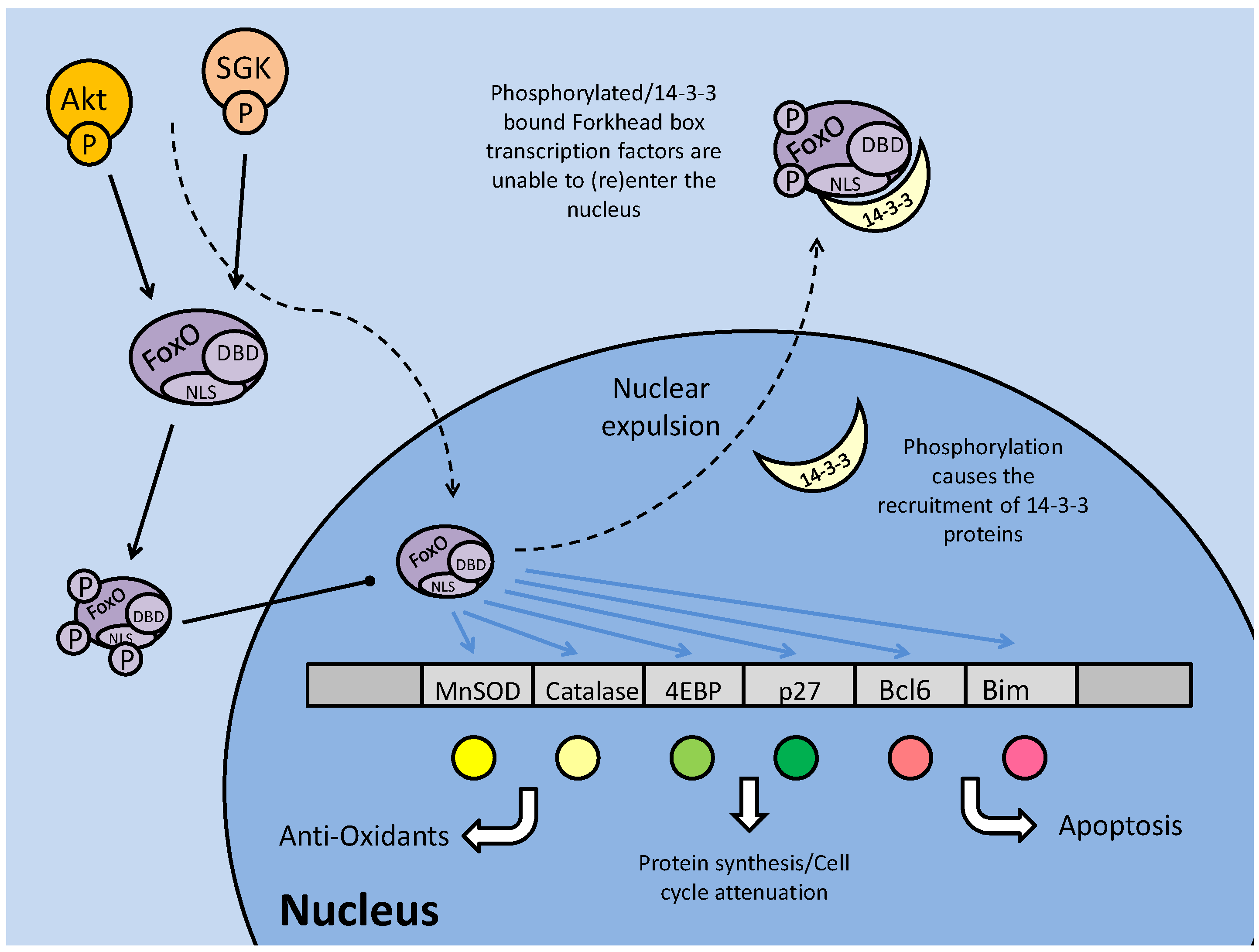
Regulation and downstream targets of the forkhead box (subtype O) transcription factors. Protein kinases (Akt and SGK) phosphorylate FoxO on up to three sites, including one proximal to the nuclear localization sequence (NLS). This prevents nuclear translocation or, in the event that the FoxO is phosphorylated in the nucleus, nuclear expulsion. Phosphorylation may stimulate binding of the 14-3-3 protein to the transcription factor, covering its NLS and DNA binding domains and blocking transcriptional regulatory capacities.
In mammals, FoxO activity is strongly linked with apoptosis through its interactions with pro-apoptotic genes including BCL6 and BIM, but may also initiate apoptosis through cell cycle arrest (Fig. 2) [27]. Mammalian FoxOs up-regulate transcription of the 'eukaryotic initiation factor 4E binding protein (4EBP), a protein that binds to the eukaryotic initiation factor 4 (eIF4e), preventing its interaction with other members of the translation initiation complex [27]. This can force cell cycle attenuation and possibly trigger apoptosis signaling. Hence, in some contexts, phosphorylation of FoxOs can be seen as an anti-apoptosis response. This however, reveals the contradictory nature of FoxO signaling, with other targets of FoxO transcriptional regulation being protective (even pro-survival) in nature. For example, FoxOs are known to regulate the expression of antioxidant enzymes such as manganese superoxide dismutase (MnSOD) and catalase that have key roles in protecting cells from the damaging effects of reactive oxygen species (ROS). This duality in the FoxO family role under stress may be a result of non-AKT mediated posttranslational phosphorylation of the FoxOs, or modification by other means such as acetylation [25].
Regulation of Glycogen Synthase Kinase
Another critical target of the Akt pathway is GSK-3 through which AKT mediates the stimulation of glucose uptake and glycogen synthesis. Glycogen synthesis is regulated in part by GSK-3 which phosphorylates glycogen synthase, inactivating it and preventing the conversion of UDP-glucose to glycogen. GSK-3 is composed of α and β subunits, both of which can be phosphorylated by Akt [20]. The interaction between Akt and GSK-3 has yet further repercussions, although Akt maintains its pro-developmental role in allowing glycogen synthesis (Fig. 1). Akt phosphorylation of GSK-3 can also relieves GSK-3 inhibition of protein synthesis at the eukaryotic initiation factor 2 (eIF2) locus and at the level of mTOR complex formation. This allows for increased protein synthesis and cell proliferation [28]. Active GSK-3 can also phosphorylate eIF2B to inactivate it, and as such, the Akt repression of GSK-3 activity prevents this. Briefly mentioned before was the interaction between Akt and mTOR, in which Akt contributes to mTOR activation; GSK-3, via Akt, may also contribute to the activation of mTOR. GSK-3, acting on the mTOR activating complex TSC1/2, is able to phosphorylate, and contribute to its activity, which in turn, negatively regulates mTOR complex formation [28]. This continues to correspond with the conflicting pro-development (Akt)/ development attenuation (GSK-3) relationship, which may be a direct link to the mTOR complex 1's (mTORC1) ability to increase ribosomal DNA transcription, which indicates increased ribosomes; allowing for increased protein synthesis and cell proliferation [28].
A further role of GSK-3 is in the regulation of cell death via c-Myc inhibition. By inactivating c-Myc, GSK-3 inhibits the induction of Bax and Bim (pro-apoptotic members of the Bcl-2 family) among other c-Myc activated apoptosis targets (Fig. 1) [29]. Furthering its role in apoptosis inhibition, the absence of GSK-3 subsequently alters glucose metabolism, specifically resulting in the dissociation of hexokinase II from the mitochondrial membrane causing its instability [29]; mitochondrial membrane integrity is one of the key factors in cell death regulation (see later section on apoptosis). Contrary to typical Akt responses that support cell development, division and survival, this would seem to allow apoptotic stimulation through c-Myc activity in a system with active Akt. However, c-Myc can be consequently down regulated through a convoluted system which involves P13K/Akt activation of E2F transcription factors [30] to regulate and counteract c-Myc apoptotic signaling. The relationship between E2F and c-Myc involves both members regulating each other, and as such, is likely dependent on other factors that will not be discussed here. Also to be taken into consideration is the role of c-Myc in cell cycle progression, through suppression of p15, p21 and p27, to allow transition through G1 into the S phase (Fig. 1) [31]. It is in this mechanism that Akt-based suppression of GSK-3 (and resultant c-Myc activity) appears to correspond best with the general pro-development role of Akt.
Regulation of the cell cycle through Akt
Akt also has numerous targets affecting the cell cycle, one of which, cyclin D, is also a target of GSK-3. In this case, active GSK-3 is able to phosphorylate cyclin D, preventing it from further interacting with cyclin dependent kinase 4 (CDK4) [31]. In terms of the cell cycle, the binding of cyclins to CDKs is essential to the formation of maturation promoting factors (MPFs), which regulate events such as microtubule formation, which allows for cellular division. Formation of these complexes in the nucleus is critical to the progression of the cell cycle past the G1 phase into the S phase, and it is here that Akt exhibits the majority of its cell cycle effects. Two inhibitors of cell cycle progression, p21 and p27, both of which can prevent the formation of the cyclin E/CDK2 complex, can be regulated by phosphorylation. Akt is capable of phosphorylating both at their NLS sites, which anchors them in the cytoplasm [31] preventing interaction with cell cycle proteins. In addition, p27 is a target of the FoxO transcription factors, and through Akt phosphorylation (preventing nuclear translocation), Akt is also able to down regulate the transcription of p27 (Fig. 1 and 2) [31]. While regulation at the G1/S phase is widely regarded as the control centre of cell cycle progression, the exit from cell cycle (M phase) into the quiescence phase (Go) is also a critically regulated step, and is also a target of Akt signaling.
The retinoblastoma family member, p130 (Rb2), is critical in signaling the exit from the cell cycle and is positively (transcriptionally) regulated by FoxO 3a [32]. While FoxO activity is negatively regulated (by Akt) to allow cell cycle progression, preventing its role in enhancing cell cycle attenuator (p27) transcription, it is seen to be active in the M phase of the cell cycle in order to promote its exit. At this stage, p130 is transcriptionally up-regulated and activated by cyclin/CDK (typically D/4) phosphorylation, which may release it from an E2F4 negative inhibitor [32]. Hence the relationship between Akt and the FoxOs appear to mediate the switch between cell cycle exit (down regulation of Akt activity) and cell cycle re-entry/progression (up-regulation of Akt activity).
In contributing to the control of critical cellular processes such as cell cycle and energy storage, Akt, like many other kinases is heavily involved in stress responses. These responses typically involve the alteration of metabolic process in order to maintain energetic homeostasis of the cell. Along with Akt, one of the major (if not the primary) energy metabolism regulators are the AMP-activated protein kinase (AMPK) family of protein kinases. They are involved in various and multiple metabolic processes (including glycolytic control, glucose transport etc.), and are triggered by a diverse range of metabolic stressors, including hypoxia, heat shock and nutrient deficiency [33]. AMPK, as well as being stimulated directly by AMP (a marker of low energy status), must be phosphorylated (at Thr 172, typically by LKB1) before becoming activated. The AMPK-related kinase (ARK5) member of this family is activated by Akt phosphorylation, which is thought to be part of an Akt-mediated response to nutrient starvation (Fig. 1) [33]. Subsequently, ARK5 can phosphorylate the tumor suppressor, 'ataxia-telangiectasia' kinase (ATM), which is a member of the PI3K family. ATM, in turn, phosphorylates the tumor suppressor transcription factor, p53, which stabilizes and activates it, leading to downstream apoptotic signaling [33]. This indicates a contradictory response in Akt signaling, since the general consensus of Akt signaling appears to support cellular survival and development.
Regulation of the Insulin response pathway from a C. elegans perspective
As mentioned above, the nematode C. elegans can enter a period of arrested development known as the dauer stage, when faced with stresses such as overcrowding, lack of food and substantial changes in temperature. Initial studies on the mutation of insulin response genes revealed that when members of the canonical IGF/PI3K/Akt pathway were mutated, dauer larvae were formed (Fig. 3). These were the daf-2 (abnormal dauer formation), age-1 and akt-1/2 genes [34]. Daf-2 encodes the tyrosine kinase receptor which binds insulin and other growth factors on the plasma membrane, and activates itself through auto-phosphorylation. AGE-1 (equivalent to mammalian PI3K) is recruited to produce PIP3 as an activator of downstream kinases, in order to propagate the insulin/IGF signaling [34]. It has been suspected that, even with the mutations in daf-2 and age-1, dauer formation may still be inhibited, through an independently activated AKT-1/2 (C. elegans Akt homologs - functionally redundant) pathway. As in the mammalian system, the DAF-2 pathway follows the canonical activation of AKT via AGE-1(PI3K) and a 3-phosphoinositide dependent kinase (PDK) homolog. Any inhibition up until the phosphorylation of AKT-1/2 typically results in DAF-16 (C. elegans forkhead box transcription factor homolog) mediated entry into the dauer state (Fig. 3) [35]. The recently discovered importance of the C. elegans PDK-1 in AKT activation indicates a further level of regulation in the DAF-2 pathway, and like its mammalian homolog, affect other critical development targets (such as p70S6K, discussed later) which affect lifespan and dauer entry [35]. Hence it is suspected that AKT, and to a lesser extent PDK-1, may be able to inhibit dauer formation through the phosphorylation of the DAF-16 transcription factors, even in the absence of functional daf-2/age-1 [36]. This is the result of PDK-1 being constitutively active under basal conditions and remaining active (even at low levels) to activate AKT [36]. Through this path, regulation of AKT, which directly affects DAF-16, is critical in controlling the switch between development into the reproductively mature state, or entry into the quiescent dauer phase.
As in mammalian systems, C. elegans AKT regulates the FoxO homolog, DAF-16, via phosphorylation, inhibiting its ability to move into the nucleus. The C. elegans FoxO also shares many of the mammalian downstream target genes, which are necessary for entry into and sustaining the dauer state (Fig. 3). DAF-16 activity centers on the attenuation of the cell cycle (affecting proteins such as p27, p130 and 4EBP) that categorizes the dauer stage of stasis [17]. The self-imposed production of antioxidants (MnSOD and catalase) in anticipation of oxidative stress, is also critical. In fact, minimization of oxidative stress is of such importance that, as in the mammalian system, the MnSOD homolog (sod3 in C. elegans) is regulated directly by DAF-16. FOXO3 has a consensus site in the MnSOD promoter region, and it is suspected that similarly, DAF-16 and sod3 are related, in order to trigger the activity of this mitochondrial antioxidant [37]. Hence, regulation of DAF-16 nuclear entry, critically affects whether the nematode will not only enter the dauer stage, but be able to endure the energetic and oxidative limitations imposed on it during this phase, in order to proceed into maturity.
One of the main physical responses to C .elegans prior to dauer entry (the 'pre-dauer' stage), is the accumulation of lipid stores, to provide a source of energy to endure the dauer stage. Hence, it could be suspected that DAF-16 has downstream targets that regulate fat metabolism during the dauer stage, in order to ration energetic expenditure. One possible target is the pantothenate kinase (C. elegans pnk-1), which is involved in coenzyme A synthesis (a key member of fat metabolism). In cases of inhibited DAF-2 signaling (as would occur during the dauer stage), pnk-1 is up-regulated, and fat storage levels are reduced, possibly indicating it as a target of DAF-16 [37]. This may be related to AMPK based responses, due to AMPK's role in the regulation of fat storage consumption rates (briefly elaborated upon in the TOR section).
Figure 3
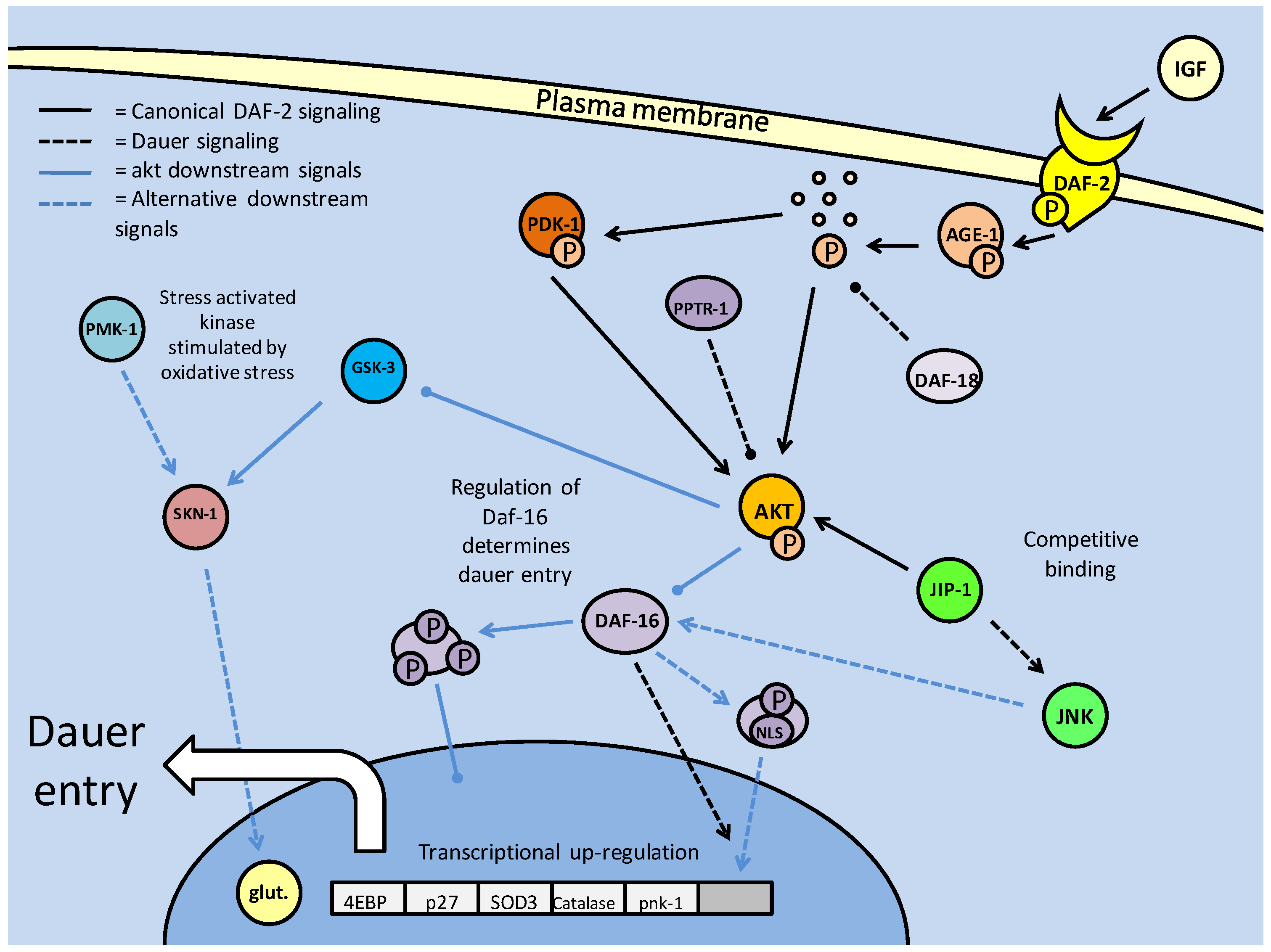
Insulin/IGF signaling in C. elegans controls entry into the dauer larval stage. Dauer entry is prevented through AKT mediated phosphorylation of the DAF-16 (FoxO homolog) transcription factor. Like the mammalian model, AKT phosphorylation is the key to canonical DAF-2 signaling, and is regulated by protein kinases (PDK-1) and phosphatases (DAF-18 and PPTR-1).
The key regulatory switch, it seems, is the phosphorylation state of AKT. As in mammalian systems, dephosphorylation via protein phosphatases (primarily PTEN, known in C. elegans DAF-18) is essential to inhibit DAF-2 signaling (Fig. 3). The DAF-18 phosphatase not only mediates dauer entry through the suppression of AGE-1 activity, but also contributes to an earlier quiescent phase. Post hatching, and prior to feeding, the C. elegans larvae are in their L1 phase, and will often enter a quiescent stage until nutrients are available [38]. Thus, it is through DAF-2 signaling, specifically with DAF-18 inhibiting AGE-1 signaling that the L1 larvae are able to go into diapause [38]. Along with PTEN phosphatase, it has recently emerged that the PP2A catalytic subunit (PPTR-1) may also contribute to C. elegans dauer regulation, acting directly on AKT-1 [39]. Active AKT requires phosphorylation in its activation loop, typically by PDK-1, but may also be phosphorylated by a TOR complex. PPTR-1 acts directly on AKT-1, but interestingly not AKT-2 or SGK-1 (the C. elegans SGK homolog, which may also phosphorylate DAF-16), both of which are phosphorylated when active, increasing lifespan through DAF-16 mediated dauer entry [39]. Regulation of AKT by PP2A in the mammalian system also reflects this (mentioned above), with the B56 family of subunits being the mammalian PPTR-1 homologs which are critical to PP2A formation. Also preventing the pro-developmental aspect of AKT signaling, B56 deficiency is a characteristic of numerous tumors. B56 isoforms also regulate the tumor suppressor p53, which furthers their role in tumor suppression [39]. Hence, the role of the PP2A subunits appears conserved between C. elegans and mammalian systems, in their anti-AKT activities which prevent excessive cellular energy expenditure and proliferation.
However, it is not only phosphatases that affect AKT in order to increase DAF-16 activity. The c-Jun N-terminal kinase (JNK) is involved in many stress response signaling cascades, and previously in mammalian systems, has been shown to interact with the IGF pathway (Fig. 3). A JNK upstream kinase (JKK) knockout inhibits C. elegans lifespan extension, indicating a positive role for JNK in dauer expression [40]. Interestingly, JNK positively regulates DAF-16 through phosphorylation (which is typically an inhibiting factor). In this case, phosphorylation appears to contribute to signaling DAF-16 for nuclear translocation, making it a unique response to stress signaling in C. elegans lifespan extension [40]. This activity of JNK in supporting lifespan extension may be in conjunction with an AKT interaction with the 'JNK interacting protein 1' (JIP1). This protein, which regulates JNK activity in mammalian systems, acting as a scaffold protein (aiding phosphorylation of target proteins) is crucial. In this context, the JIP1 protein may act as a scaffold protein for both AKT and JNK, assisting both in their enzymatic activities [41]. Hence, a competition forms for binding with this co-factor, between AKT and JNK. JNK requires both phosphorylation and JIP1 binding to become active. Binding of AKT to JIP1 may actively prevent JNK activation of DAF-16, while increasing its own capacity to inhibit DAF-16.
Supplemental to the IGF pathway, which acts primarily through PI3K/Akt activity, are a number of insulin receptor substrates and adaptors which can help to propagate the insulin response signals, or indeed, signal secondary pathways through insulin receptor activity. In C. elegans, this may result in an alteration in dauer signaling, with DAF-2 activity central to its regulation. The insulin receptors (typically tyrosine kinases) phosphorylate downstream 'insulin receptor substrates' (IRSs), such as the aap-1 PI3K adaptor subunit which attaches to the catalytic subunit, and the IST-1 IRS, which may also aid in formational function of AGE-1 [42]. While these two elements are not absolutely essential for IGF signaling, they are required for full DAF-2 activity. Although mutations in both still resulted in DAF-2 signaling, as revealed through levels of DAF-16 protein expression (and resulting numbers of dauer larvae), it is suspected that in conjunction with mutations of more critical members of the pathway (AGE-1 and AKT), dauer arrest was enhanced [42]. This also opens up the possibility of alternate DAF-2 signaling, possibly via homologs of mitogen-activated protein kinases (MAPKs).
Whereas the dauer stress response is the most explored, analysis of C. elegans responses to oxidative stress has also revealed a number of targets and responses that are less studied, but possibly contribute to dauer survival (Fig. 3). Oxidative stress, and its critical role in aging, has made the study of its response elements critical in such fields as diabetes and cancer research. One of the primary targets in protective responses against oxidative damage (ROS based) is the SKN-1 transcription factor [43]. This transcription factor targets 'phase II' detoxification genes such as SOD and Glutathione-S-Transferase (GST), which are antioxidant enzymes, preventing oxyradical damage to membranes and DNA [43]. The SKN-1 protein is a C. elegans homolog of the mammalian NF-E2 related factor (Nrf) family, which is also a critical oxidative response element, and is regulated both positively and negatively by phosphorylation. Under normal conditions, when SKN-1 activity is not necessary, it is anchored in the cytoplasm by gsk-3 (C. elegans GSK-3 homolog) mediated phosphorylation (likely at Ser-393) [43]. Under conditions of oxidative stress, however, the p38 MAPK ortholog (PMK-1), as in mammalian systems, begins its stress response cascade by phosphorylating and activating SKN-1 as one of its targets [43]. The SKN-1 transcription factor may also be activated by a number of less well-documented stress response kinases such as the NEKL-2 (mammalian NEK-8)and IKKε-1(mammalian NF-κB inhibitor) kinases, involved in cell cycle regulation and host immunity, respectively [44]. Survival duration, in response to oxidative stress, drops drastically in the absence of these particular kinases, likely indicating that their roles are critical process regulators, aside from their interactions with SKN-1 [44]. However, their role in SKN-1 activation may be more dominant than expected, when looking at the role AKT plays in this pathway. Under typical dauer conditions, of which one of the contributing factors is oxidative stress, AKT is suppressed, allowing DAF-16, and more pertinently, GSK-3 activity. This, hypothetically, would result in negative regulation of SKN-1 in a period where it would likely need to be active.
Transforming Growth Factor β Signaling: a Role in Dauer Regulation
The transforming growth factor β (TGF-β) signaling pathway is involved in both proliferation and cell differentiation. It responds to neuronal signaling, which is in turn responsive to external signals including environmental cues. The TGF-β pathway has parallels with the Insulin/IGF pathway and its expression is also typically seen under conditions conducive to growth that involve considerable energy expenditure. Pro-TGF-β signaling has been implicated as a component and target of tumorigenesis. TGF-β signaling and its role in cancer signaling is well known to be paradoxical, with apparent functions in both cell cycle attenuation/apoptosis and the promotion of tumor formation. It is still unclear as to how the switch in activity is made, but in different types of cancer, increased TGF-β production and downstream Smad dependent activity are seen to induce tumorigenesis. Consequently, anti-TGF-β responses can be partially classified as anti-proliferative or anti-developmental.
The TGF-β superfamily in mammals is a large and varied grouping of growth factors and developmental elements, which include the TGF-β ligands as well as 'bone morphogenic proteins' (BMPs), activins and 'growth and differentiation factors' (GDFs). The general pathway of signaling involves ligand binding at the cellular receptor (serine/threonine kinase receptors), followed by activation and downstream messaging via the 'mothers against decapentaplegic' (Smad) family of transcription factors [47]. In mammalian systems, the ligands require dimerization before binding and activating the type II receptor. The ser/thr kinase receptors must complex in order to initiate downstream signaling, with the type II receptor able to recruit the type I receptor, after interacting with the ligand (Fig. 4) [47]. The type II receptor, which has higher affinity for ligands, has the capacity to autophosphorylate, which is likely a critical factor in its own activation as well as subsequent activation of the type I receptors (via phosphorylation) [48]. There are multiple type I (designated ALK1-7) and type II receptors which provide specificity to both different ligands (upstream via the type II receptor), and different Smads (downstream via the type I receptor) [48]. Regulation of the phosphorylation state of the type I receptor's is critical to the success of TGF-β signaling, and there are numerous interactions with the receptor which can lead to its activation or can facilitate its inactivation/degradation. One such inhibitory interaction is with the prolyl isomerase FKBP12 protein. FKBP12 can bind the type I receptor, and through structural changes, make it unable to interact with the type II receptor (Fig. 4) [48]. De-phosphorylation of the type-1 receptor is another common mechanism of TGF-β inhibition, with phosphatases such as PP2A (although, specifically the Bδ subunit) targeting the type-I receptor. Disabling the receptors prevents signaling of the major downstream targets of the TGF- β pathway, the Smad transcription factors [48].
The primary effectors of TGF- β signaling, the Smad transcription factors
Downstream of the TGF-β receptor kinases is the Smad family of transcription factors. In mammals, Smads are categorized in three types: receptors (Smad 1,2,3,5 and 8) that require double phosphorylation in order to become active and translocate into the nucleus, complexing (Smad 4), and inhibitory (Smad 6 and 7) [47]. Phosphorylation of the receptor (r-) Smads can either activate or inhibit the transcription factors, depending on where the protein is phosphorylated. The SXS motif at the C-terminal is critical to Smad activity, with double phosphorylation at this site necessary, prior to complexing and entry into the nucleus [48]. Active r-Smads, forming complexes with themselves (Smad2/3 and Smad1/5/8) or remaining monomeric (typically Smad 2), will further complex with Smad 4 and move into the nucleus (Fig. 4) [47]. Another means of moving into the nucleus is to complex with other transcription factors, relying upon their nuclear localization sequences (NLSs) in order to move out of the cytoplasm. An example of this is the Smad 2 interaction with the tumor suppressor p53, activated upstream by a receptor tyrosine kinase [47]. Once in the nucleus, Smad complexes are able to bind TGF-β promoter elements, and in some instances (with the Smad2/3), bind co-factors such as TAZ or p300 (also a p53 co-factor) which stabilize the complex and partially prevent nuclear expulsion (Fig. 4). The primary member of Smad signaling is Smad 2, which can, but does not require homodimerization with other r-Smads (although it still requires conjunction with Smad4). Its phosphorylation and presence in the nucleus is a significant indicator of successful TGF-β signal transduction [47]. Smad 2 activation may also be increased by an interaction with the 'Smad anchor for receptor activation' (SARA), which in complexing, relocates the Smad to the type I receptor for phosphorylation [48].
Mentioned above, the phosphorylation of Smads can also lead to their inability to complex and enter the nucleus, and in the cases of complexes already in the nucleus, their expulsion and subsequent degradation. The linker region is a fairly unconserved region sandwiched between two 'MAD homology' (MH1 and 2) domains and, like the SXS region, is susceptible to phosphorylation. Unlike the SXS domain, phosphorylation at the linker site is known to decrease Smad transcription factor activity and is mediated by a series of kinases implicated in anti-proliferative and stress response procedures. The cyclin dependent kinases (CDKs), GSK-3, and MAPKs including ERK and JNK, which can all show anti-proliferative or energy conservation roles, can phosphorylate the linker sites of Smad proteins [48]. In the cytoplasm, phosphorylation of linker sites is linked to ubiquitin ligase mediated degradation of the proteins, whereas in the nucleus it leads to Smad expulsion (Fig. 4). The degradation response that is mediated by E3 ubiquitin ligases (Smurf 1 and 2) appears to be almost exclusively signalled by phosphorylated residues on the Smad proteins, which not only implicates linker phosphorylation, but likely the SXS motif phosphorylation as well [48]. The primary targets for this activity are Smad 1, Smad 4 and the Smad2/3 complex [48]. The nuclear expulsion response can be regulated in at least two ways. One is dephosphorylation of the complexes, which can be accomplished by several phosphatases. Interestingly, the protein phosphatase PPM1A, which can directly bind phosphorylated Smad2/3 complexes and promote their nuclear expulsion, may be stabilized by the PTEN phosphatase [48]. An alternate means of expulsion affects Smad 4 which is exported after sumoylation of the protein [47].
Figure 4
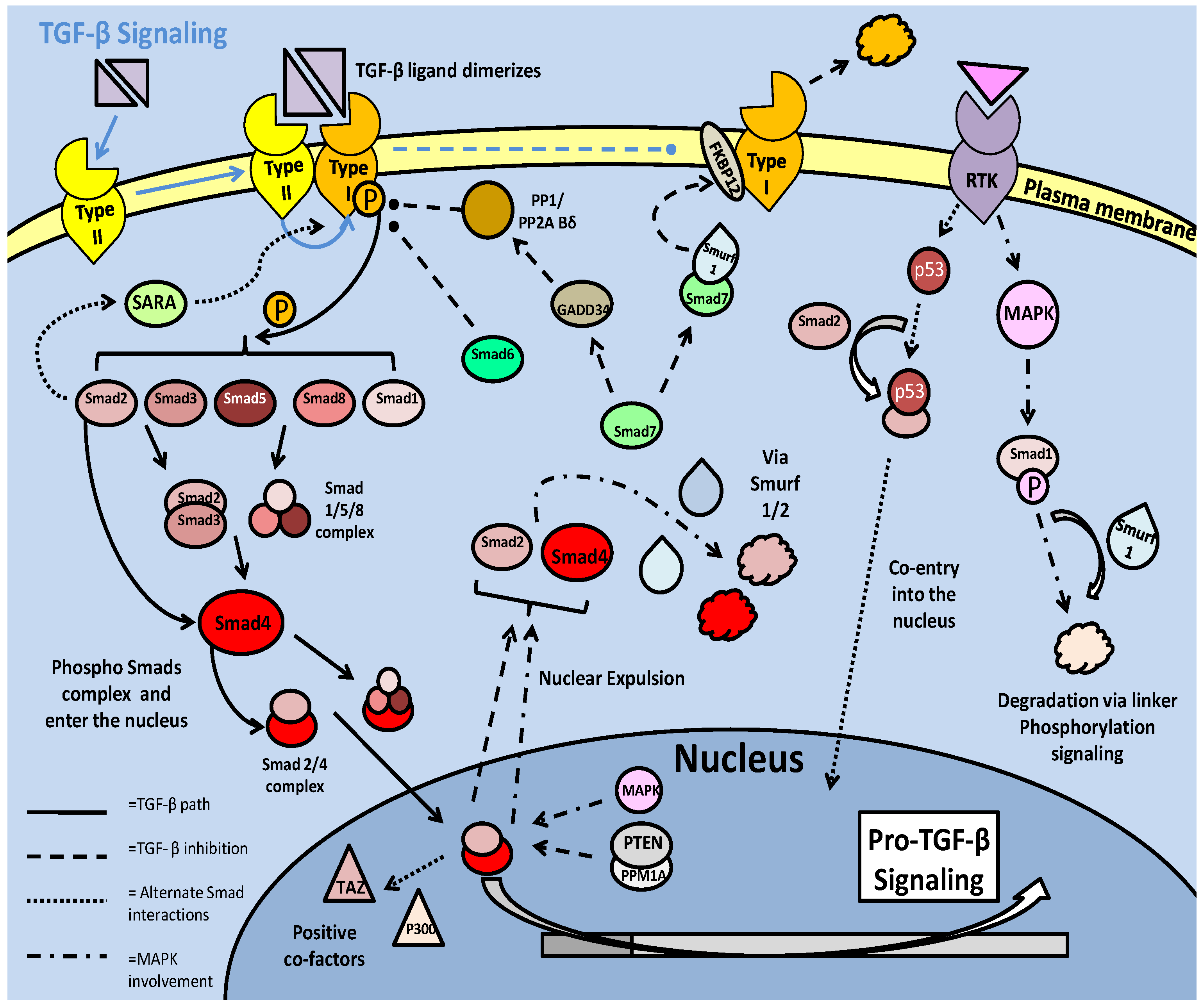
TGF-β signaling in mammalian models. Signaling begins with ligand dimerization and interaction with the type II receptor. This recruits and activates (via phosphorylation) the type I receptor. The active type I receptor phosphorylates downstream Smad transcription factors, which form complexes and enter the nucleus. There are numerous inhibitory pathways that act on both the type I receptor, and the Smad transcription factors, typically resulting in loss of phosphorylation state or ubiquitin ligase mediated degradation.
Negative regulation of the TGF- β pathway through inhibitory Smads
Whereas the r-Smads are the primary effectors of the TGF-β pathway, the inhibitory Smads, which rely on protein-protein interaction more so than transcriptional activity, play a large role in regulating r-Smad signaling. As part of this role, the inhibitory Smad7 may mediate direct type I receptor degradation through the recruitment of the ubiquitin ligases Smurf1/2. This particular degradation signal may also be enhanced by the binding of FKBP12 to the receptor (mentioned above), with a subsequent interaction between the Smad7-Smurf-1 complex aiding ubiquitination of the receptor (Fig. 4) [48]. Along with degradation, Smad 7 is able to prevent type I receptor signaling through dephosphorylation. Recruiting protein phosphatase 1 (PP1), through an interaction with GADD34, Smad 7 is able to prevent downstream phosphorylation of the pro-TGF-β Smads [47]. While the role of PP1 here is to inactivate Smad phosphorylation via the receptor, it is also capable of preventing Smad 2 phosphorylation through binding (inactivation) of the SARA protein that facilitates relocation of the Smad to the receptor [48].
As a protective mechanism, degradation of the receptors, signaled through Smad 7-Smurf-1/2 interactions, can be partially compensated for if the receptors bind the heat shock protein, HSP 90, which maintains receptor structure, preventing ubiquitin-mediated degradation [48]. The inhibitory Smads may act as a feedback mechanism during excessive TGF-β signaling, or may become active dependent on the developmental stage of the cell.
The pro-development, anti-dauer TGF- β pathway in C. elegans
The TGF -β pathway in C. elegans is one of the major signaling pathways associated with entry into the dauer stage, and is often thought of as a parallel system to the the insulin/IGF pathway, although secondary path to it. Like the insulin response/IGF pathway, the active TGF-β pathway acts to suppress entry into the dauer stage [18]. The TGF-β pathway functions through ligand binding to cellular receptors; type I (DAF-1) and type II (DAF-4) (Fig. 5). This system purports cell-cell communication and eventual signal transduction, which could lead to a consensus event; of importance in the context of C. elegans, entry into dauer [49]. In this sense, the parallel pathway of Insulin/IGF signaling, which is also cell receptor based, may produce converged signals through cellular cross-talk, in order to suppress dauer entry. Downstream of the cellular receptors is the TGF-β (DAF-7) protein which in turn regulates production of downstream Smads. The Smads must be phosphorylated to become active, which in the case of pro-TGF-β Smads is done at the cellular receptor complex (via DAF-1) followed by entry into the nucleus. Phosphorylation activation of the Smads, as in mammal systems, occurs at the C terminal, but while it has not been studied in depth, some phosphorylations of C. elegans Smads can result in a reduction of expected Smad responses [50]. This is a possible indication of linker site regulation, used to negatively regulate TGF-β signaling. Once in the nucleus, the pro-TGF-β Smads are able to stimulate the transcription of genes, but they are also capable of acting in protein-protein complexes as cofactors for each other, to prevent dauer signaling [49]. Pro-dauer signaling is also, interestingly, mediated by Smad proteins. Hence, the Smads can be divided into pro- (DAF-3/5) and anti- (DAF-8/14) dauer members, with the anti-dauer members acting upstream of the pro-dauer members to regulate dauer entry. During successful TGF-β signaling, a relationship forms where DAF-8/14, through directly inhibiting DAF-3/5 in the nucleus, prevents dauer entry (Fig. 5). In experimental conditions, it has been shown that DAF-3 mutants are unable to form dauer larvae, and as such, have a severely reduced lifespan [18].
Similar to the insulin response pathway, TGF-β signaling comes through cell membrane receptors from upstream neuronal signals [51]. These neuronal signals function throughout the lifespan of C. elegans in order to regulate which developmental stage the nematode will transition into. In cases where environmental signals do not necessitate entry into the dauer stage, the transition between juvenile and reproductively mature nematodes occurs over a period of a few days, shortening overall lifespan significantly in comparison to dauer animals. One critical moderator of reproductive maturity/capability is the 'Egg Laying Deficient' (EGL) family of proteins, which have many roles in C. elegans development. The egl-4 member of the family (which encodes a cyclic GMP-dependent protein kinase) functions in conjunction with the TGF-β pathway, with its mutation resulting in pro-dauer DAF-3/5 (Smads) activity and entry into dauer [51]. However, while egl-4 mutations, by indirectly inhibiting TGF-β, cause entry to dauer, the dauer formed is abnormal with development severely altered. Along with this, the mutation in egl-4 (and resultant TGF- β signaling) affects critical developmental processes such as neuronal signaling, body size and the ability to lay eggs [51]. In terms of its effect on dauer formation, the mutation of egl-4 which causes deficiencies in neuronal signaling, links egl-4 mutation to the inhibition of TGF-β, since the TGF-β receptors are dependent on receiving these neuronal signals. This response indicates that active egl-4 could be critical to TGF-β signaling, and resultantly, dauer entry [51]. In this respect, age-1/akt pathway mutations show similar phenotypic responses to egl-4 mutants, notably in increased size (possibly linking this to the LKB1/AMPK activated fat accumulation), which is a pre-dauer event. An interesting response to egl-4 mutations is that chemosensory signaling disruption may be reversed by subsequent mutations in daf-3/5 (Smads) suggesting that they may work downstream of egl-4. This could implicate the Smads as feedback inhibitors of TGF-β signaling, similar to the actions of Smad 6/7 members in mammalian systems, in order to select for dauer entry [51].
An alternative TGF-β pathway, which functions through the type II receptor (DAF-4) and alternate downstream Smads (SMA-2, 3 and 4), appears to a lesser extent than the standard DAF-7/ DAF-1:DAF-4 led pathway, to be involved in development such as body size determinacy [49]. These Smads, which have been suggested to form a heterotrimer complex, are activated by phosphorylation of a consensus sequence at the C terminal end of the protein. The C. elegans SMA-2 and 3 (apparent homologues of r-Smad 2 and 3) are thought to complex individually or together with SMA-4 (a potential co-Smad, Smad-4 homolog) after phosphorylation. The complexes subsequently move into the nucleus in order to interact with transcriptional co-factors [50]. While in all models of Smad activity, phosphorylation is critical, it is suggested that in the C. elegans system, phosphorylation is more relevant to binding transcriptional co-factors than to nuclear translocation [50]. Mutations in either sma-2 or -3 lead to a shortened body size defect, which shows their role in normal body size development, and also possibly a necessity in pre-dauer accumulation of fats.
Figure 5
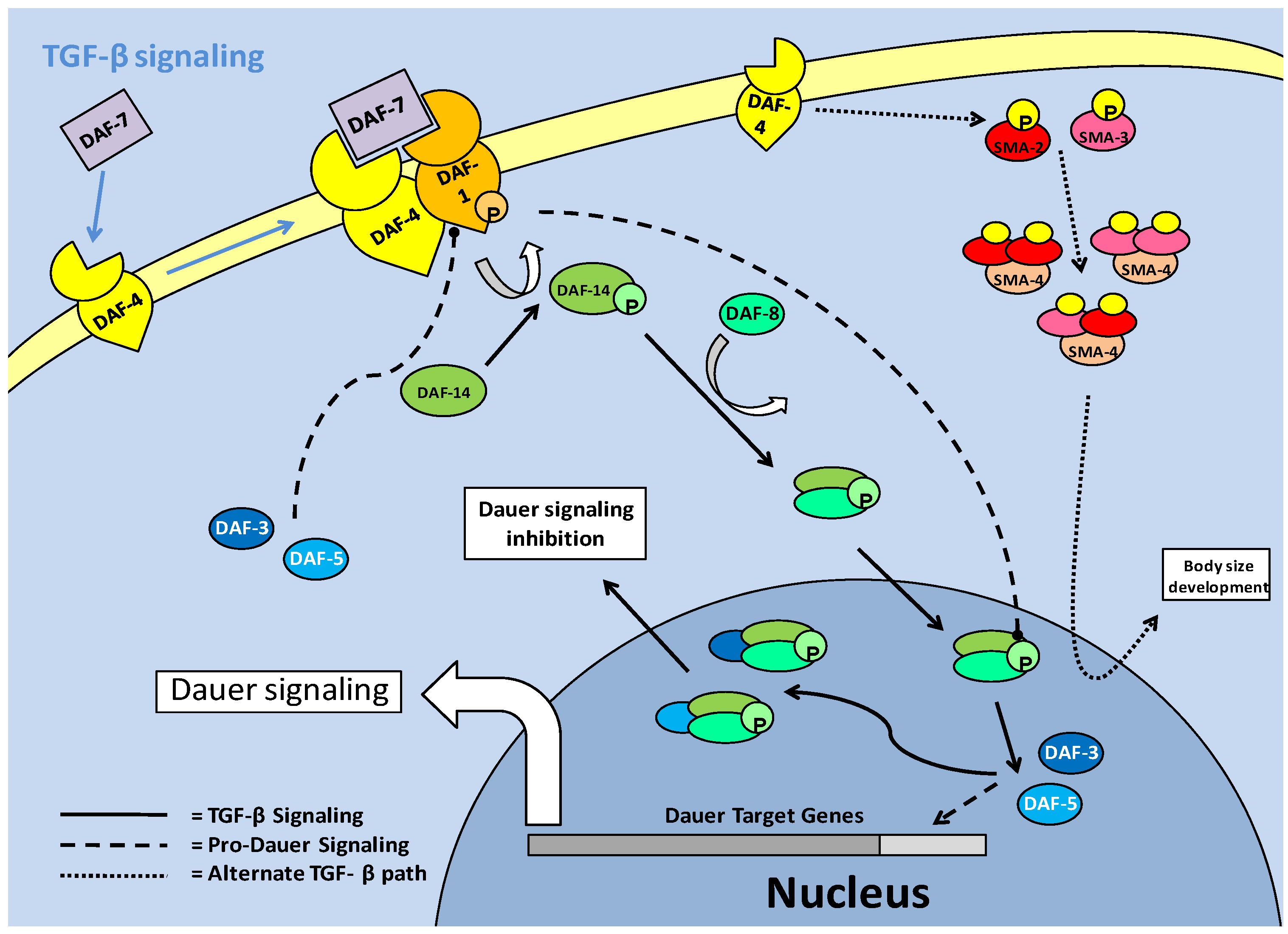
TGF-β signaling in C. elegans. The TGF-β ligand (DAF-7) stimulates the type II receptor (DAF-4), which in turn, recruits and phosphorylates the type I receptor (DAF-1). DAF-1 phosphorylates (activates) downstream Smads (DAF-14), which complex with DAF-8 and enter the nucleus. In the nucleus, the DAF-8/-14 complex inhibits pro-dauer Smads (DAF-3/-5) and promotes development, rather than dauer formation. An alternate Smad based pathway involves hetero-trimer formation of a combination of SMA-2/-3 and 4. These trimers enter the nucleus, associate with transcriptional co-factors and regulate body size development of C. elegans.
The TGF-β pathway appears to repress dauer entry, in conjunction with its roles in growth and development, but it has been suggested that it may only function this way at the L2d (pre-dauer)/Dauer state junction [49]. Since TGF-β (DAF-7) signaling is prevalent in many tissue types of C. elegans, including hypodermis (which is critical for regulation of body size), intestine and pharynx, it may be involved in the physiological preparation during transition into 'pre-dauer' (L2d) larvae [49]. To further this, there is an alternate target of the TGF-β pathway which counteracts AKT activity rather than mirroring it. In the L2d stage, DAF-7 signaling has been shown to increase daf-18 (PTEN) expression [24]. Once DAF-18 is active, the production of phosphatidylinositol-3, 4, 5-trisphosphate, by AGE-1 (PI3K) is reversed and AKT phosphorylation is subsequently lowered. This is manifested as an increase in DAF-16 in the nucleus and the entry of C. elegans larvae into their dauer stage [24].
Target of Rapamycin Kinases: Interactions with the Insulin Response Pathway and Regulation of Dauer Entry
The Target of Rapamycin (TOR) kinases are highly conserved in multicellular organisms and are involved in numerous critical cell processes including protein synthesis, cell cycle regulation and autophagy. In mammals, mTOR (mammalian target of rapamycin) regulates protein translation and the cell cycle, primarily by promoting protein synthesis [52]. The PI3K/Akt cascade, as part of the insulin response pathway activates mTOR through phosphorylation. The primary function of mTOR is to regulate translation by phosphorylating two key proteins, p70-S6 protein kinase (P70S6K) and the eIF-4E binding protein (4E-BP). P70S6K is activated by mTOR whereas phosphorylation of 4EBP inhibits its binding to eIF4E. However, mTOR is also known to interact with PP2A which, in turn, helps to sustain protein synthesis by dephosphorylating protein synthesis inhibiting machinery [52].
Figure 6
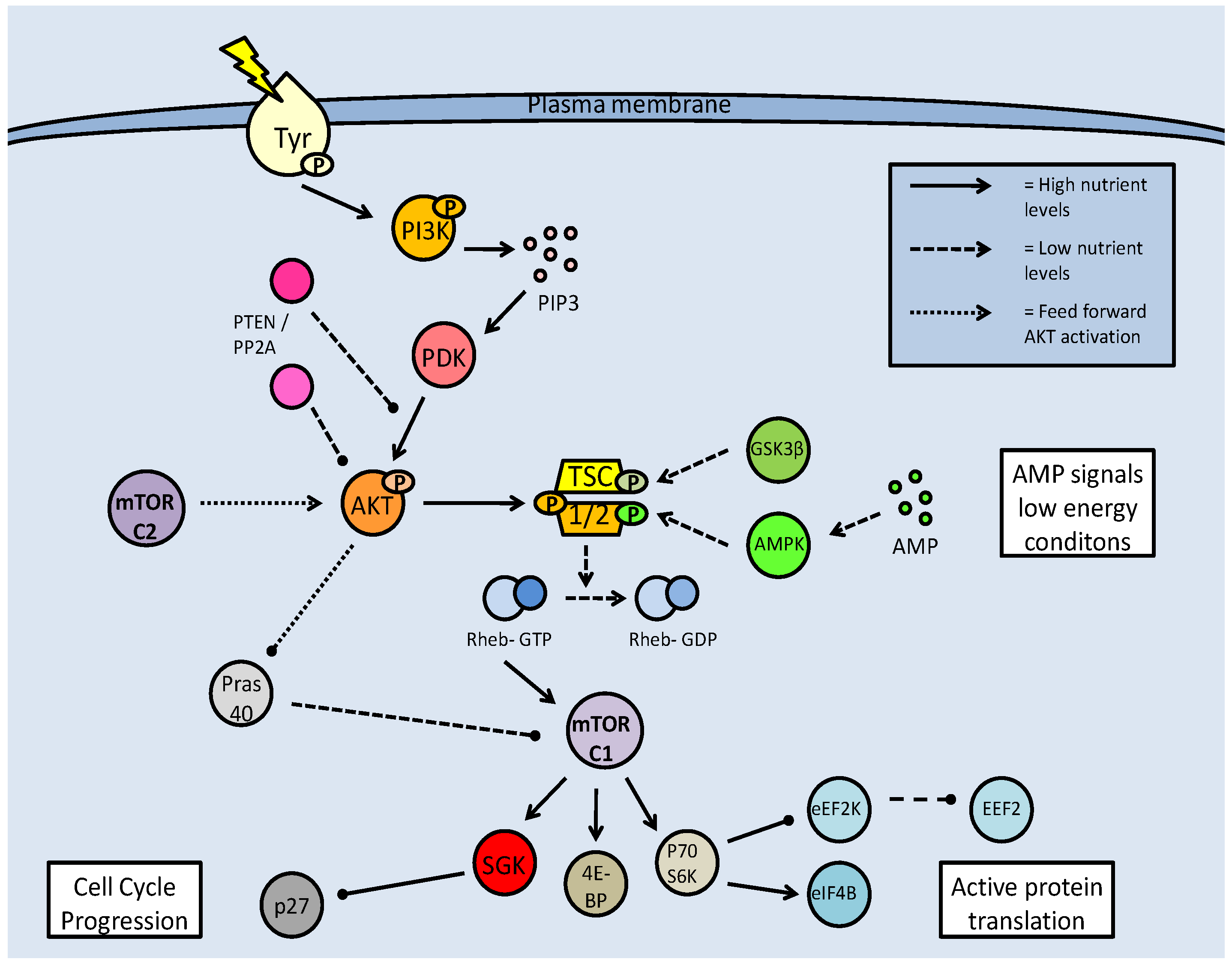
Mammalian target of rapamycin (mTOR) activation pathway. Formation of the mTOR complexes (mTORC1 and C2) is dependent on regulation of the tuberin/hamartin (TSC1/2) complex. This is regulated by numerous kinases, which can inhibit (AKT) or activate the complex (GSK-3β and AMPK). Active TSC1/2, interacting with Rheb-GTP, inhibits mTOR complex formation, and subsequent downstream activities. mTOR activation is often based on nutrient availability.
The action of mTOR in regulating the 4E-BP1 is well known. When eIF4E is bound to 4E-BP1 it is prevented from joining the translation initiation complex but upon phosphorylation by mTOR, 4E-BP1 releases eIF4E which is then phosphorylated by p70S6K and can join the active initiation complex [52]. The eukaryotic elongation factor (eEF2) is also regulated by mTOR. eEF2 is phosphorylated and inactivated by eEF2 kinase (eEF2K) which is, in turn, phosphorylated and inactivated by p70S6K as a result of mTOR activity (Fig. 6). As a consequence of mTOR action, therefore, p70S6K is activated, eEF2K is inactivated, and eEF2 is activated. This allows the elongation complex to form and support active protein translation. mTOR is also seen to promote the translation of calmodulin dependent kinase (CAMK) subunits and microtubule associated proteins (MAP2), both of which are involved in maintaining progression in the cell cycle [52]. Under these conditions, calmodulin-dependent protein kinase (CAMK) acts upstream of ERK1, which in turn, acts upstream of p27, the cell cycle attenuator. CAMK phosphorylates and activates ERK, and ERK phosphorylates and inactivates p27. The MAP2 protein is active in the G2 phase of the cell cycle, stabilizing the microtubules by cross-linking them with themselves.
Maintaining active TOR complexes through the TSC1/2 barrier
Two mTOR complexes are found in mammals, mTORC1 and C2, both with distinct and varied functions. The mTORC1 complex combines mTOR and the 'regulatory associated protein of mTOR' (Raptor), whereas in mTORC2, mTOR complexes with the 'rapamycin insensitive companion of mTOR' (Rictor) and a stress activated protein kinase interacting protein (SIN). Both complex with a G-protein subunit (GβL) (Fig. 7) [52]. mTOR signaling essentially mediates amino acid availability for protein synthesis. Under high amino acid availability, a Class III PI3K (Vsp34) is activated and through the recruitment of PIP3 and PDK, activates Akt, which then has two means of activating mTOR. Akt can phosphorylate either Pras 40 or the tuberin/hamartin (TSC1/2) complex to prevent their inhibitory effects on mTOR (Fig. 6) [52]. In this, albeit complicated, way, Akt activity maintains mTOR activity.
Figure 7
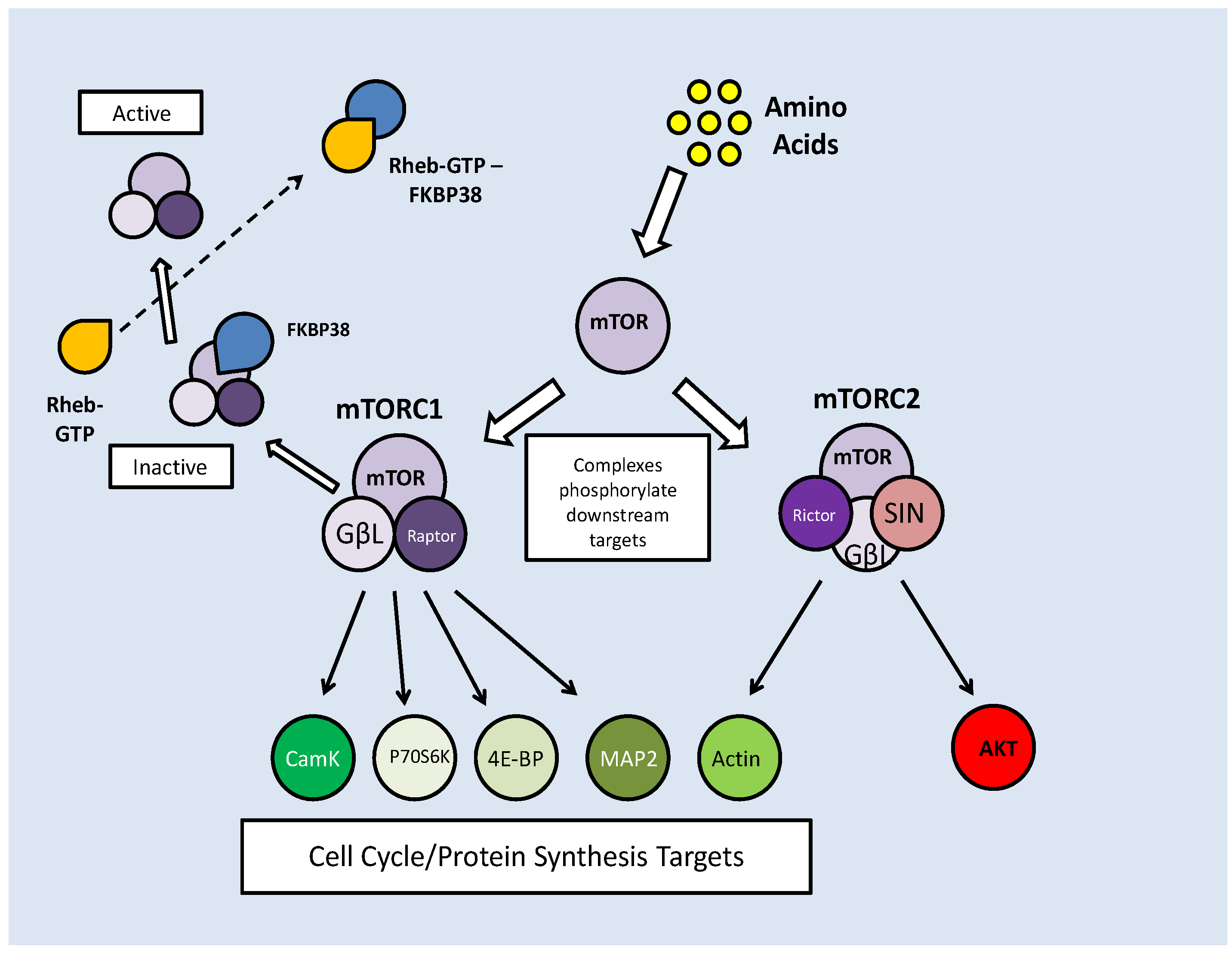
mTOR complex formation combines either Raptor (C1) or Rictor (C2). Inhibition of mTORC1 by FKBP38 can be reversed by direct interaction with Rheb-GTP. Both complexes have downstream targets to promote cell cycle progression and protein translation, but mTORC2 also has feedback activation of mTOR activation through AKT phosphorylation.
Along with their AKT maintained activity, mTOR complexes also require protection from prolyl isomerases (which are indicated in protein folding), which bind to mTOR, preventing their phosphorylation capacity. FKBP38 binds and inhibits the mTORC1 complex [53]. To counteract this, a Ras like GTPase, Rheb (Ras homolog enriched in brain), binds to GTP making it an effector of FKBP38. When Rheb-GTP binds FKBP38, it is released from the mTORC1 complex, thereby nullifying its effects (Fig. 7) [53]. The stimulation of mTOR activity by high amino acid levels is reflected in a reduced level of FKBP38-mTOR interactions and subsequently increased Rheb-FKBP38 binding [53].
At the TSC1/2 branch point, there are a number of kinases that can act, and while phosphorylation from Akt prevents the complex from inhibiting mTOR, phosphorylation at alternate sites can activate it. Under energetically stressful conditions, AMPK signaling increases (due to high AMP) and along with glycogen synthase kinase beta (GSK-3β), can phosphorylate the TSC1/2 complex and lead to mTOR inhibition (Fig. 6) [52].
As part of its actions in cell cycle regulation, mTOR maintains a novel interaction with the serum glucocorticoid regulated kinase (SGK). Acting on the p27 member of the 'kinase inhibitor protein' (KIP) family, which incidentally can also be phosphorylated directly by Akt, mTOR maintains the progression of the cell cycle (Fig. 9) [54]. The direct binding of p27 to E-type cyclin-CDK2 complexes prevents the cell cycle from progressing to the S phase. Phosphorylation of p27 by mTOR prevents p27 from entering the nucleus and it accumulates in the cytoplasm (Fig. 9) [54]. The mTORC1 complex also acts upstream of SGK-1 to phosphorylate it in the hydrophobic (H) motif, prior to SGK1 being phosphorylated by PDK1 and becoming fully activated [54]. The mTOR complexes also act as H motif kinases to P70S6K (through mTORC1) and Akt (through mTORC2). The mTOR/SGK interaction not only maintains cell cycle progression but has important implications in the entry into the dauer stage for C. elegans, which are elaborated below.
Figure 8
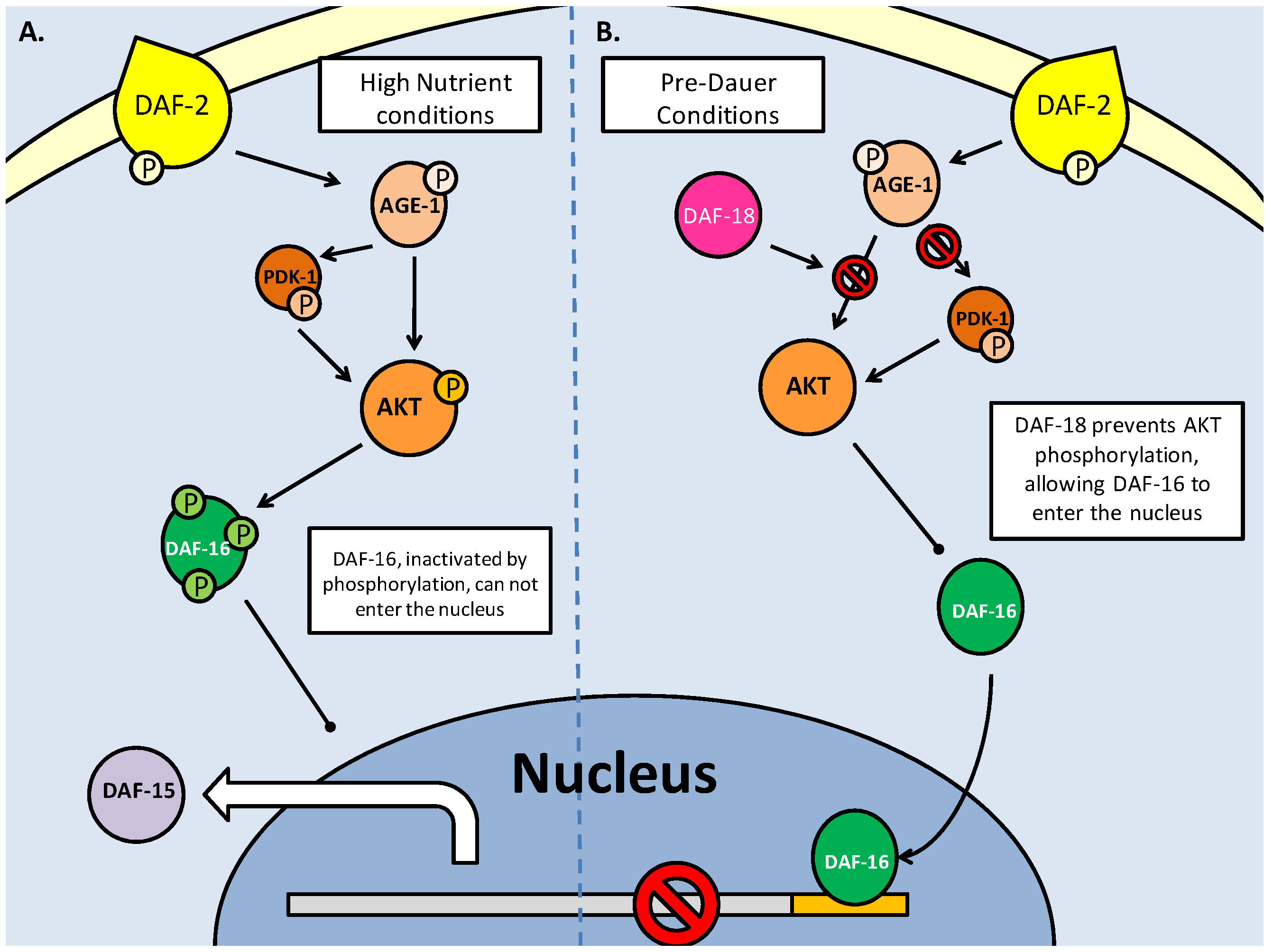
The C. elegans pathway of mTOR production is dependent on insulin/IGF response. TOR homologs DAF-15 (Raptor) and LET-363 (TOR) are regulated by DAF-16 (FoxO) transcription factors which are also responsible for entry into dauer. A. Shows high nutrient conditions, with insulin signaling, through active AGE-1(PI3K) and AKT phosphorylating DAF-16, impeding its entry into the nucleus and allowing transcription of the TOR genes. B. When phosphorylation of AKT is blocked by DAF-18 (PTEN), DAF-16 is able to enter the nucleus, and can inhibit the transcription of TOR genes.
Figure 9
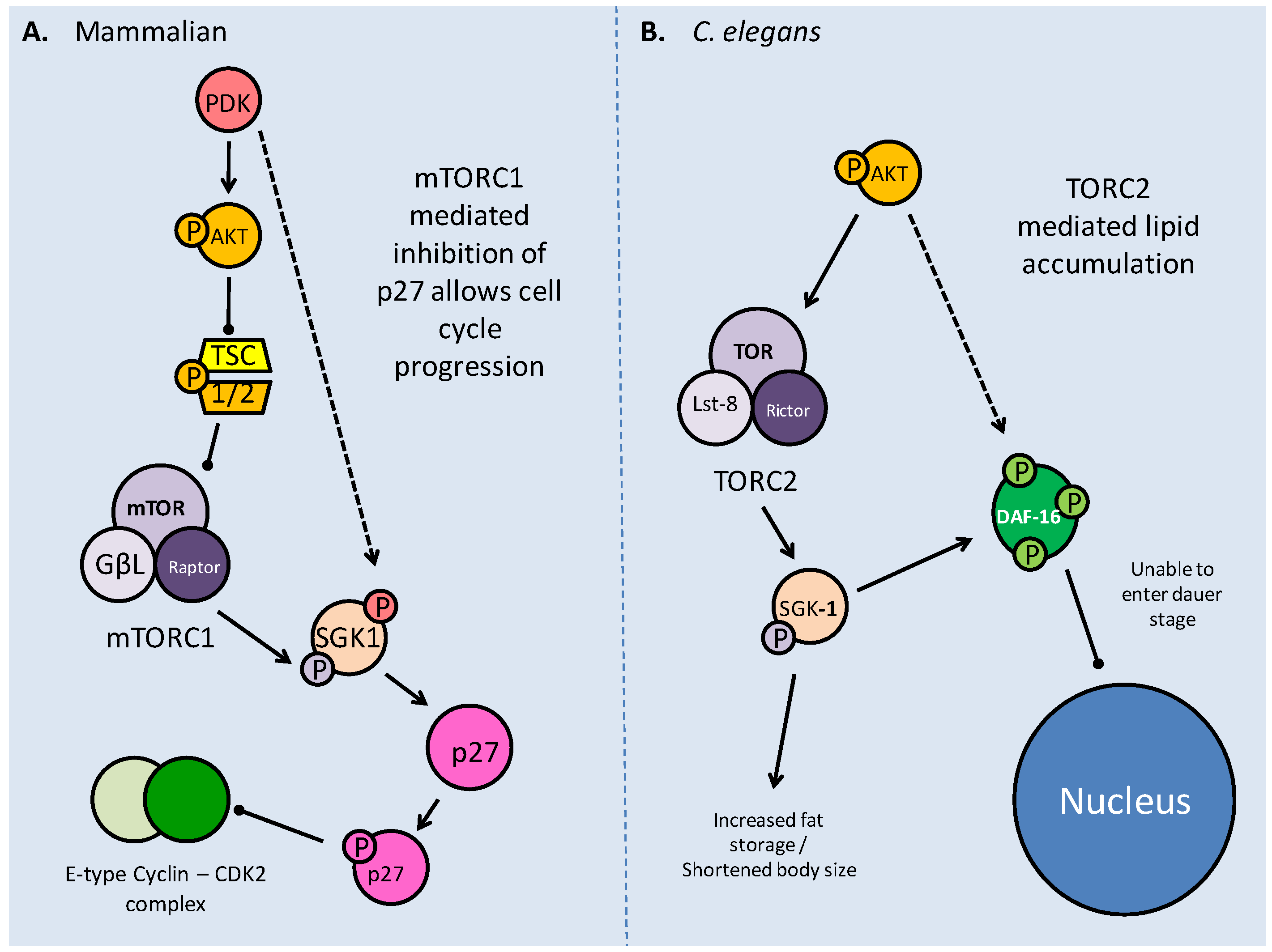
TOR interactions with SGK1 in both mammalian and C. elegans models. A. Phosphorylation of SGK1 by mTORC1 and then PDK activates the kinase, before it acts upon p27. Phosphorylated p27 is unable to bind E-type Cyclin - CDK2 complexes, which in turn allow progression into the S phase of the cell cycle. B. The C. elegans TORC2 complex phosphorylates SGK-1 which modifies DAF-16 to prevent entry into dauer, and increases lipid stores, possibly as a preparation for dauer.
The activation or inhibition of mTOR also appears to have an effect on autophagy; the process of recycling cellular components and reducing the size of the cell. In periods of energetic deficit such as during MRD, reduced mTOR activity and increased AMPK activity could lead to autophagy. Oppositely, under conditions of increased amino acid availability, which correlate with elevated mTOR activity, autophagy is suppressed [52]. The topic of autophagy will be further elaborated later in this review.
The translational effects of mTOR are typically necessary to the progress of the cell cycle, but when an organism transitions into a hypometabolic state, it is important to shut down energetically expensive processes, such as protein synthesis. This typically leads to a suppression of insulin signaling (and other growth factor) pathways, leading to reduced Akt and mTOR activities. Such is the case in C. elegans as it enters the dauer stage of larval development. The dauer stage, which is triggered by stressful conditions (e.g. temperature rise, lack of oxygen, lack of food), is characterized by pre-accumulation of lipid stores, the suppression of the PI3K/Akt (Age-1/akt in C. elegans) pathway and the up-regulation of FoxO transcription factor (Daf16 in C. elegans) activity.
The anti-dauer activity of C. elegans TOR
In the C. elegans model, the TOR family is represented by two members, DAF-15 (a C. elegans ortholog of Raptor) and LET-363(TOR), which have been shown to play a role, by their suppression, in entry into the dauer stage [55]. The two TOR proteins associate to function as one, with DAF-15 acting as a bridging protein between TOR and its targets. Suppression of this complex results in enhanced fat storage, possibly in preparation for the dauer period [55]. As in mammalian systems, TOR functions in the AKT controlled insulin-response pathway in C. elegans and its activation pattern mirrors that of the AKT family of kinases (AKT-1 and 2) (Fig. 8) [55]. Typically, when nutrient levels are high, and reproductive growth is possible, the genes mediating reproduction and increased metabolism are activated by TOR which in turn is regulated by AKT. In this system, AKT inhibits DAF-16 through phosphorylation which prevents DAF-16 entering the nucleus and binding the regulatory region of daf-15, preventing its transcription (Fig. 8). DAF-16 may also inhibit TOR through a series of currently unknown downstream transcription factors [55]. Thus, under limited nutrient conditions, associated with the preliminary conditions of dauer formation, AKT activity is minimized, leading to active DAF-16, and then decreased TOR (let-363/daf-15) activity. The DAF-16 downstream effectors are activated, while the inactivity of TOR leads to an accumulation of fat [55]. In the pre-dauer stage, fat accumulation in the intestinal and hypodermal cells takes place, providing an energy source throughout their anti-senescence period.
The regulatory profile of TOR and DAF-16 is also affected by the PTEN phosphatase (DAF-18). It is suspected that DAF-18 and DAF-16 have complimentary activities, with DAF-18 able to directly de-phosphorylate DAF-16, as well as prevent the upstream kinase, AKT, from becoming active. Indeed, DAF-18 is able to dephosphorylate PIP3, which if active, would activate (phosphorylate) first PDK and then AKT (Fig. 6 and 8) [55]. In a similar manner, PP2A affects TOR activity by directly de-phosphorylating akt, preventing its downstream activity. This PP2A effect has been put forward as a mechanism to support DAF-16 entry into the nucleus and initiate the dauer response (Fig. 6) [56]. Hence a correlation between high phosphatase activity and low TOR activity could be predicted as a marker of entry into the dauer stage.
The relationship between TOR and SGK (SGK-1 in C. elegans) in mammalian systems maintains cell cycle progression via inhibition of p27 [54], but in C. elegans a novel role of the TORC2 complex is seen that affects fat accumulation, a critical preparatory factor for prolonged dauer periods [57]. The C. elegans Rictor homolog, while not apparently necessary for early development, has a role in pre-dauer development, primarily as an effector of AK T, as part of the TORC2 complex. It is thought that TORC2 recruits SGK-1 to affect fat accumulation and body size [57]. Mutations in both C. elegans Rictor and sgk-1 showed the same phenotypic response of increased lipid stores and shortened body sizes, indicating their direct relationship (Fig. 9) [57]. This indicates a novel role of SGK-1, which had previously been implicated in suppression of DAF-16 (via phosphorylation) as an anti-dauer response.
The relationship between TOR and AMPK
To conclude this section, a short discussion of AMPK effects is needed. As mentioned above, the inverse relationship between the activity of TOR and AMPK is critical for long term survival. AMPK is not critical to dauer entry, but its absence causes premature death in C. elegans as a result of a rapid consumption of energy stores and consequent organ failure while in the dauer stage [58]. AMPK, identified in many systems as a critical metabolic regulator, maintains energetic homeostasis by regulating the consumption of stored lipid reserves during the dauer period [58]. Key to lipid consumption is regulation of the activity of adipose triglyceride lipase (ATGL, ATGL-1 in C. elegans), that is phosphorylated by AMPK at multiple sites [58]. AMPK activity is regulated in two ways; post-translationally and by translation of its catalytic subunit, AAK. The hetero-trimeric AMPK complex is dependent on AMP signaling, but can attain increased activity through phosphorylation. The serine/threonine kinase, LKB1 (C. elegans PAR-4) in a complex with two cofactors, phosphorylates AMPK, likely at the α2 (AAK-2) subunit to significantly increase its activity. In cases where AMPK catalytic subunit expression is suppressed, primarily AAK-2, rapid lipid depletion is seen and the life span of these nematodes drops to about one-third of control values; this can be reversed with resumed AAK-2 production [58].
The STAT Family: Regulation of Development and Interactions with TGF-β to regulate C. elegans Lifespan
The 'Signal transducer and activators of transcription' (STAT) family of transcription factors are a highly conserved family of proteins involved in developmental signaling. The STAT pathway has parallels with the TGF-β pathway in that it is largely based on responses to growth-factors and cytokines, and is primarily involved in development. The STAT transcription factors are also involved in host immunity, formation of blood cells and inflammation responses [59]. The mammalian STAT family contains seven members, with STAT-1 and STAT-3 being the prominent members [59]. STAT activation is a conserved process and requires tyrosine phosphorylation (tyr 701), typically by the Janus kinase family (JAK), which allows nuclear translocation and subsequent transcription factor activity. STATs are also able to interact in a protein-protein fashion in order to enhance/suppress target protein activity.
Regulating the transcription factors are numerous inhibitors, which act on the STATs at different stages of their activity. They include the 'suppressors of cytokine signaling' family (SOCs), which prevent STAT phosphorylation by JAK by binding the receptor site on the transcription factor, and the 'protein inhibitors of activated STATs' (PIAS), which bind phosphorylated STATs and prevent transcriptional activity (Fig. 10) [60]. These inhibitors, while acting on mainly on STATs, also have roles in apoptosis suppression and cell cycle regulation.
STATs in stress response and their interactions with known SREs
As part of their role in development, the STATs aid in determining the survival state of cells that have been exposed to ischemia (restricted blood flow, cutting off oxygen and nutrient delivery and waste removal from cells) and reperfusion (resumption of blood flow). Ischemia/reperfusion (I/R), a significant factor in the damage done during stroke or heart attack, triggers divergent responses by members of the STAT family; specifically via STAT-1 and STAT-3 [61]. Interestingly, however, these two have contradictory responses, STAT-1 mediating apoptosis signaling whereas STAT-3 mediates protective responses. Following I/R insult, phosphorylation of STAT-1 by JAK or MAPKs leads to nuclear translocation, the up-regulation of pro-apoptotic genes such as Fas, Fas-L (the 'death receptor' and its ligand, respectively) and Caspase-1, and the repression of anti-apoptotic proteins such as Bcl-2 and Bcl-xL (Fig. 10) [61]. In keeping with a pro-apoptotic role, STAT-1 also binds directly to the tumor suppressor protein, p53, which aids p53 binding to pro-apoptotic protein promoter sites. STAT-1 also binds the MDM2 promoter, repressing its expression (Fig. 10) [60]. This repression enhances the stability and activity of p53, which may otherwise be bound and inactivated by MDM2 (see further discussion below in the p53 section).
Figure 10
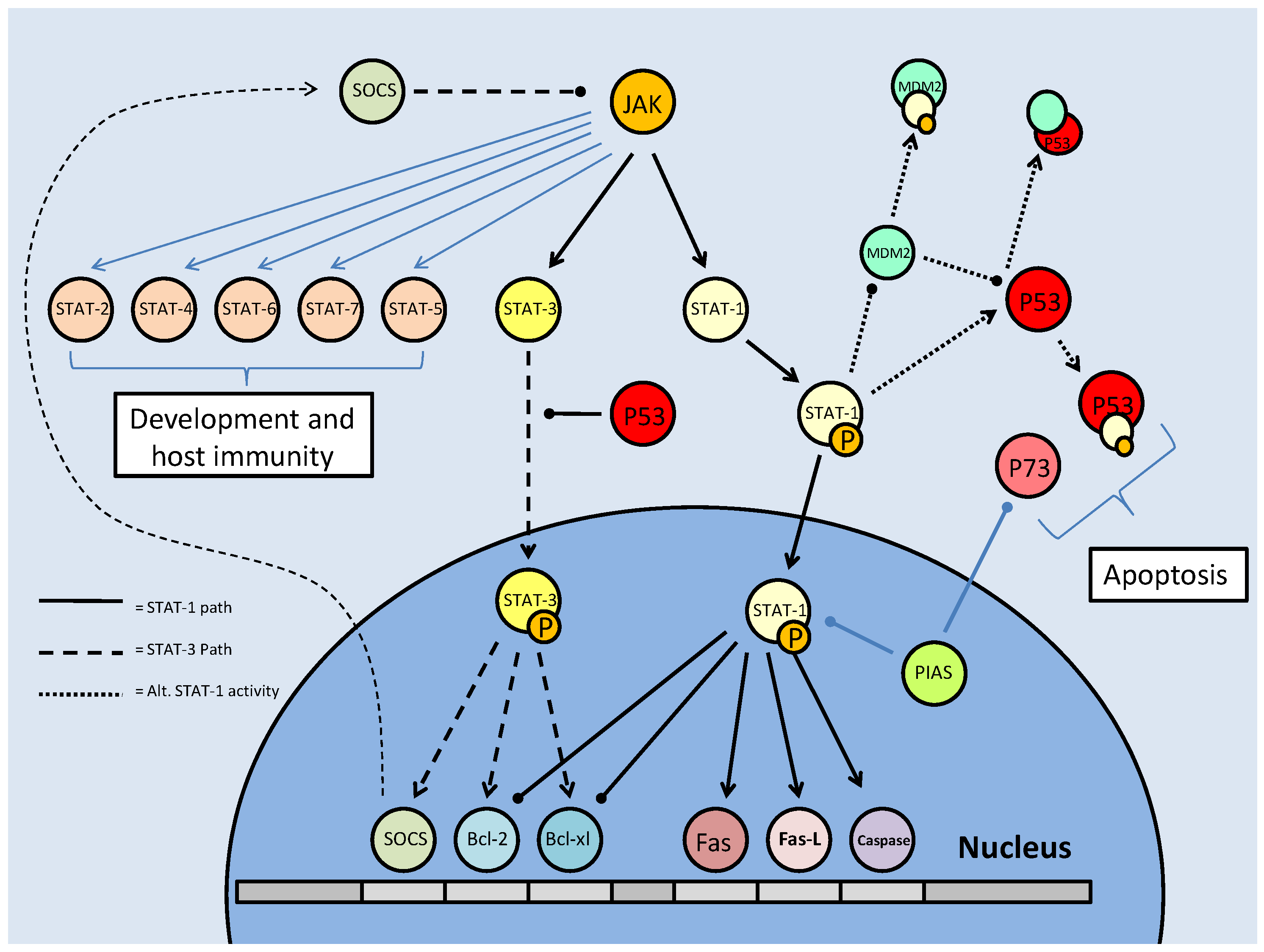
Mammalian STAT signaling controls multiple developmental processes. Key members of the STAT transcription factors, STAT-1 and 3, also regulate apoptosis. STAT-1 transcriptionally up-regulates apoptosis signaling genes, as well as interacting with the P53 tumour suppressor. This interaction increases P53 activity. Active P53 may also inhibit STAT-3 activation, which would prevent the downstream up-regulation of anti-apoptotic/anti-STAT-1 proteins.
The pattern of positive regulation of STATs by p53 is not uniform, however, since active p53 promotes the inhibition of STAT-3 (Fig. 10). STAT-3 may be classified as an oncogene due to its constitutive expression in numerous cancerous tissue types and its downstream transcriptional targets that inhibit apoptotic signaling or maintain cell cycle progression [62]. Although the mechanism is not clear, p53, possibly by repressing JAK expression or increasing tyrosine phosphatase activity, prevents STAT-3 phosphorylation and subsequently affects its DNA binding capacity [62]. As a result, this may act as a pro-apoptotic signal, since STAT-3 is able to increase the expression of anti-apoptotic Bcl-2 and Bcl-xl, the same proteins that are repressed by active STAT-1 (Fig. 10) [60]. Similarly, p53 affects the cytokine-induced STAT5 as part of a pro-apoptotic mechanism [62]. In conjunction with P53/STAT-1 interactions, PIAS-1 (the STAT-1 inhibitor) is also known to bind p73 (a member of the p53 tumor suppressor family) and sumoylate it (Fig. 10). This prevents critical downstream transcriptional effects on Bcl-2 family members and cell cycle regulators such as p21 [63].
In addition to I/R, hyperosmotic shock is another stress that can trigger STAT responsive signals. Hyperosmotic stress (e.g. due to dehydration or hypersalinity) trigger the p38 MAPK, through its upstream activator MKK6, which in turn is able to regulate STAT activity. Under these conditions p38 MAPK phosphorylates the critical tyrosine residue on STAT-1, activating the transcription factor, while also phosphorylating the tyrosine phosphatase SHP2 [64]. This should inactivate the phosphatase (however this remains undetermined [64] which would otherwise act to repress STAT phosphorylation.
Since stress activated protein kinases like p38 MAPK can activate the STATs, a link between STAT activation and one of the major stress responsive pathways, the insulin/IGF pathway, may be expected. Hence, in a relatively recent discovery, it has been shown that the Insulin/IGF pathway, which typically signals via the PI3K/Akt cascade, also signals the JAK/STAT pathway. The SOCs STAT inhibitors are able to repress this via direct interaction at the insulin-like growth factor I receptor (IGF-IR), which may also affect the canonical insulin response pathways [65]. In addition, SOC production can become part of a feedback inhibition loop, with STAT-3 able to up-regulate their transcription [66].
The role of STA-1 in development and dauer
Whereas in vertebrates, the STAT family of transcription factors are important in many biological processes such as cytokine signaling and host immunity, in C. elegans they may also play an important regulatory role in the transition into the dauer stage. The STA-1 transcription factor, a STAT ortholog, regulates larval development parallel to and in conjunction with TGF-β. Along with the DAF-16 and Smad (DAF-8/14) transcription factors, STATs are emerging as key regulatory switches through their actions in repressing dauer formation and promoting senescence.
As in mammalian systems, STA-1 is regulated by tyrosine phosphorylation, which is required for entry of the transcription factor into the nucleus and DNA binding (Fig. 11). In C. elegans, sta-1 expression is seen in most of the same tissues as TGF-β (including intestine, pharynx, and critically for development, muscle), once again indicating a parallel response [67]. Though STA-1 only holds a 22% amino acid homology to mammalian STATs, it maintains the conserved regions that characterize tyrosine phosphorylation and transcriptional regulation. STAT phosphorylation in mammals is typically mediated by the JAK family, but although STA-1 is known to be phosphorylated, the kinase responsible has not yet been identified [67]. While STA-1 responses could be characterized as anti-dauer (in the line of TGF-β), its expression is seen throughout the dauer period as well as prior to it. Its activity remains unchanged throughout the latency period and into subsequent developmental stages [67]. This would typically indicate a role less focussed on dauer determinacy than continuous development. However, the continuous activity profile is likely what allows STA-1 to act as a compensatory element in TGF-β signaling.
Figure 11
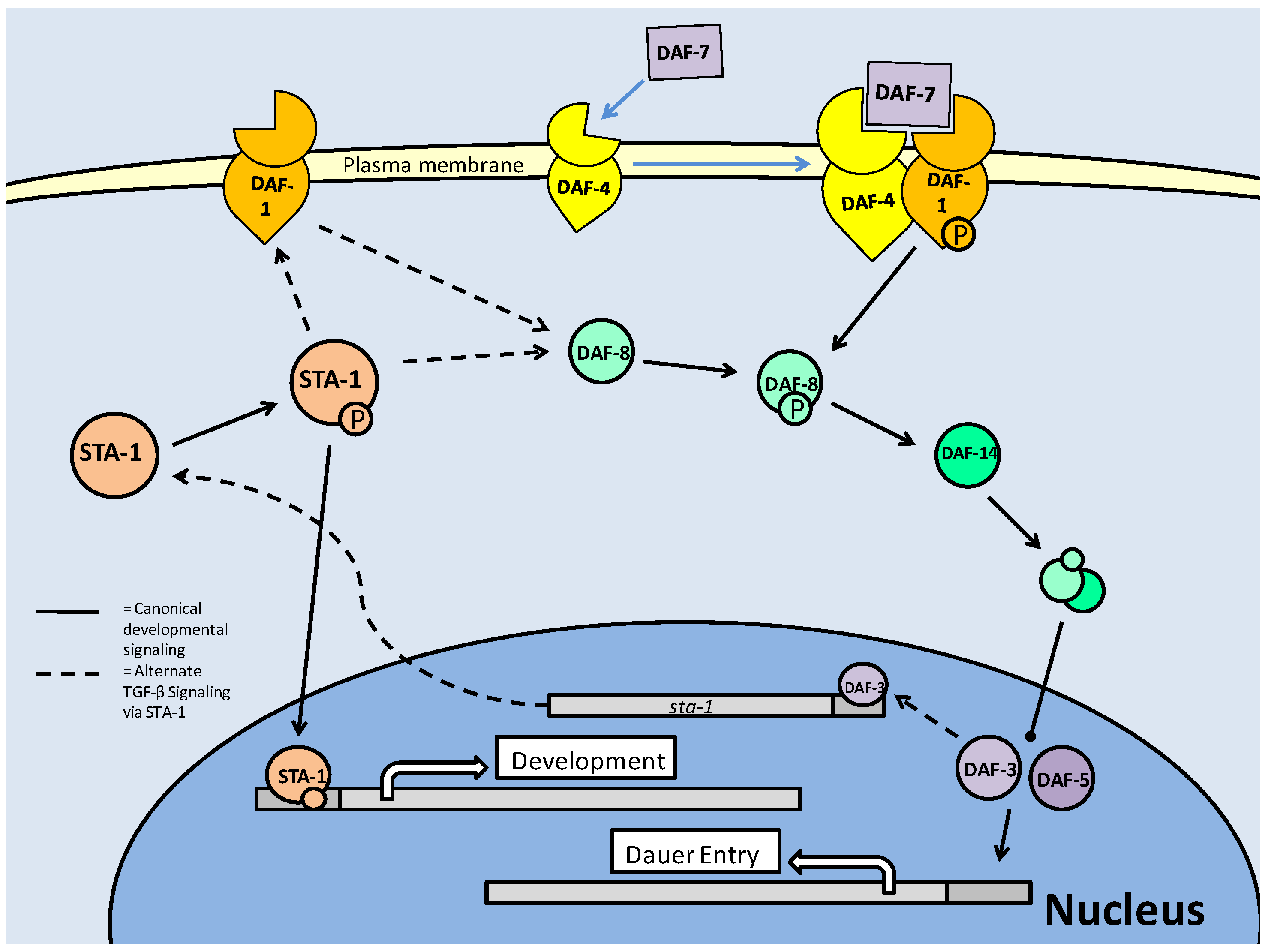
STA-1 Signaling in C. elegans. Standard STA-1 activity mirrors the TGF-β pathway, in terms of contribution to development, but there is an alternate mechanism in which STA-1 directly interacts with members of this path. In the absence of DAF-7, 4 or 14, STA-1 is able to directly contribute to TGF-β signaling through interactions with the DAF-1 receptor or the downstream Smad, DAF-8. There is also a proposed feedback inhibition of dauer initiation through (pro-dauer) DAF-3 regulated transcription of sta-1.
Like the TGF-β pathway, STA-1 signaling is heavily influenced by neuronal signaling. The absence of STA-1 at raised temperatures (which would possibly be conducive to dauer signaling conditions) results in ~30% higher rates of dauer entry [67]. Thus, like TGF-β, the C. elegans STAT has both developmental and dauer responsive roles which it performs in conjunction with TGF-β. STA-1 can act to repress dauer through two paths; one compensatory and one feedback [67]. In the first mechanism, STA-1 itself can activate anti-dauer signaling through interactions with the type I receptor (DAF-1) and downstream Smad (DAF-8) (Fig. 11) [67]. These interactions indicate a break from the canonical TGF-β signaling, and possibly indicate a compensatory activation in the absence of DAF-7(TGF-β ligand), DAF-4(type II receptor) or DAF-14 (Smad). The secondary mechanism involves up-regulation (via increased transcription) of sta-1, and then activity through the first mechanism. The sta-1 sequence contains Smad binding regions, which in a mechanism of feedback inhibition, allows interaction with the pro-dauer Smad, DAF-3 (Fig. 11) [67].
Programs of Cell Death or Degeneration in Response to Stress: Apoptosis or Autophagy?
Apoptosis
Apoptosis or programmed cell death is a process by which cells are destroyed in a regulated manner; often this occurs as a response to extreme stress but it is also an integral part of programs of development and senescence. Apoptosis is characterized by chromatin damage and resultant DNA cutting [68]. The primary mediators of apoptosis are cysteine proteases of the 'caspase' family that are targeted directly or indirectly (i.e. through second messengers or pro-apoptotic proteins) and which, in turn, trigger a host of proteins such as endoribonucleases and proteases.
The control of apoptosis through mitochondrial integrity
Apoptosis can be signaled by a number of different stress conditions such as DNA damage, oxygen/energy debt or starvation, but the response pathway remains fairly conserved, with the mitochondria being the key to its initiation. Regulating the mitochondrial response is the B-cell lymphoma 2 (Bcl-2) family of proteins, and within this group, specifically, the BH3-only members of the family [69]. The BH3 (Bcl-2 Homology 3) domain and its complementary binding cleft are critical to the activity of these proteins, with the majority of their protein-protein interactions occurring at these sites. The Bcl-2 family contains both pro (Bax, Bak, Bim) and anti (Bcl-2, Bcl-xl, Bcl-3) apoptotic members, which through the BH3 domain are able to interact with each other [69]. These bindings either result in the stimulation of apoptosis (i.e. the homo-dimerization of pro apoptotic members) or its inhibition (i.e. the hetero-dimerization of a pro- and anti-apoptotic member). Pro-apoptotic members of the family, primarily Bax or Bak, translocate to the mitochondrial membrane when activated, and stimulate outer mitochondrial membrane permeability (OMMP). This allows the release of proteins/secondary messengers from the inter-membrane space (IMS) into the cytosol; of particular importance is cytochromes C release. Subsequently, caspases are activated through cleavage, dimerization, and auto-proteolysis reactions, and go on to trigger downstream cell death proteins such as DNAases, proteases and 'inhibitor of apoptosis' (IAP) antagonizers [69]. Hence the idea of mitochondrial membrane maintenance as the key to apoptotic regulation is formed.
Figure 12
Apoptotic signaling in C. elegans (A) and mammalian (B) models. The anti-apoptotic Bcl-2 family members (CED-9, Bcl-2 and Bcl-xl) bind pro-apoptotic proteins attempting to locate to the outer mitochondrial membrane (OMM), neutralizing their activity. At the OMM, pro-apoptotic BH3 containing proteins will form a pore (OMMP) which allows mitochondrial proteins and second messengers to enter the cytoplasm, signaling downstream caspase activity (though it is unknown if cytochrome C release is a factor in C. elegans apoptosis). In both models, apoptosis and autophagy (via Bax/DRP-1 and Beclin-1) may be inhibited by Bcl-2/CED-9 binding. In the mammalian model, this can be reversed through the phosphorylation of Bcl-2 by JNK.
The physical dynamics of the mitochondrial membrane are either stabilized through fusion events where mitochondria form interconnections, or destabilized through mitochondrial fragmentation (or 'fission'), which destroys the integrity of the membrane and leads to apoptotic signaling. The proposed Bax/Bak interaction which occurs at the OMM is suspected to disrupt the lipid bilayer, creating an OMM pore [69]. To regulate the formation of these pores, the Bcl-2 and Bcl-xl anti-apoptotic members of the Bcl-2 family form hetero-dimers through a protein-protein interaction, preventing the Bax/Bak homo-dimer interaction, nullifying their OMM pore formation capacity (Fig. 12). By contrast, the tumor suppressor p53 is shown to promote apoptosis primarily through transcriptional activation of pro-apoptotic genes (PUMA, Noxa, Bax), but is also speculated to bind directly to the Bax/Bak oligomer and activate it. This correlates with role of p53 in apoptosis signaling, but is a novel, non transcription-factor mechanism [69].
While we tend to focus on the interactions of the Bcl-2 family as a means of apoptosis suppression, the interactions function both ways, with a pro-survival signal equally likely to provoke an inhibitory response from the pro-apoptotic members; possibly overwhelming the balance and eventually triggering apoptosis [69].
The pathways of programmed cellular death in C. elegans
C. elegans have three major cell death pathways; apoptotic, autophagic and necrotic. The pathway triggered is dependent on the stress stimulus. Apoptosis signaling is largely characterized by DNA damage and fragmentation of the chromatin, which triggers the 'C. elegans p53 like' (CEP-1) transcription factor and EGL-1 (C. elegans 'BH3-domain-death activator' homolog) responses [70]. Apoptosis in C. elegans can occur in two phases. First, it is seen as part of the developmental process occurring at the embryonic stage and in the L2 larvae; resulting in the removal of muscular, hypodermal and neuronal cells as well as a large proportion of oocytes (reproductive/egg cells) from mature gonads [70]. Secondly, apoptosis may occur when induced by genotoxic stress, seen in the germline cells of hermaphrodites. Necrosis only occurs under prolonged periods of cellular stress [70]. Acidification of the cell is the primary cause of necrotic cell death. Signaled by excess Ca2+ release, as a result of modification and degeneration of the calcium channels/exchangers, cellular calpains (which recruit ROS's to the lysosomal membrane) trigger lysosomal rupturing [69]. With the physical degradation of the lysosome, cellular pH drops as H+-ATPases are damaged, and the release of cathepsins (proteases) which are activated by the lowered pH cause the destruction of cellular components [70].
Apoptosis in C. elegans
Apoptosis signaling in C. elegans, as in mammalian systems, can be initiated by the p53 tumor suppressor, CEP-1. CEP-1 transcriptionally up-regulates production of the 'egg laying abnormal' (EGL-1) protein [70]. EGL-1, like the BCL2 family of proteins, contains a BH3-only-domain, and when activated, is thought to locate to the mitochondrial surface, possibly in response to the presence of CED-9/CED-4 complexes. CED-9 and CED-4 are C. elegans homologs of Bcl2 and 'apoptotic protease activating factor 1' (APAF1), and are anti- and pro-apoptotic, respectively. Once at the mitochondrial membrane, EGL-1 causes membrane opening, and subsequent release of apoptosis signaling components (Fig. 12) [70]. This may mirror the BH3-only protein (Bax/Bak) activity in mammals, which causes the formation of the OMMP. CED-4, in a sense, acting like caspase 8 in the mammalian caspase cascade, activates the downstream C. elegans caspase (3/9) homolog, CED-3. CED-3 starts as monomeric subunits which require dimerization and auto-proteolysis, and when functional acts as the 'executioner kinase', triggering major cell destruction events such as protease activity [71]. Dimerization is mediated by CED-4, and is followed by auto-proteolysis which creates the functional subunits (large and small). Interestingly, in C. elegans (and mammalian models only), there is a protein which mimics the small subunit of CED-3, preventing functional caspase activity by binding the CED-3 monomer. While this may inhibit CED-3 activity to a degree, its action can be overwritten by the induction of dimerization (of functional units) by CED-4 [71]. This level of control could be used to regulate non-specific apoptosis events which could harm larval development. Concurrent with the CED4/3 pathway of apoptosis initiation, a series of CEDs including 7, 1, 6 and the cytoskeletal modifier, CED-10, modify the structure of a neighbouring cell in order to 'engulf' the apoptosed cell, for eventual degradation [70].
Mitochondrial fission, leading to fragmentation, is thought to be the major event in apoptosis signaling, with the integrity of the outer membrane susceptible to pro-apoptotic factors. This indicates that the activity of the BCL2 homolog, CED-9, is critical in maintaining mitochondrial structure. As in the mammalian system, pores formed in the outer membrane are a result of binding by pro-apoptotic BH3-only proteins. The BH3 domains are also critical in the inhibition of these pro-apoptotic proteins, with BCL-2 able to bind Bax, and similarly a suspected CED-9: DRP-1 interaction in C. elegans [72]. DRP-1, the 'death associated protein kinase' related protein kinase, is a serine/threonine kinase in C. elegans, and its over-expression is known to cause mitochondrial fission, which can lead to the release of important pathway signaling molecules such as cytochrome C, which is seen to trigger caspase activity in mammalian systems, and ROSs. Conversely, over-expression of the CED-9 protein causes mitochondrial fusion [72]. The fusion event may be suppressed by up-regulation of DRP-1, which further indicates that binding of BH3-only proteins in their BH3 domain inactivates them, possibly resulting in a regulatory system of mitochondrial fission/fusion dependent on the relative activity levels of CED-9 and DRP-1 (Fig. 12) [72]. Whether this is important in apoptosis/caspase signaling or not remains a contentious issue [73, 74].
As mentioned above, the importance of the CED-9/DRP-1 interaction in regulating mitochondrial responsive apoptosis is in debate, but there appears to be a non mitochondrial role for DRP-1 in apoptosis signaling. The fission-1 (FIS-1) homolog, fis-2, in its standard role recruits DRP-1 to the mitochondrial membrane, promoting alteration of the outer membrane and the potential signaling of downstream caspases CED-3 and 4. However, in knockouts experiments of ced-3 and 4 where increase in the total number of viable cells indicated their roles in apoptosis, additional mutations in drp-1 and fis-2 further increased the numbers of cells which survived. This indicates that these proteins associated with mitochondrial fission have (slight) apoptotic activity independent of the downstream caspases (CED-3/4) that fission would signal [73]. To further this, the non-mitochondrial fission role of DRP-1 appears to also require cleavage from CED-3 [73]. Thus, while it appears that as in mammalian systems, the majority of apoptosis responses arise from mitochondrial fission and caspase signaling, there may be alternate routes for the apoptosis machinery to initiate cell death.
Autophagy
An alternative to complete cell death that is an option under some metabolic stress conditions is the relatively recently discovered process called autophagy. Autophagy is the process whereby cells effectively “down-size” themselves by degrading portions of their organelles and cytosolic components, recycling them to obtain precursors and energy, and minimizing cell volume, in an attempt to prolong viability under stressful conditions. There are multiple forms of autophagy, the most prominent being 'macro-autophagy'. This follows the conserved model of autophagosome formation from free membrane. The autophagosomes section large portions of the cytoplasm and transport them to the lysosomes, where after a fusion event between lysosome and autophagosome, the contents are transferred [70]. While macro-autophagy is the main form, alternate forms of autophagy include chaperone mediated autophagy (CMA) in which cytosolic proteins are guided into the lysosomes by chaperone proteins, and micro-autophagy which involves direct lysosomal membrane invagination, cytoplasmic sectioning and subsequent degradation [70]. The common theme in autophagy is the engulfing of cytoplasmic fractions followed by their degradation.
Macro-autophagy involves the multi-step formation of autophagosomes which grow to encase a section of cytosol (that may also include organelles) and then fuse to lysosomes. The fused structure, known as an autophagolysosome, carries out the degradation of its contents with hydrolases [68]. Formation of the autophagosome requires coordinated actions of multiple autophagy proteins, the Atgs, which are largely involved in complexing in order to bind and elongate the autophagosomal membrane (Fig. 13). The process begins by binding 'free cellular membrane' to the initiating Beclin 1 (Atg6) complex. Next, Atg proteins 5 and 12 begin a crucial conjugation mechanism mediated by Atg7 and 10. Atg16 binds to the Atg12-Atg5 complex, which localize at the isolated membrane and initiate the elongation of the pre-autophagosomal membrane (Fig. 13) [68]. This process is extremely rapid, with the Atg12-Atg5 complex formed immediately after individual Atg protein synthesis. The next stage, completion of the autophagosome, is mediated by Atg8 (LC3), a microtubule associated protein. Atg8 is cleaved (Atg8c) and activated by Atg4 and is then conjugated to phosphatidyl-ethanolamine (PE) which then binds to the membrane and completes its formation (Fig. 13) [68]. Once formed, the autophagosome fuses with a lysosome, and its contents are degraded by hydrolases (Fig. 13) [68]. The target cell remains 'minimized' until the stress has abated, after which, it can begin restructuring.
Inducing autophagy by caloric restriction and tumor profiling
One of the key initiating conditions of autophagy is starvation. When faced with prolonged caloric restriction, autophagy is triggered to recover precursor molecules, limiting the energetic expense of resynthesis. Along this line, another critical role of autophagy would be in the removal of nonessential proteins so that their amino acids can be recycled to meet synthesis demands for essential proteins. Under normal caloric intake, autophagy is naturally inhibited with the pro-survival Bcl-2 complexed with the autophagy initiator Beclin-1 [75]. Similar to its pro-survival role in binding pro-apoptotic proteins, Bcl-2 is suggested to bind Beclin- 1 at its BH3 domain. The dissociation of this complex, in order to induce autophagy, is mediated by mitogen activated protein kinases (MAPKs) and JNK (Fig. 12). By phosphorylating Bcl-2 at three sites within the critical 'non-structured loop' its physical conformation is altered, preventing it from binding to Beclin-1 [75]. This regulatory mechanism poses an interesting compromise between autophagy and apoptosis, with Bcl-2 binding both the pro-autophagy Beclin-1 and the pro-apoptosis Bax. With JNK being activated by starvation, the selection of autophagy or apoptosis is based upon duration of the exposure. For example, HeLa cells subjected to short periods (up to 4 hours) of nutrient deprivation showed dissociation of the Bcl-2/Beclin-1 complex, indicating an early onset stimulation of autophagy. However, after 16 hours, and approaching maximal levels of Bcl-2 phosphorylation, Bcl-2/Bax complex dissociation along with Caspase 3 activation occurred, indicating apoptotic signaling [76].
Figure 13
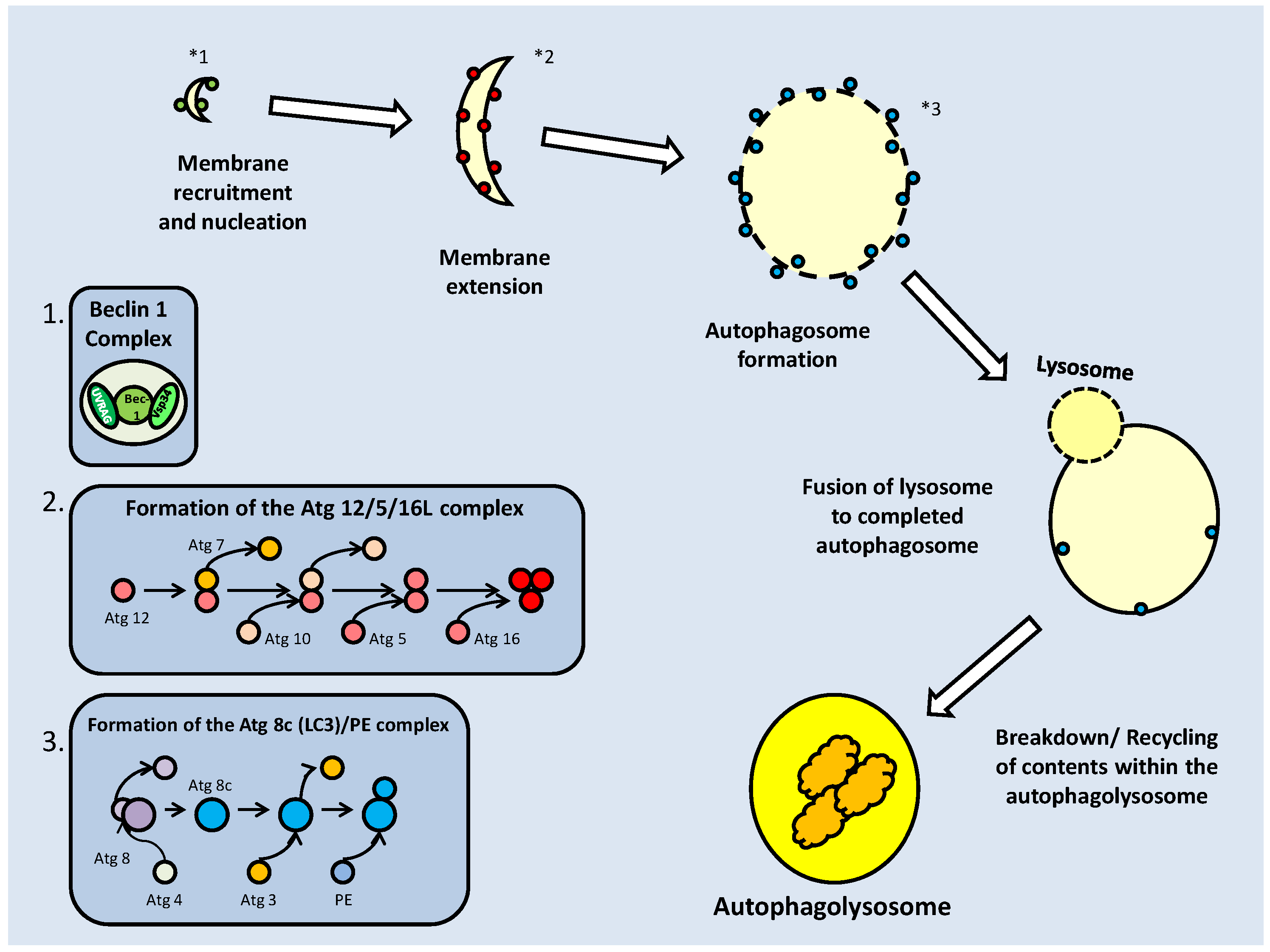
Formation of the autophagosome, and subsequently, the autophagolysosome. Formation begins with the binding of isolated membrane by the Beclin-1 complex. This recruits further Atg proteins to the membrane, via nucleation, and initiates the elongation of the membrane by the Atg 12/5/16L complex. The cleaved Atg8 (LC3)/Phosphatidyl ethanolamine (PE) complex finalizes the formation of the autophagosome. Binding to and incorporation of the lysosome creates the autophagolysosome which is then able to degrade/recycle its contents through hydrolase activities.
The control of the Bcl-2 family by its own members, in order to form active homo-dimers or inactive hetero-dimers, is not the only level of control on these proteins. Controls by JNK and p53 directly on individual members, or complexes, function to effect a selection between autophagy and apoptosis. Like p53, the tumor suppressor p14 (ARF) is able to exert both p53 dependent (primarily through its inhibition of MDM2) and independent activities to determine the survival condition of the cell [77]. Beclin-1, as mentioned previously, is able to interact with the Bcl-2 family members through its own BH3 domain (interacting with the BH3 binding groove of the BH3-only-protein members). In a cell survival mechanism, both anti-apoptosis Bcl-2 and Bcl-xl can complex with Beclin-1, and inactivate autophagy [77]. Despite its positive interactions with the pro-apoptotic p53, ARF induces autophagy (as a function of its tumor suppressor capacity) by binding Bcl-xl, allowing isolated Beclin-1 to initiate autophagy. When active, ARF localizes to the mitochondria, where it is able to interact with Bcl-xl proteins at the outer membrane. The physical complex between ARF and Bcl-xl can both sequester free Bcl-xl and unbind Beclin-1-bound Bcl-xl; this results in large increases in autophagy [77]. Throughout, p53 maintains its role as an autophagy suppressor, by strongly repressing the transcription of ARF. However, this repression is largely seen in unstressed cells, suggesting that p53 regulates ARF in order to prevent unwanted autophagy [77].
In the case of tumor suppression, both autophagy and apoptosis are largely able to facilitate the process and, hence, the ability of p53 to also stimulate autophagy does not come as a surprise. While the cytoplasmic form (i.e. inactive for transcriptional regulation) inhibits autophagy via direct protein interaction, its transcriptional activities have been seen to up-regulate both PTEN and TSC2, which lead to the inactivation of mTOR, a potent autophagy inhibitor. Specific to autophagy, active p53 also increases the transcription of 'damage regulated autophagy modulator' DRAM, which encodes a lysosomal protein that induces autophagy [78].
The relationship of mTOR and Beclin-1 in determining the state of autophagy
To return to one of the aforementioned points, one of the major regulators of autophagy is upstream inhibition by the mTOR kinase. This kinase which is responsible for maintaining protein synthesis and cell cycle progression is linked with pro-survival activity, and acts in competition with Beclin-1 to inhibit autophagosome formation. Beclin 1(Vps 30) forms a class III PI3K complex, along with a tumor suppressor UVRAG and Vps34, which regulates the formation and nucleation of the autophagosome. Interestingly, both Beclin-1 and mTOR compete for Vps34 (Fig. 14). Signaled by high amino acid availability, Vps34 triggers the activity of mTORC1, which can affect another critical initiator of autophagy, the Atg1 kinase (Atg1k) complex [79]. By hyper-phosphorylating Atg13, mTOR is able to prevent the complexing of Atg13 and Atg1, which is a critical step in the formation of the Atg1k complex [79]. Subsequently, the autophagosome cannot be formed (Fig. 14). Beclin-1, which contains a conserved binding site for Vps34, can have its interaction broken by Bcl-2 family member binding, in order to prevent autophagy (Fig. 13). Thus, the initiation of autophagy is, to a significant degree, dependent on which protein Vps34 complexes with. In nutrient rich conditions mTOR is likely to preferentially complex with it, and along with Bcl-2 proteins, represses high levels of autophagy that would be detrimental to the cell. Conversely, under low nutrient conditions, it is likely that Vps34 preferentially binds free Beclin-1, in order to maintain energetic homeostasis.
Basal and induced autophagy in C. elegans development
In C. elegans, the transition into the dauer stage is mediated by a number of critical changes which are imposed to allow survival, mainly without food. In the L2 larval stage, prior to dauer entry, autophagy proteins accumulate, and subsequently autophagosomal bodies are formed [80]. Indeed, the initial step of autophagy, mediated by BEC-1 (Atg6, Homolog of Beclin-1) is crucial to correct dauer formation. In the absence of bec-1, abnormal dauer formation results, often resulting in the premature death of the larvae [81]. Whilst BEC-1 appears to be the critical member in C. elegans dauer formation, deletions of autophagy proteins involved in complex formation (Atg1, 7 or 8) also resulted in abnormal dauers, but not to the extent seen when bec-1 was absent [81]. Autophagy, in C. elegans, like apoptosis maintains a basal level of activity acting as a cellular quality control mechanism; this prevents the accumulation of protein aggregates and damaged organelles as well as mediating developmental transitions [82]. This role of cellular protection is in contrast to the levels of expression during energetic stress, which turns autophagy from cellular protector to cellular recycler. In the case of the dauer stage, ATG genes are not crucial to entry, but their absence causes abnormalities that may prevent dauer transition, and even survival of the dauer stage [82].
Figure 14
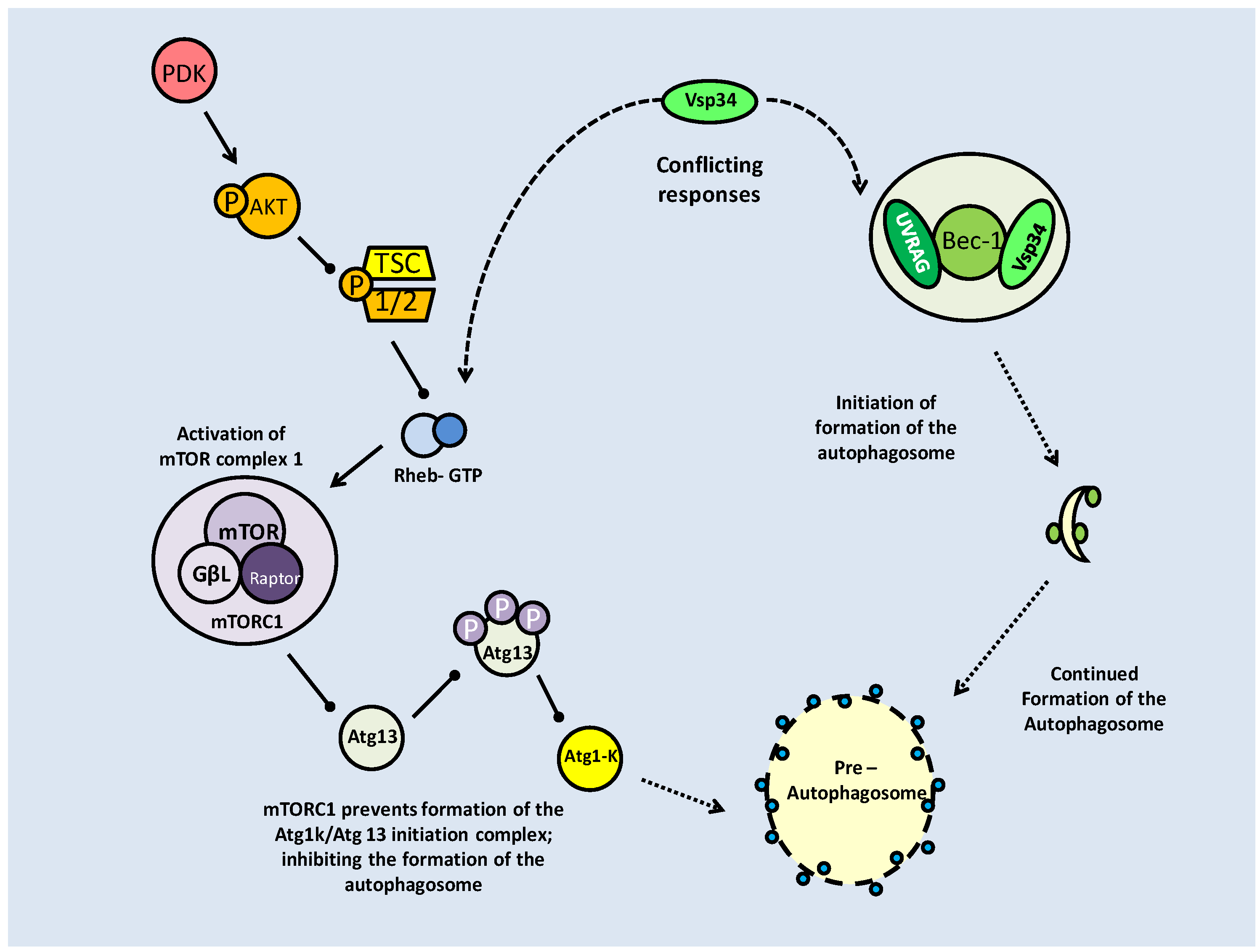
Regulation of autophagy by mTOR and Beclin-1. Both compete for Vsp34, with Beclin-1 requiring it for its complex formation, and mTOR requiring it for its Rheb-GTP based activation. While Beclin-1 promotes autophagy, mTOR blocks autophagosome formation via Atg-1 kinase inhibition, by phosphorylating Atg13.
To a degree, regulation of autophagy in C. elegans and mammalian systems mirrors each other, as is also seen for their mechanisms of apoptosis regulation. The link between TOR and autophagy appears to be intact, if activation patterns at the entry of dauer are compared. The link between autophagy initiation and mTOR suppression at the L2 stage, suggests that like in the mammal system, TOR suppression allows autophagy. Experimental evidence shows mutations in C. elegans TOR cause significant lifespan increase, lipid accumulation as well as altering nutrient absorption to create a caloric restriction profile. Caloric restriction, and the resulting slow down of metabolic processes, has been strongly linked to lifespan extension, and is an initiator of autophagy [70].
The interaction between Beclin-1 and pro-survival Bcl-2, used to inhibit autophagy, is conserved in the nematode model, with BEC-1/CED-9 protein interactions disrupting the formation of the Beclin-1 initiation complex (Fig. 12). With Bec-1 bound, it is not able to bind the C. elegans Vps34 homolog [82]. Another mechanism also seems to occur where EGL depletion, along with the induction of starvation, indirectly induces autophagy [82]. Speculatively, this could indicate a mammalian-like Bax-Bcl-2-Beclin-1 compromise in C. elegans where under starvation conditions, CED-9 may bind pro-apoptotic EGL-1, leaving BEC-1 unbound. Interestingly in C. elegans, the key autophagy complex protein, ATG5, may be proteolytically activated to translocate to the mitochondrial membrane, triggering cytochrome C release and resulting caspase activity [82], indicating once again, the high degree of cross-activation between autophagy and apoptosis signaling.
Tumor Suppressor p53: Regulation of Apoptosis and Lifespan of C. elegans
The tumor suppressor p53 is a stress activated transcription factor that regulates a number of downstream targets involved with cell cycle control and apoptosis. Its activity, which requires it to be uncomplexed from the mouse double minute 2 (MDM2) protein, is triggered primarily by DNA damage signaling [83]. The MDM2-p53 complex is broken by a number of post-translational modifications to both proteins that stabilize p53. Alternatively, if p53 remains bound to MDM2, it is a target for ubiquitin ligase and subjected to proteasome-mediated degradation. The primary means of isolating p53 from its complex with MDM2 appears to be phosphorylation, but it has over 24 sites for posttranslational modification by mechanisms including phosphorylation, acetylation, sumoylation, methylation and ubiquitination [83]. To this end, both the p53 target protein and MDM2 may be post-translationally modified, in order to break the complex (Fig. 15). The 'mutated in ataxia-telangiectasia' (ATM) kinase (possibly regulated in part by Akt), responds to double-stranded DNA breaks and phosphorylates p53 on multiple serine residues, but p53 may also be phosphorylated by major stress response kinases such as p38 MAPK and JNK, as well as cell cycle checkpoint (CHK) kinases. A further stabilization mechanism of p53 occurs with the phosphorylation of its ubiquitination sites, releasing it from MDM2 and further, preventing its degradation. This can be seen with DNA damage induced JNK activity (Fig. 15) [83]. The p53 C-terminus is the key regulatory region for its activity, with post-translational modification in this area helping to increase the DNA binding transcriptional activity of p53 [83].
Figure 15
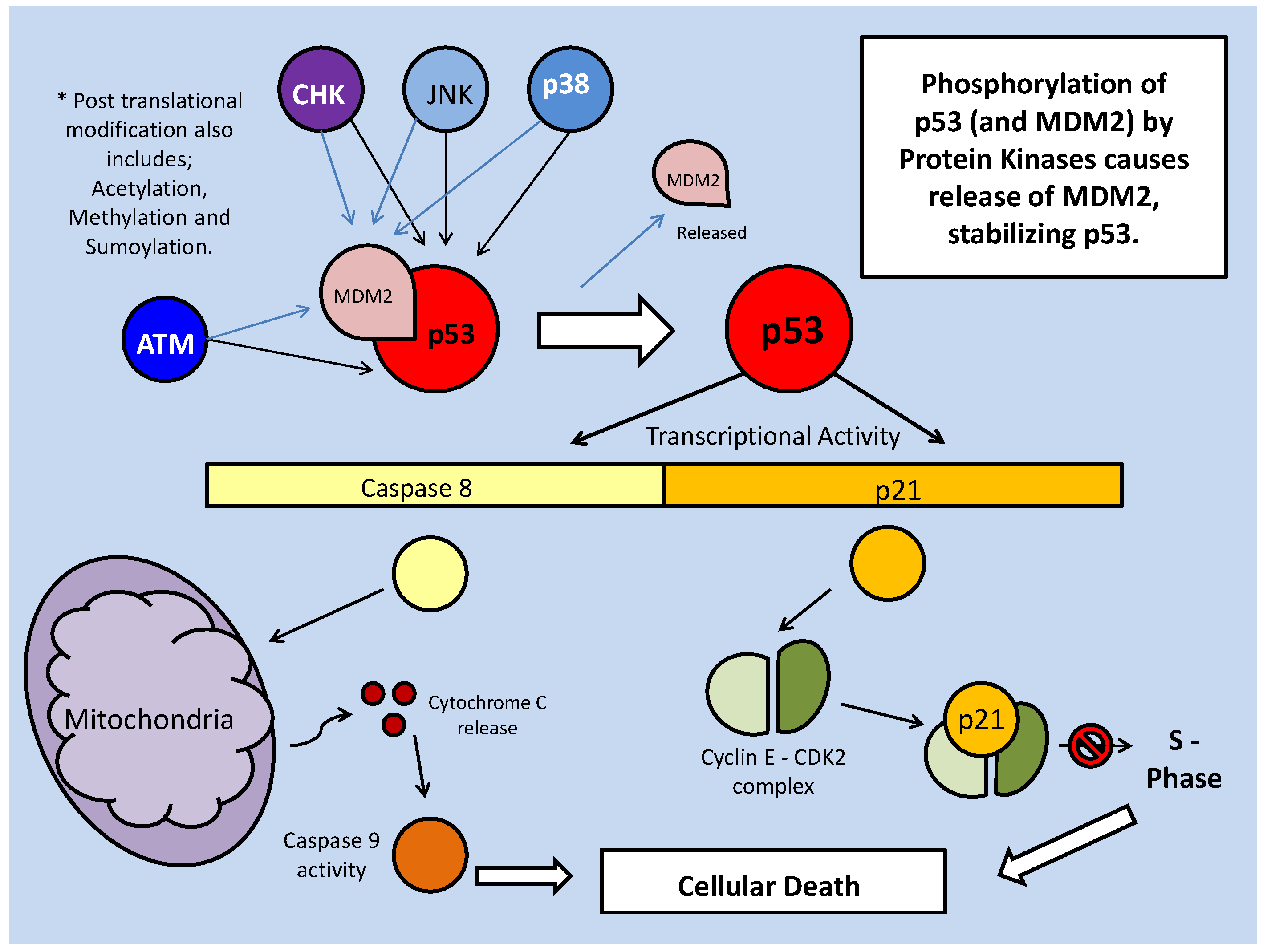
Activation of the p53 transcription factor begins with stress signaling (primarily DNA Damage). This triggers post-translational modification of the (inactive) p53-MDM2 complex, releasing and activating p53. P53 triggers death by halting the cell cycle, through p21, and triggering the caspase cascade, through Caspase 8.
Co-factor binding is also known to increase p53 transcriptional activity. Phosphorylation of critical sites on the transcription factor can lead to the recruitment of cofactors such as p300, CBP and P/CAF, which in turn promote acetylation of the p53 C-terminus. This is a protective mechanism which prevents p53 ubiquitination, stabilizing the protein [83]. In cases of hypoxia, HIF1- and α p53 compete for the binding of p300 as a stabilizing co-factor, despite having opposite roles under hypoxic conditions. Whereas hypoxia causes the up-regulation of both of these transcription factors, they have roles in cell survival versus apoptosis signaling, respectively [84]. It has also been suggested that along with the competition to bind p300, the release of MDM2 from p53, can then cause the degradation (via ubiquitination) of HIF-1α [84].
The role of p53 in programmed cellular death
The role of p53 signaling has frequently been associated with programmed cellular death, via control of the caspase cascade (through Caspase 8), but also includes regulation of the cell cycle. This role, however, can be used for different results, with long term attenuation of the cell cycle used to signal apoptosis, and short term attenuation to correct mistakes occurring during the cell cycle and provide recovery from stress conditions. The cell cycle regulatory activity of p53 comes at the critical G1/S phase, prior to G2 and mitosis. When stable (most often as a result of phosphorylation via ATM kinase), p53 acts to up-regulate p21, which then binds to the cyclinE-CDK2 complex (Fig. 15). This binding prevents downstream phosphorylation, and entry into the S phase, halting the cell cycle [83].
Figure 16
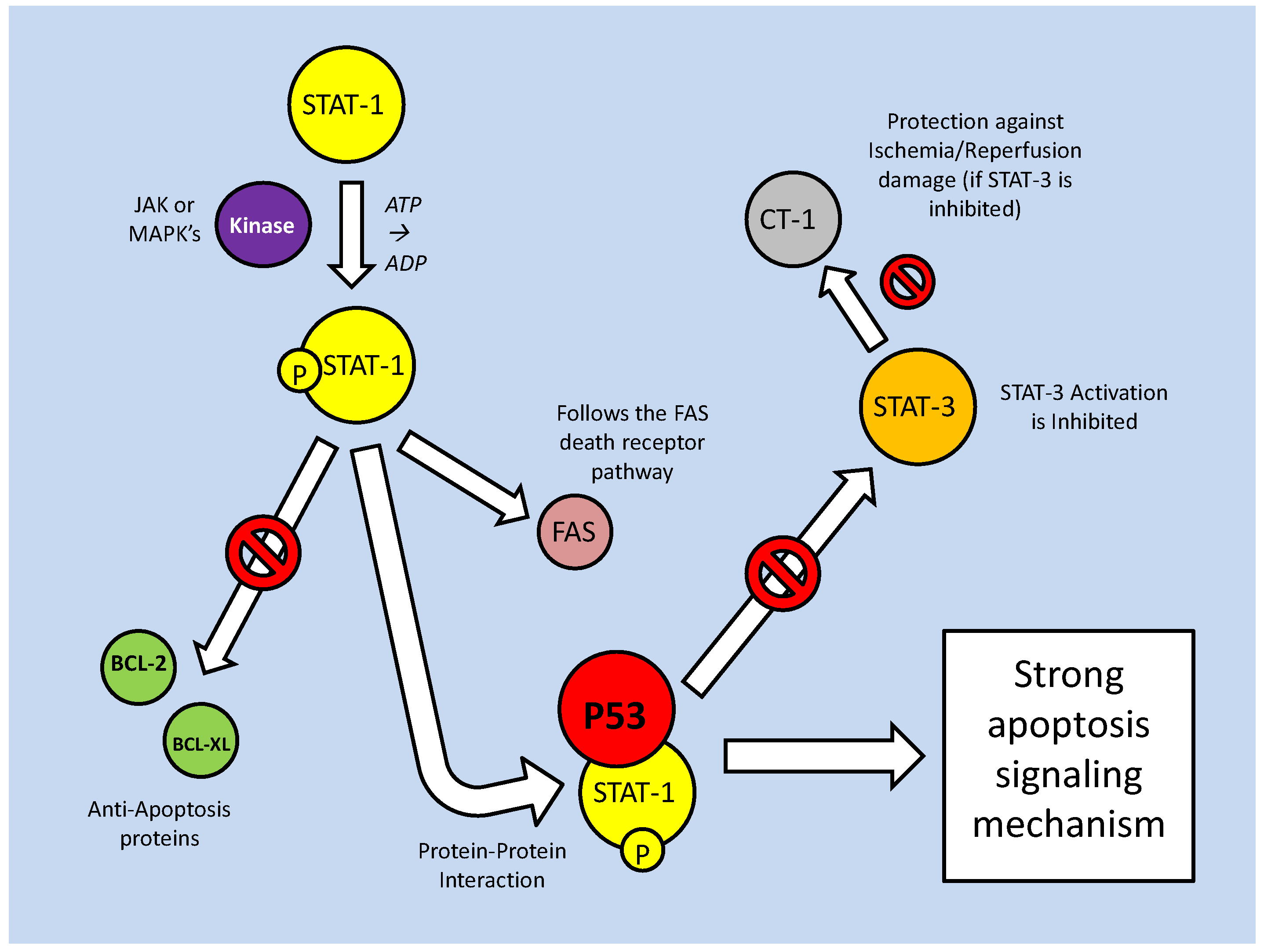
The protein-protein interaction between P53 and STAT1-p induces apoptosis and suppresses STAT3 protective mechanisms. STAT1-p inhibits transcription of anti-apoptosis BCL genes and enhances apoptosis signaling by p53 through binding.
The STAT family of transcription factors has roles in both cell death and survival but in an interaction with p53, they show a forcible move to apoptosis (Fig. 16). STAT1, when activated by phosphorylation, inhibits transcription of the anti-apoptosis BCL-2 family genes whereas p53 itself, when signaled into action by DNA damage, can activate downstream cysteine proteases, specifically, Caspase 8. This enters the mitochondria, triggering cytochrome C release, which triggers an apoptosis signaling cascade through Caspase 9 [62]. STAT-1 acts in conjunction with p53, when phosphorylated by p38 MAPK or Janus kinase (JAK), in a novel protein-protein interaction and the combined activity of p-STAT-1 and p53 is more efficient in apoptosis signaling than their independent activities [62]. Conversely, STAT-3, which acts to protect against apoptosis under ischemia/reperfusion conditions, is inhibited (upstream) by p53 (Fig. 16) [62].
The association of p53 with apoptosis has long been known, but it has recently become apparent that p53 is also involved in autophagy control. In mammals, the p53 family consists of three homologous transcription factors; p53, p63 and p73 [85]. Whereas p53 is known to promote apoptosis (via caspase cascades), p73 has a novel interaction with autophagy genes (Atg5 and 7). The mTOR protein kinase, which is a critical kinase in many stress response models, is a positive regulator of p53 (via phosphorylation activity), but a negative regulator of p73. Acting upstream of p73, mTOR inactivates the transcription factor, which in turn is unable to increase the transcription of autophagy proteins. As a result, loss of p73 function caused necrosis in some tissues [85]. Possibly in a negative feedback response, p53, via the recruitment of AMPK (likely acting on the TSC1/2 complex), has been shown to inactivate mTOR (Fig. 17).
Figure 17
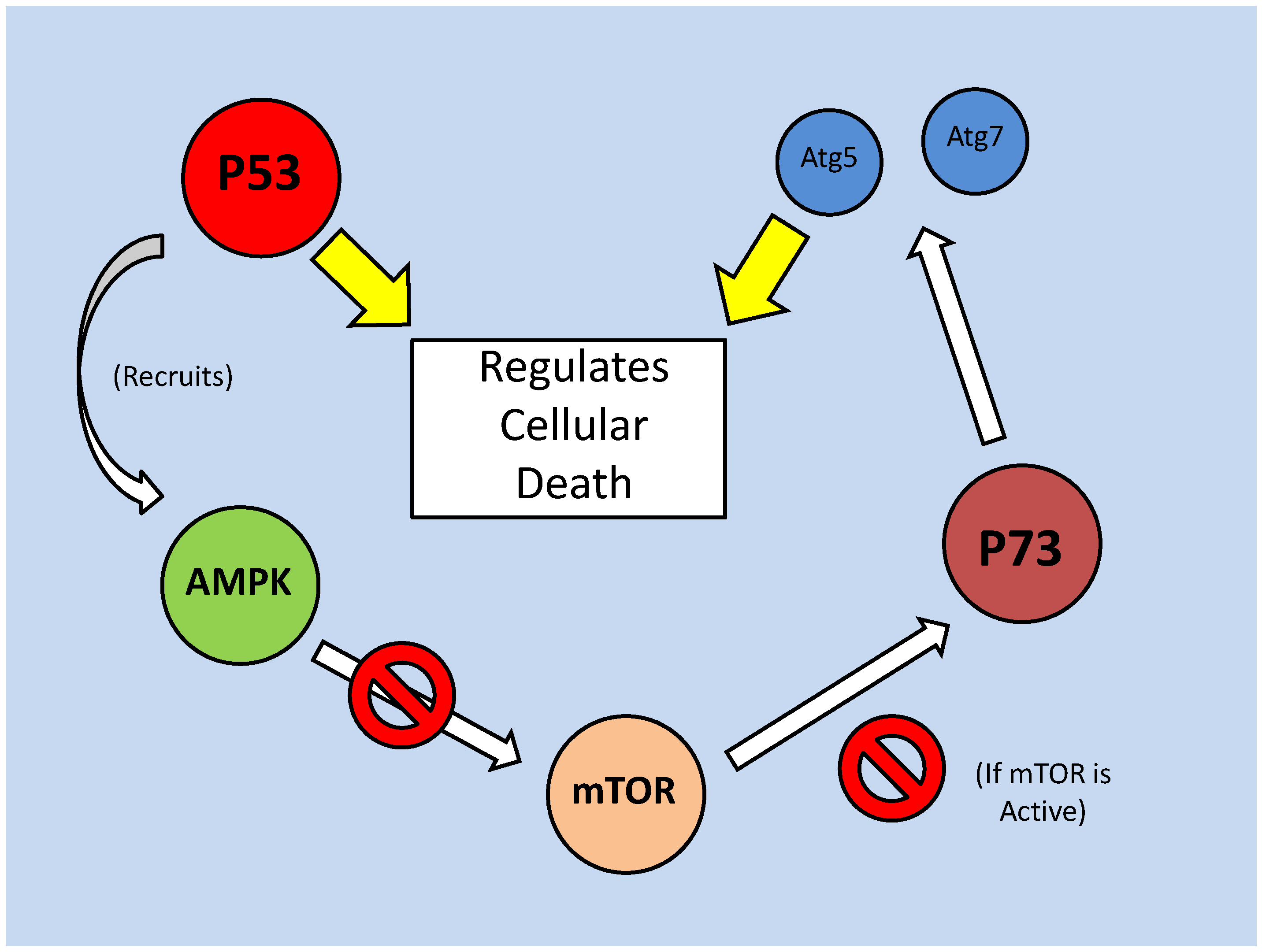
P53, via recruitment of AMPK, regulates mTOR which in turn, is unable to inhibit the p53 family member, p73. P73 is known to up-regulate transcription of autophagy genes.
The DNA damage responsive Cep-1 in C. elegans
There appears to be only a single member of the p53 family represented in C. elegans, encoded by the cep-1 gene. However, with the importance of regulating autophagy during the dauer phase of life, CEP-1 is an important target for study. As in mammals, CEP-1 is a tumor suppressor signaled by DNA damage and participates in apoptosis signaling. Specifically, CEP-1 is triggered by ionizing radiation (IR)-induced DNA damage, and typically cep-1 deletions have only been seen to be lethal in the embryonic stage [86]. This likely represents the necessity of CEP-1 mediated apoptosis that is predominantly for development and houekeeping. The relationship between CEP-1 activity and cellular survival appears to follow the mammalian system, with a lack of CEP-1 making the nematode unable to assess DNA damage which causes substantial cellular damage. On the other hand, a forced over-expression of cep-1 can increase caspase activity, leading to apoptosis [87]. Ectopic apoptosis in the somatic tissue of C. elegans is not dealt with well, and often ends in death of the organism. CEP-1, which with excess stress signaling can trigger egg laying defective (EGL-1) protein mediated apoptosis, is also able to interact directly with the CED (C. elegans caspase orthologs) family of proteins [87]. Active CEP-1 up-regulates the transcription of a number of pro-apoptotic genes, particularly ced-13 and egl-1. CED-13 and EGL-1 act to bind anti-apoptotic (BCL2 homolog) CED-9 protein, preventing its activity (Fig. 18) [88]. Interestingly, though, another interesting point is the ability of CEP-1 to induce apoptosis in the absence of typical apoptotic machinery (CED3/4), suggesting that while CEP-1 may act upstream of the CEDs, it is not essential [86]. While in the mammalian system, a clear regulating protein (MDM2) has been established as controlling P53, a similar model in C. elegans is only suspected at this point. The Skp1/cullin/F-box (SCF) E3 ubiquitin ligase (SCFFSN-1) has been identified as a potential CEP-1 regulator. In germline cells it has been discovered that the SCFFSN-1 inhibits CEP-1, but through an unknown intermediate protein [89]. Also like in the mammalian system, CEP-1 activity is controlled by post-translational modification. The C. elegans sirtuin homolog SIR-2.1 is a deacetylase, and as such is capable of destabilizing CEP-1 by deacetylating it (presumably at the C-terminal), significantly affecting lifespan (Fig. 18). In both mammalian and yeast systems, sirtuins are seen to have anti-apoptosis roles, and over-expression of sir-2.1 in C. elegans is seen to significantly increase lifespan, although it also requires DAF-16 activity [90]. Interestingly, in germline experiments, SIR-2.1 activity was seen to run parallel (or possibly upstream) to CEP-1, in inducing apoptosis, after DNA damage [90]. This runs counter to the deacetylase activity of sirtuins, but is perhaps a selective event; only being seen in early apoptotic signaling [90]. Generally, though, reductions in cep-1 expression have been shown to correlate with increases C. elegans lifespan. Furthering this, the DAF-16 protein, which is active and essential for entry into the dauer period, is suspected to repress Cep-1 transcription [87]. This system is predicted on the foundation that lifespan extensions (approximately 10%) seen with cep-1 RNAi requires daf-16 activity, and that CEP-1activity does not inhibit DAF-16 nuclear localization [87]. Hence, cep-1 could potentially be a downstream target of DAF-16. Along with this, there is also thought to be a link between CEP-1 depletion and autophagy, suggesting that the resultant lifespan increase with CEP-1 absence is mediated, in some fashion, by autophagy [91].
Figure 18
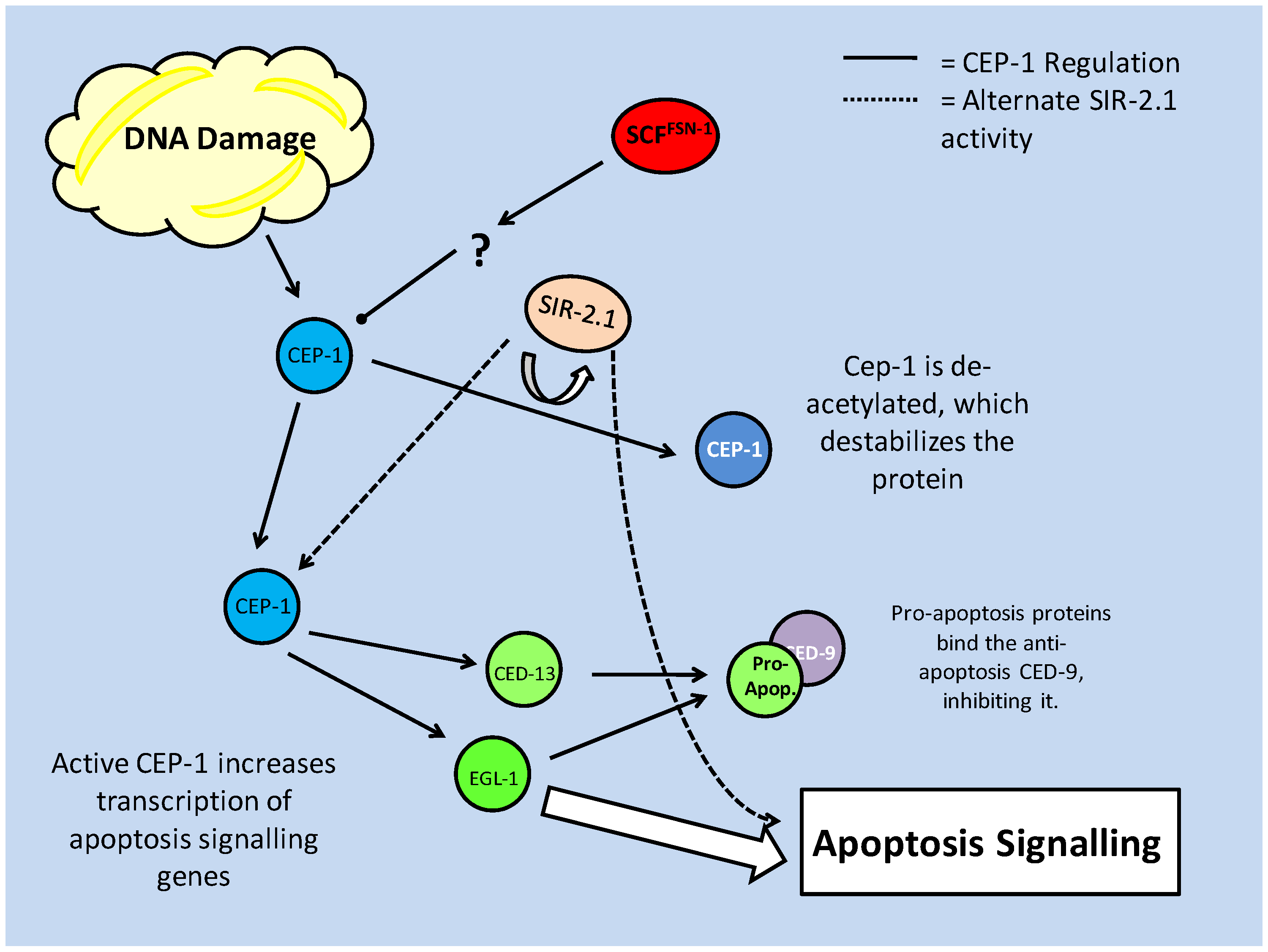
The C. elegans p53 ortholog, CEP-1, is stimulated by DNA damage, but can be inhibited by both SIR- 2.1 (deacetylase) and SCFFSN-1 (through an unknown). Its transcription factor activity is controlled by post-translational modification. While the sirtuin like protein, SIR-2.1, can deacetylate and destabilize Cep-1, under certain conditions, it is also seen to trigger germline apoptosis; either upstream of or parallel to CEP-1.
The Cell Cycle: Regulation of cellular proliferation and quiescence
The cell cycle is a complex, highly regulated process through which critical events such as aging and cellular proliferation (cancer) are affected. The standard cell cycle is composed of four stages which accomplish goals including DNA replication, segregation of chromosomes and cytokinesis. As illustrated in Fig. 19, the cell cycle is typically described as beginning with a gap phase (G1) which then proceeds into the DNA synthesis phase (S) and is followed by a second gap phase (G2). Subsequently, the cell enters the final mitosis (M) phase. The cell cycle can be continuous or cells may exit from continuous cycling into a phase of quiescence (Go), a temporary exit that leaves the cycle at the G1 stage (Fig. 19).
In mammals, cell division and the rate cell division rate are largely determined by genetic profile, which controls differentiation and transformation state. On a protein level, progression of the cell cycle is mediated by complexes of cyclin proteins and 'cyclin dependent kinases' (CDKs) that form maturation promoting factors (MPFs). These MPFs, via kinase activity, go on to phosphorylate microtubule forming proteins and chromatin remodeling complexes, amongst others, in order to progress the cell cycle. The relationship between specific CDKs and cyclins is critical to activate the enzymatic properties of the subsequent complex.
Figure 19
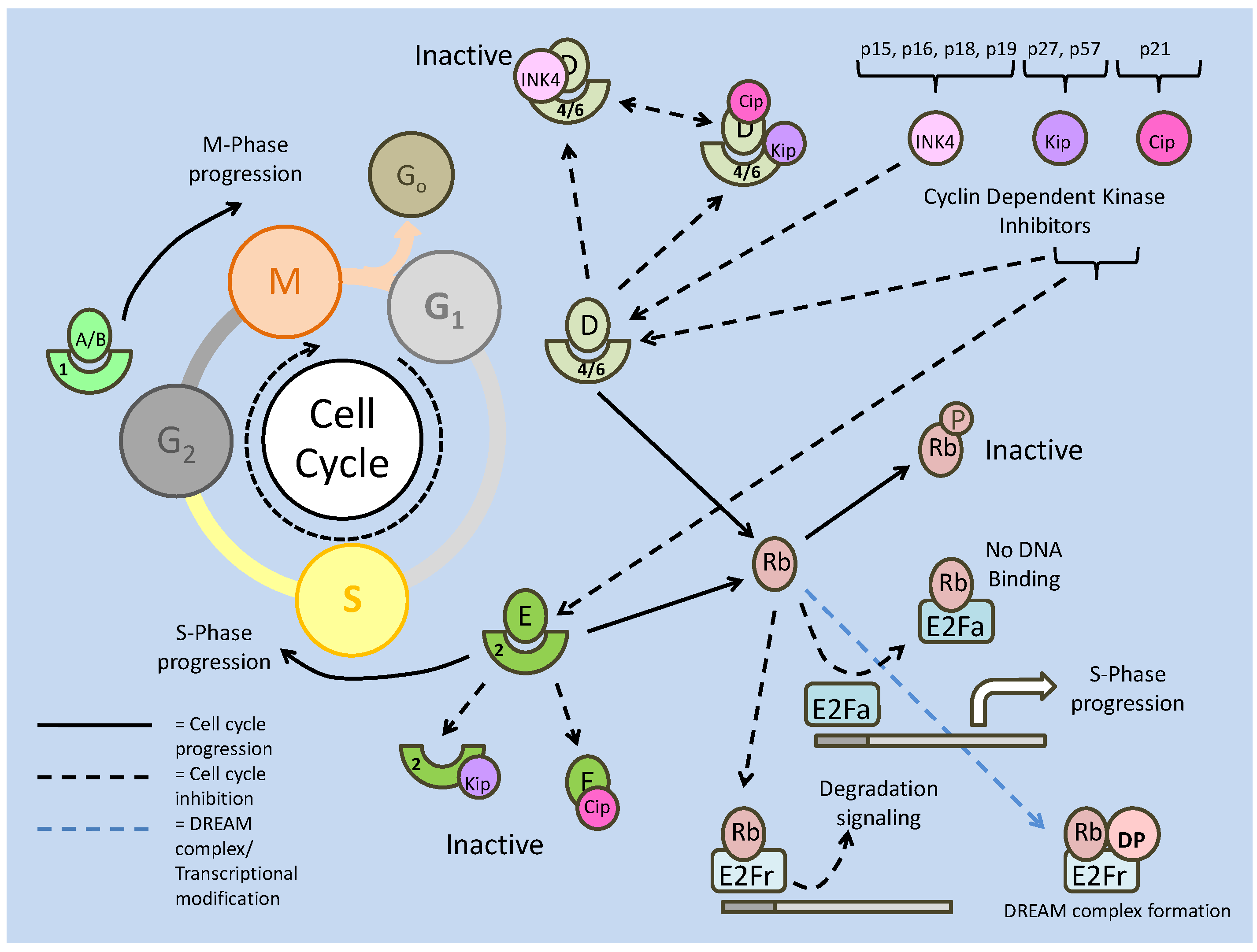
Basic regulation of the mammalian cell cycle. The cyclin/CDK complexes which act to push the cell cycle across the G1/S barrier act primarily on the Rb protein. The complexes themselves are subject to inhibition from CKIs. Rb phosphorylation prevents its binding (and inhibition) of E2Fa (activating) transcription factors. It similarly inhibits its activities with E2Fr (repressing) transcription factors, which would compete with the E2Fa factors for promoter binding sites on critical S phase genes.
Typically, cyclins for complexes with specific CDKs: cyclin A/B complex with CDK1, cyclin D complexes with CDK4/6, and cyclin E complexes with CDK2. These cyclins make the kinases enzymatically active, and determine their subcellular locations [92]. Cyclins A/B are present and necessary from the time a cell enters S phase until cell cycle completion, whereas cyclin D and E are primarily involved in regulating the G1 phase [92]. The presence and importance of both cyclin D and E in the mammalian G1 is indicated clearly when these are over-expressed; this results in shortened (or accelerated) G1 phases [92]. Their requirement in G1 also makes them key targets for cell cycle attenuation. There are numerous ways in which cyclin activity may be inhibited, but two are most common: post-translational modification via reversible phosphorylation, and inhibition of binding. Phosphorylation of cyclin D, for example, may be carried out by glycogen synthase kinase (GSK-3) and causes cyclin D export from the nucleus (as well as targeting the protein for ubiquitin-mediated degradation) which prevents its associations with CDKs [93]. Both cyclin D and E are also regulated by inhibitory proteins (see below) which bind them (and their respective kinases), preventing complex formation. The effects of this type of inhibition, however, can be reversed by the production of additional cyclins, and also in some cases, through compensatory activities of cyclin A.
Blocking the activity of the cyclin/CDK complexes are the so-called CDK inhibitors (CKIs). These proteins may be divided into two classes, the 'inhibitor of CDK4' (INK4) and the cyclin inhibiting protein/kinase inhibiting protein (Cip/Kip) families [94]. The INK4 group contains four members; p15, p16, p18 and p19. The INK4 inhibitors function to bind CDK 4/6 and prevent its association with cyclin D, thereby negating its downstream, kinase effects [94]. The primary members of the Cip/Kip group are p21, p27 and p57 and these are disrupters of cyclin D and E activity as well as CDKs 2, 4 and 6. Both p27 and 57 are Kips, whereas p21 is a Cip. These inhibitors function by binding their targets and preventing their downstream enzymatic activities.
Whereas both INK4s and Cip/Kips are inhibitors of the cyclin D/CDK4 complex, they may also act competitively. The ratio between INK4 bound complexes and Cip/Kip bound complexes varies with the amount of INK4 protein present [94]. Displacement of Cip/Kips by INK4s releases the Cip/Kips to bind cyclinE/CDK2 components, and attenuate the cell cycle at the G1 phase. It is this dynamic between the two CKI families that also provides the Cips/Kips with an interesting dual role with their targets (Fig. 19). While these proteins can bind cyclin or kinase (depending on the family member), to inhibit the binding of the other and prevent cyclin/CDK complexing, there are many cases where these CKIs have been found bound to active cyclin D/CDK4 complexes. In this sense, the cyclin D/CDK4 complex may have a role in sequestering CKI, preventing p27 and p21 from inhibiting the cyclin E/CDK2 complex [94]. Another more interesting role for the Cip/Kip family is in aiding the formation of cyclin D/CDK4 complexes. The p21 and p27 proteins are capable of stabilizing cyclin D and CDK4, respectively, in order that their complex assembly is promoted [94]. While this may appear to further the cell cycle, it is likely a role in conjunction with their ability to 'deliver' complexes for INK4 inactivation. Due to the important role of CKIs in regulating the cell cycle, and their own internal dynamics, they are often the target of cell cycle progression proteins, and may be phosphorylated and tagged for ubiquitin mediated degradation. However, their positive regulation may also be used in order to prevent uncontrolled proliferation, illustrating the fine level of balance and regulation in the cell cycle.
The cyclin/CDK, Rb/E2F dynamic in control of the cell cycle
Within the mammalian cell cycle, the major stages of regulation occur at the gap phase barriers (G1/S and G2/M), and while many of the same elements are present at these cross-points, their function and necessity may differ. In G1 phase, the primary event needed to promote progression into the S phase is the phosphorylation of the retinoblastoma protein family (Rb; p107 and p130) (Fig. 19). When active, these proteins (also known as 'pocket proteins') bind members of the E2F transcription factor family [95]. The E2F family members have binding sites for Rb proteins and 'differentiation regulated transcription factor proteins' (DP), which affect their activity depending on whether they are transcriptional activating or repressing members of the E2F family. There are cases where all three (Rb, DP and E2F (amongst others)) form a complex, but this will be covered in greater detail later. E2F family members 1-3 (E2F1-3) are transcription activators that target genes in order to progress the cell cycle into S phase. They are negatively regulated by Rb binding. E2F4 and 5 members, however, are transcriptional repressors that competitively bind the same promoter regions of the S phase progression target genes [95]. Binding of Rb to the repressor E2Fs increases their ability to bind the same promoters, and subsequently, signal transcriptional repressing (chromatin modeling enzymes) machinery [95]. If Rb activity succeeds, then cells may not progress into the S phase but can instead be held in a quiescence phase (Go).
Hence, the regulation of the Rb/E2F dynamic is seen throughout the cell cycle, but is of notable importance at the G1 phase. With phosphorylation of the Rb protein causing unbinding from the transcription factor, it is the cyclin/CDK complexes that determine its activity. While both cyclin E/CDK2 and cyclin D/CDK4 complexes are able to phosphorylate Rb, it is suspected that cyclin D/CDK4 is primarily responsible. In double-knockouts of CDK 2 and 4, using mouse embryo fibroblasts (MEFs), a steady loss in Rb phosphorylation was duly seen, resulting in decreased E2F target transcription, including that of CDK1 (which can be an upstream inhibitor of Rb) [96]. A positive feedback loop in this circuit leads to the induction of cyclin E transcription by the E2Fs, which in turn bind and activate CDK2 for Rb phosphorylation.
While their seemingly critical role in the regulation of the cell cycle would suggest against it, the presence of all cyclins and CDKs are not necessary for cellular replication. For example, cyclins D and E do not seem to be required in embryonic cells in mammalian systems, neither was the presence of the cyclin D/CDK4 complex in embryonic stem cells [96]. In numerous mouse knockout models it was discovered that CDK4/6 was not necessary to complete the cell cycle [96]. The absence of CDK4 caused a 4 hour delay in S phase entry, but this was suspected to be a result of impaired cyclin E/CDK2, via uninhibited p27 activity. It was subsequently shown through CDK4/p27 double mutants that CDK2 activity and the S phase entry were restored [96]. Similar to the condition of CDK4 absence, were the results of CDK2 mutations, which also caused a 4 hour delay in S phase entry but did not prevent cell cycle progression. It is suspected that CDK1 takes the role of 'cyclin E binding partner', allowing the cell to pass the G1/S barrier [96]. Combined, this indicates that the specifics of cyclin/CDK presence at given stages may show functional redundancy, but also maybe indicative of recovery or compensatory mechanisms when these proteins are targeted for inhibition.
Post-translational control of the cell cycle
Control of the cell cycle is heavily regulated by post-translational modifications (Fig. 20). Cyclins are phosphorylated and ubiquitinated for degradation, Rb is phosphorylated by cyclin/CDK complexes and dissociated from E2Fs. The CDKs are also subject to regulation via phosphorylation. The highly conserved Wee family of kinases (originally discovered in Schizosaccharomyces pombe; including Wee1, 2 and Myt1) is involved in phosphorylating threonine and tyrosine residues of the 'cell division cycle' 2 (Cdc2) and CDKs [96]. Like the CDKs, Cdc2 forms complexes with cyclins (A and B) to initiate mitosis, and is phosphorylated (at Tyr15) and inactivated by Wee1. Similarly, CDK2 may be phosphorylated on both Tyr15 and Thr14, likely in order to regulate the G1/S transition [97]. Counteracting these inhibitory phosphorylations is dephosphorylation mediated by the cdc25 phosphatase. These cyclin/CDK complex activators are able to dephosphorylate both Tyr15 and Thr14 residues, and are commonly seen to be over-expressed in human cancers, making them viable drug targets [98]. The cdc25 phosphatases not only act to repress inhibitory phosphorylation, but are targets of it themselves. DNA checkpoints in the cell cycle preserve accurate replication and report DNA damage. The key kinases in these checkpoints, including ATM kinase, activate downstream checkpoint kinases (Chks), which often act to stop the cell cycle from progressing, allowing repairs to be made (Fig. 20) [99]. Specifically, Chk1 can inhibit cdc25 via phosphorylation of a c-terminal residue (at a homologous site to a human 14-3-3 binding site). This phosphorylation inactivates cdc25, and while it is not required for inhibition, may recruit 14-3-3 proteins to sequester the phosphatases (see previous discussion of 14-3-3 proteins) [99]. This system of phosphorylation control occurs across organisms, indicating a high level of conservation and importance in cell cycle regulation.
While there are numerous cell cycle inhibiting proteins, there are also targets which seek to enhance cell cycle progression. The oncogenic activity of c-Myc which seeks to create cellular proliferation through increased cell cycle activity, acts at the G1/S barrier. Like over-expression of the cyclins D and E, over-expression of c-Myc results in a shortened G1 profile. This is suspected to be due to gene targets that it up-regulates in order to progress to the DNA replication phase; targets that may include cyclin D, p21 and E2F2 [100]. In mammalian (rat) cell lines, it was subsequently shown that the absence of c-Myc significantly reduces cyclin D/CDK4 activity, which in turn delays the formation of CDK2 complexes [100]. The absence of c-Myc was also notably correlated with an increase in the p27 kinase inhibiting protein [100]. Thus, as in other systems controlling proliferation, c-Myc is seen to exert its influence (Fig. 20). Interestingly, like many other components regulating the cell cycle, c-Myc has a role in apoptosis stimulation and is subsequently, negatively regulated by E2F1. Protein kinase B (Akt), is signaled by E2F1 over expression, and inhibits the signaling of target apoptosis proteins by c-Myc [101]. E2F1 has its own apoptotic functions, which are also interestingly regulated by Akt, via up-regulation of the DNA binding topoisomerase II β binding protein (TopBP1) [101].
The variation and regulation of the C. elegans cell cycle
The C. elegans model has been invaluable in the study of the cell cycle. The ease of culturing, ability to isolate the different replication cycle phases, and the application of genetic manipulation has helped discover a number of firsts in cell cycle regulation. Cell cycle regulation in C. elegans is defined by the type of cell; embryonic, somatic, endoreduplication, and meiotic. Each of these cell types, while largely undergoing the same processes, can differ in cell cycle profile. Embryonic lines, for example, lack the gap phases, and as such do not require the regulatory controls on these stages [102]. Similarly, the endoreduplication cycles are without mitosis, following DNA replication [102]. The somatic cells, however, undergo the full cell cycle, which means that controls in these cells are most similar to mammalian controls.
Cell cycle control in nematodes is again primarily influenced by the activities and regulation of cyclin/CDK complexes (Fig. 21). In C. elegans, there are both cyclin E and D homologs (CYE-1 and CYD-1, respectively) as well as a CDK4 homolog (CDK-4). Currently, no definite C. elegans CDK2 homolog is known, but a likely candidate gene (k03e5.3) with ~40% amino acid homology to the human protein, has been identified [104]. Also similar to mammalian cell cycle regulation is the presence of CKIs such as homologs of the p27 kip (CKI-1), Myt1/Wee-1 kip (WEE-1.3) and critical regulating phosphatases Cdc25 and 14 (CDC-25.1 and CDC-14) (Fig. 21) [102, 104]. Once again, the G1/S phase and G2/M barriers are critical targets of regulation via the cyclin/CDK complexes. Whilst there are members of the C. elegans CDK family that are involved in multiple phases of the cell cycle, like CDK-1, there are also stage specific interactions like the cyclin/CDK complexes, which again mirrors mammalian systems.
Figure 20
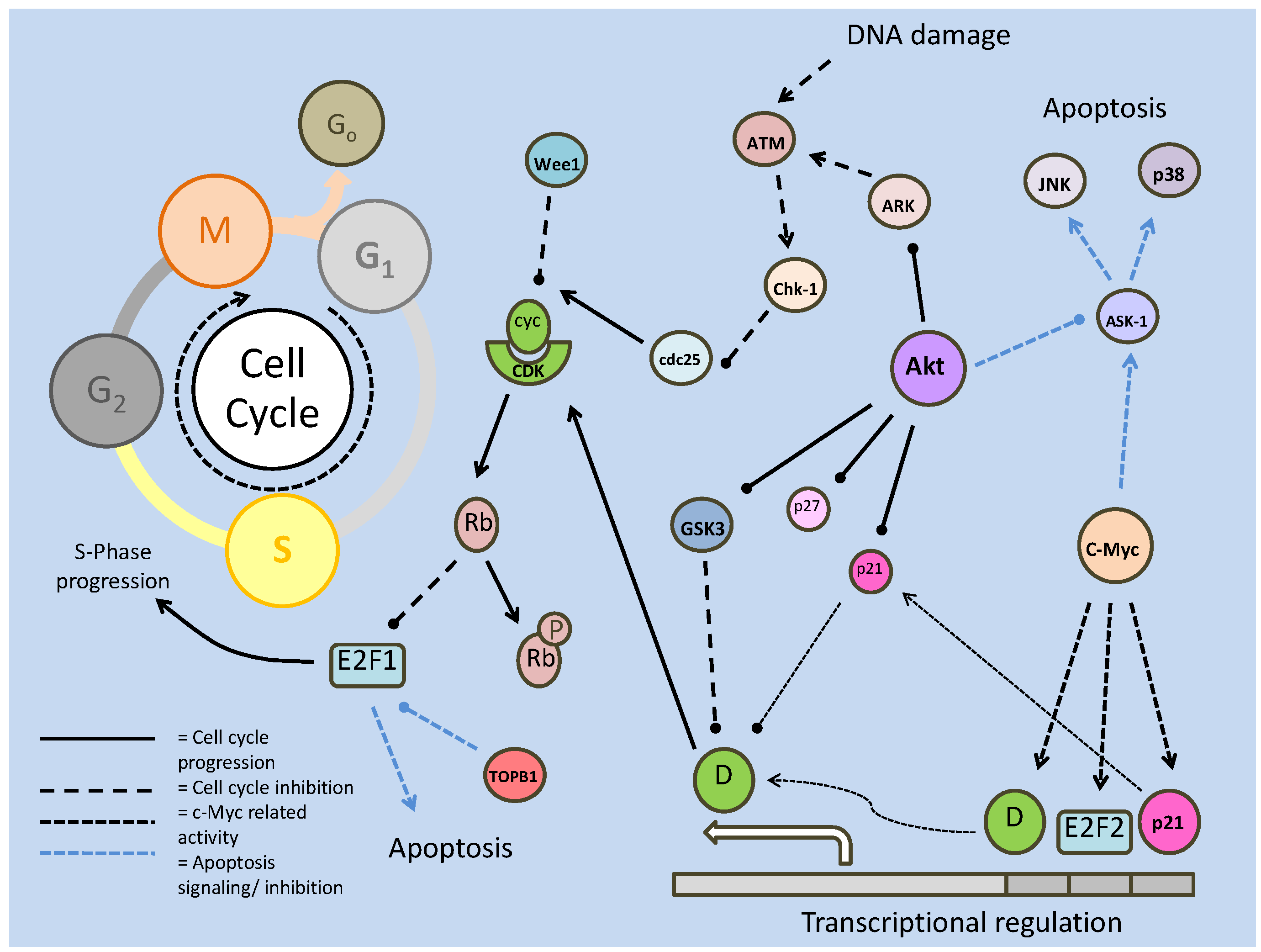
Reversible phosphorylation regulation of the cell cycle. Cyclin/CDK complexes may be inactivated through phosphorylation, mediated by the Wee kinase family. Phosphorylation may be reversed by the Cdc-25 phosphatase. This phosphatase may also be inactivated through Chk-1 mediated phosphorylation. DNA checkpoint kinases are regulated by the ATM kinase, which in turn may receive signals from DNA damage and the insulin response pathway. The Akt kinase of the insulin signaling pathway, phosphorylates numerous cell cycle targets, including Cip/Kips and GSK-3 (which targets cyclin D).
Figure 21
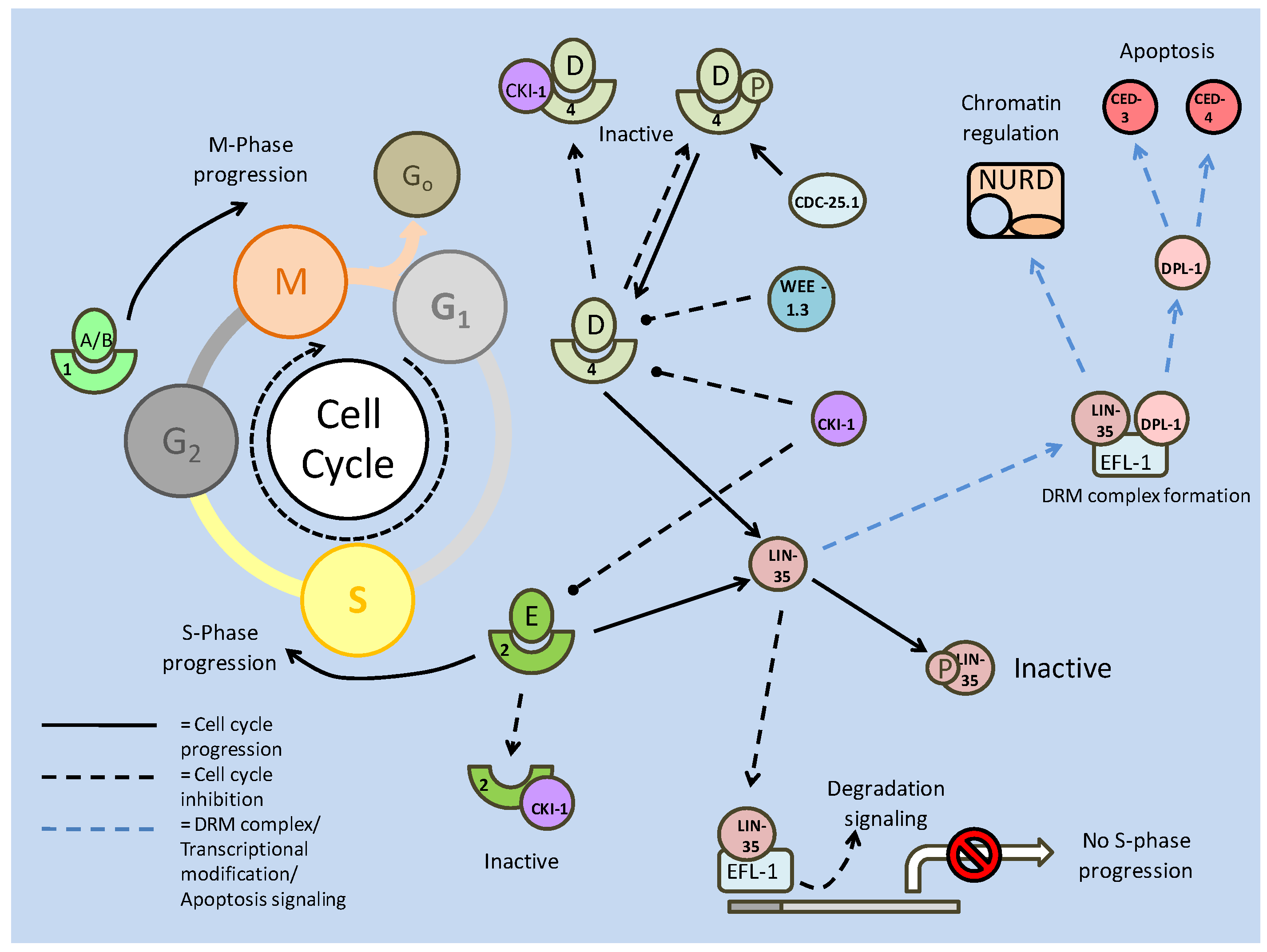
Regulation in the C. elegans cell cycle. Like the mammalian system, the primary role of the cyclin/CDK complexes is to phosphorylate the retinoblastoma protein (LIN-35), which negatively regulates S-phase progression genes. The cyclin/CDK complexes are susceptible to phosphorylation (which can be reversed by CDC-25.1) and inhibition by CKIs, which act to attenuate the cell cycle.
In the embryonic cycle, while there are no gap phases, interactions between cyclins and CDKs still regulate completion. The absence of cyclins A and B, which typically interact with CDK-1, was tested using RNAi. The resultant arrest in embryonic cell division indicated that these cyclin/CDK complexes are necessary to prevent cell cycle attenuation [102]. Because of its role in the G1 phase, the CYD-1/CDK-4 complex is not often seen in embryonic cell development [102], but post embryonic development, the necessity of cyclin D/CDK4 (CYD-1/CDK-4) complexes becomes apparent.
The G1 phase, critical in cell cycle regulation in many models, integrates a number of signals; including the cyclin/CDKs, their cyclin/kinase inhibitors, retinoblastoma proteins and E2F transcription factors, and microRNAs. In identifying C. elegans cell cycle regulators, vulval cells have contributed significantly. Part of this is due to the cell cycle attenuation, or in this case quiescence, that occurs throughout the L1 and 2 larval stages, and only resumes mid-way through the L3 stage, which makes them an intriguing model of regulation [103]. Of the vulval cell types, a number of critical cell cycle regulators were discovered in the 'multivulva' (Muv) phenotype [104]. The Muv phenotype requires the expression of multiple classes of 'synthetic' multivulva genes (synMuv), and amongst these (synMuv class B) were discovered a number of critical genes that associate together to regulate the cell cycle (Fig. 21). The retinoblastoma (pRB) family of tumor suppressors (mentioned above) is represented in the 'abnormal cell lineage' 35 (LIN-35) protein, acting as a C. elegans p107 homolog [102]. The E2F family of transcription factors are also represented through the E2F family like (EFL-1) protein, which closely resembles the transcription inhibiting E2F4. Another known factor to associate with pRb is the 'differentiation-regulated transcription factor 1 polypeptide' or DP-like protein (DPL-1) [95].
As in mammalian G1 regulation, the role of nematode CYD-1/CDK-4 appears to function primarily in regulating cell cycle attenuators, and not specifically in cell cycle development. By inhibiting the Cip/Kip CKI-1 and LIN-35, the CYD-1/CDK-4 complex allows the CYE-1/CDK-2 to function, and propel the cell cycle into the S phase [104]. Through the use of RNAi, it was revealed that while both lin-35 and efl-1 individually have disruptive effects on G1 progression, and when their deletion was put in conjunction with a cki-1 deletion, the resultant increase in cellular proliferation was far greater [104]. This earmarks the Cip/Kip homolog, CKI-1 as the likely controlling factor in G1/S phase progress. In regards to this, the promoter region of the cki-1 gene has numerous 5' upstream sequences, which would allow the gene to be regulated by multiple signals [104]. In vulval precursor cells (VPCs), the cki-1 promoter region was found to have over 20 consensus binding sites for ETS and forkhead box transcription factors [103].
Signaling and interactions between the cyclin/CDKs and CKIs are typically done through post-translational modification, using phosphorylation as a means of altering protein conformation, and as a result, activity. Hence dephosphorylation, through cell cycle phosphatases, CDC-25 and 14, is of equal importance. These two major cell cycle phosphatases work in opposition to each other, with CDC-25 maintaining active cyclin/CDKs, and CDC-14 acting upstream of CKIs to maintain their activity. In C. elegans, the cdc-25 phosphatase has four members (CDC-25.1-4), and dephosphorylates cyclin/CDK complexes that have been phosphorylated (and inactivated) by the Wee1/Myt kinase homolog, WEE-1.3 [102]. It is suspected that WEE-1.3 primarily targets CDK-1 (from mutational data), but the C. elegans CDK-2 (K03E5.3) contains a similar ATP binding site that may also be negatively regulated by phosphorylation [102]. The CDC-14 phosphatase on the other hand, is suspected to maintain CKI-1 stability by dephosphorylating it, stimulating cell cycle arrest. It is thought that CKIs, from mammalian to C. elegans, are targeted by ubiquitin for proteasomal degradation, after being phosphorylated [104].
The DREAM/DRM complex and its role in development
A further similarity to the mammalian system, but on the periphery of core cell cycle regulation, is the complexing of C. elegans Rb related proteins. Complex initiation, which likely begins with a heterodimerization between EFL-1 and DPL-1 proteins, later aggregates with LIN-35. This trimer, along with other myeloblastosis (MYB) proteins (which themselves are able to regulate cell proliferation/differentiation), forms what is known as the DREAM complex (C. elegans DRM) [95]. DREAM complexes are suspected to manipulate chromatin in order to regulate transcription of specific genes. Whereas the mammalian model of the DREAM complex is thought to directly target promoter regions of developmental genes, the C. elegans DRM complex is thought to possibly act in conjunction with the nucleasome remodeling and deacetylase (NuRD) complexes; in order to regulate chromatin structure of epidermal growth factors (EGFs) (Fig. 21) [95]. Consequently, this would regulate Ras/EGF receptor/MAPK pathways. Interestingly, while the LIN-35/EFLl-1/DPL-1 complex is likely to repress transcription, the DPL-1 protein is thought to also complex an activating transcription factor member of the E2F family in order to promote the S phase [104]. Like many cell cycle regulators, the DPL-1 protein has the capacity to both progress the cell cycle and initiate apoptosis. For example, DPL-1 can also promote the expression of ced-4 and 3, which are caspase activator and caspase homologs, respectively.
The combined activities of LIN-35 (including its associated factors) and CKI-1 result in an attenuation of the cell cycle at the G1/S phase, but they are not the only members to cause arrest at this stage. The abnormal cell lineage members 19 and 23 (re-named for the 'cullin' gene family, cul-1, and lin-23, respectively) encode for members of the ubiquitin ligase complex, which would be able to degrade G1/S phase regulators [102]. The Skp-1-Cul1-Fbox (SCF) complex particularly targets CYE-1. The cullin family, with six members in C. elegans, is involved in the degradation of numerous proteins; specifically those involved in cell cycle progression. Targets include mei-1 (involved in microtubule severing which allows chromosome segregation) and cdt (involved in DNA replication), which, if active could allow the progression and completion of the cell cycle [102].
Developmentally, apoptosis is necessary for C. elegans at the embryonic and L2 larval stages, and as seen through many of the regulators of the cell cycle (and throughout the preceding sections) apoptosis and proliferation mediation are often closely linked. Apoptosis mediates the removal of muscular, hypodermal, reproductive (egg) and neuronal cells, when necessary, to promote development. The cells targeted for apoptosis subsequently exhibit cell cycle arrest prior to cell death. Thus, it comes as little surprise that the C. elegans p53 tumor suppressor homolog, CEP-1, interacts with the cell cycle machinery. DNA checkpoints ensure that DNA is properly replicated, as well as undamaged, and are able to arrest the cell cycle if its conditions are not met [102]. In the germline, genotoxic stress can induce CEP-1 mediated apoptosis, and can also regulate cellular proliferation. CEP-1 is suspected to be an upstream activator of the cell cycle associated phg-1 (a C. elegans 'Growth Arrest-Specific1' (GAS1) equivalent) [105]. Upstream of this relationship, the CLK-2 protein, in C. elegans, is essentially an S-phase checkpoint regulator, which is suspected to transmit DNA damage signals to CEP-1 (and PHG-1), and resultantly control rates of proliferation and DNA repair [105]. Another example of checkpoint control in C. elegans can be seen in the germline of mature hemaphroditic adults, in pre-oocyte cells (Pachytene stage germ cells). These cells routinely undergo apoptosis, and in response to checkpoint signaling (via MRT-2 and HUS-1), CEP-1 activity is triggered [106]. Downstream, CEP-1 activates pro-apoptotic BH3-only proteins (EGL-1 and CED-13), which allows the expression of caspases (ced4 and 3) via inhibition of anti-apoptotic proteins (primarily CED-9). Interestingly, parallel to this, LIN-35 (via ced-9 inhibition), EFL-1 and DPL-1(both via ced4/3 activation) may also be involved in pre-oocyte apoptosis [106].
The novel role of mirco RNAs in metabolic regulation
Whereas the patterns of cell cycle regulation, in terms of the key proteins, are fairly well conserved and understood from C. elegans to mammalian systems, the fairly novel role of micro RNAs (miRNA) in transcriptional regulation has proven to have substantial effects on the cell cycle. Micro RNAs, first discovered in C. elegans, are short (approximately 20-25nt) RNA sequences that repress gene expression by binding to 3' untranslated regions (UTRs) of target mRNAs to prevent their translation [107]. Once bound, the miRNAs aggregate with other protein members of a 'RNA-induced silencing complex' (RISC). This can signal RNase activity, which cleaves the target mRNA, nullifying its expression. Micro RNAs are highly conserved across species and have been found to be involved in numerous critical cellular processes such as the regulation of metabolism, apoptosis signaling and, relevant here, cell cycle control [107]. Whereas miRNAs can function in clusters, their regulation patterns are individual, despite often having common (larval) stage specific expression patterns with the other members of the complex. There are hundreds of identified miRNAs, each able to affect multiple mRNA targets, which indicates that miRNAs are major players in cellular regulation.
Although some miRNAs are suspected of participating in cellular 'housekeeping' functions, others show patterns of up-regulation at significant developmental transitions such as from L1 to L2, and L4 to the mature adult stages of C. elegans larval development, respectively [108]. These include lin-4 and let-7. The let-7 family of miRNAs, along with the miR-15a/16-1 cluster, has recently come to light as important regulators of the cell cycle and tumor suppression, from C. elegans to human systems. The tumor suppressing capacities of let-7 in human models have significant implications in cancer research, with deletions of let-7 producing loci seen in many cancers [108]. Let-7 has numerous targets, including the c-Myc oncogene, CDK6 and CDC25A. Regulation of the cell cycle members would prevent progression in both G1/S and G2/M phases, and indeed over-expression of let-7 resulted in an accumulation of cells in the quiescence (Go) phase [108]. Similarly, enforced expression of miR-16 (which also targets CDK6) results in cells accumulating in the Go phase. Along with the cyclin dependent kinases, miRNAs have also been seen to target their inhibitors; the Cip/Kip family of proteins. One of the major G1/S phase regulating CKIs, p27, is regulated in mammalian systems by the miR-221/miR-222 cluster. Along with p27, p21 is also suspected to be regulated by miRNAs, the miR-17 family, in response to a p53 mediated cell cycle arrest signal which uses p21 to mediate the arrest [108]. Recently, it has been shown that members of the 'inhibitors of CDK4' (INK4), p15, p16, p18 and p19 can be regulated by miRNAs (notably p16 can be repressed by miR-24), and the absence of miRNA regulation of p27 by miR-221/222 has been linked to tumorigenesis [108]. Hence, regulation of CKIs by miRNAs, appears to be an important step in G1/S phase progression, and as such has possible implications in cancer development.
The miRNA interaction with c-Myc, alluded to briefly, above, affects the positive relationship between c-Myc and the activating E2Fs, which if left unregulated may lead to apoptosis (through excess proliferation signals), and as such, is a target for inhibition. In a feed-forward inhibition mechanism, c-Myc is also able to activate the expression of the miR17-92 complex of miRNAs, which in turn inhibits the translation of E2F1-3 genes [108]. Similarly, the E2F1 transcription factor is able to repress its own translation, to prevent excess cell proliferation, via the activation of the miR-106b-25 complex. The Rb proteins are also targeted by miRNAs to affect their interactions with the E2Fs. The p130 member, which typically binds the repressor E2F4, may be directly targeted by miR17 [108]. Subsequently, it is down-regulated, which indicates that miR17 may act to promote G1 progression.
Conclusion
Studies with C. elegans provide the most detailed and comprehensive information about the biochemistry and molecular biology involved in regulating dormancy in a eukaryote. The signaling and regulatory mechanisms described in this review suggest the blueprint for transitioning into a hypometabolic state and the “balancing act” between three states: growth/development, stasis/life extension, and degradation. They illustrate the intricacies of regulatory responses to nutrient and environmental stresses and the wide-ranging interactions that have to be considered when planning a coordinated suppression of cellular metabolism that achieves homeostasis and life extension over long periods of time spent in dormancy. From studies to date we know that many principles of metabolic arrest are conserved across phylogenetic lines; from controlling energetic expenditure via post-translational control of glycolytic proteins and ATPases, to protein-protein and protein-DNA interactions regulating cellular proliferation and apoptosis. The C. elegans model suggests additional mechanisms that could be fundamental principles of metabolic suppression across phylogeny and gives us many new roads to explore to determine the common and disparate mechanisms that underlie hypometabolism in its many forms. Similarly, with C. elegans as a marker for other models of hypometabolism, new avenues of regulation to conditions such as ischemia/reperfusion damage, insulin resistance, neurodegenerative diseases and cancer are opened to us in the search for modern clinical treatments.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Yamashita O, Hasegawa K. Embryonic diapause. In: (ed.) Kerkut G.A, Gilbert L.I. Comprehensive Insect Physiology, Biochemistry and Pharmacology, vol. 1. Oxford: Pergamon Press. 1985:407-434
2. Nakagaki M, Takei R, Nagashima E, Yaginuma T. Cell cycles in embryos of the silkworm, Bombyx mori: G2-arrest at diapause stage. Roux's Archives of Developmental Biology. 1991;200:223-229
3. MacRae T.H. Molecular chaperones, stress resistance and development in Artemia franciscana. Semin Cell Dev Biol. 2003;14(5):251-258
4. Renfree M.B, Shaw G. Diapause. Annu Rev Physiol. 2000;62:353-375
5. Lopes F.L, Desmarais J.A, Murphy BD. Embryonic diapause and its regulation. Reproduction. 2004;128(6):669-678
6. Storey K.B. Out cold: biochemical regulation of mammalian hibernation. Gerontology. 2009 in press
7. Hochachka P.W, Lutz PL. Mechanism, Origin, and Evolution of Anoxia Tolerance in Animals. Comp Biochem Physiol B. 2001;130(4):435- 459
8. Denlinger D.L. Regulation of Diapause. Annu Rev Entomol. 2002;47:93- 122
9. Clegg J.S. Cryptobiosis - A Peculiar State of Biological Organization. Comp Biochem Physiol B. 2001;128(4):613- 24
10. Storey K.B. Life in the Slow Lane: Molecular Mechanisms of Estivation. Comp Biochem Physiol A. 2002;133(3):733- 754
11. Storey K.B, Storey JM. Metabolic Rate Depression and Biochemical Adaptation in Anaerobiosis, Hibernation and Estivation. Quart Rev Biol. 1990;65(2):145- 174
12. Storey K.B, Storey JM. Metabolic Rate Depression in Animals: Transcriptional and Translational Control. Biol Rev Camb Philos Soc. 2004;79(1):207- 233
13. Heldmaier G, Ortmann S, Elvert R. Natural Hypometabolism During Hibernation and Daily Torpor in Mammals. Respir Physiol Neurobiol. 2004;141(3):317- 329
14. Berk A.J. Regulation of Eukaryotic Transcription Factors by Post-Translational Modification. Biochim Biophys Acta. 1989;1009(2):103- 109
15. Johnson E.S. Protein Modification by SUMO. Annu Rev Biochem. 2004;73:355- 382
16. Vanfleteren J.R, Braeckman BP. Mechanisms of life span determination in Caenorhabditis elegans. Neurobiol Aging. 1999;20:487- 502
17. Burgering B.M.T, Kops GJPL. Cell cycle and death control: long live forkheads. Trends Biochem Sci. 2002;27(7):352-360
18. Beall M.J, Pearce EJ. Transforming growth factor-beta and insulin like signaling pathways in parasitic helminthes. Intl J Parasit. 2002;32:399- 404
19. Barbieri M, Bonafe M, Franceschi C, Paolisso G. Insulin/IGF-I-Signaling Pathway: An Evolutionarily Conserved Mechanism of Longevity from Yeast to Humans. Am J Physiol. 2003;285(5):E1064- E1071
20. Ramnanan C.J, Groom A.G, Storey KB. Akt and its Downstream Targets Play Key Roles in Mediating Dormancy in Land Snails. Comp Biochem Physiol B. 2007;148(2):245- 255
21. Osaki M, Oshimura M, Ito H. PI3K-Akt Pathway: Its Functions and Alterations in Human Cancer. Apoptosis. 2004;9:667- 676
22. Jiang B.H, Liu LZ. PI3K/PTEN signaling in Tumorigenesis and Angiogenesis. Biochim Biophys Acta. 2008;1784:150- 158
23. Tremblay M.L, Giguere V. Phosphatases at the Heart of FoxO Metabolic Control. Cell Metab. 2008;7(2):101- 103
24. Lee R.Y.N, Hench J, Ruvkun G. Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the Daf-2 insulin-like signaling pathway. Curr Biol. 2001;11:1950-1957
25. Van der Horst A, Burgering BM. Stressing the role of FoxO Proteins in Lifespan and Disease. Nat Rev Mol Cell Biol. 2007;8(6):440- 450
26. Obsilova V, Silhan J, Boura E, Teisinger J, Obsil T. 14-3-3 Proteins: a Family of Versatile Molecular Regulators. Physiol Res. 2008;57(3):11- 21
27. Van Gorp A.C, Pomeranz K.M, Birkenkamp K.U, Hui R.C, Lam E.W, Coffer PJ. Chronic Protein Kinase B (PKB/c-Akt) Activation Leads to Apoptosis Induced by Oxidative Stress-Mediated Foxo3a Transcriptional Up-Regulation. Cancer Res. 2008;66(22):10760- 10769
28. Proud C.G. Signaling to Translation: How Signal Transduction Pathways Control the Protein Synthetic Machinery. Biochem J. 2007;403(2):217- 234
29. Kotliarova S, Pastorino S, Kovell L.C, Kotliarov Y, Song H, Zhang W, Bailey R, Maric D, Zenklusen J.C, Lee J, Fine HA. Glycogen Synthase Kinase-3 Inhibition Induces Glioma Cell Death Through c-MYC, Nuclear Factor-Kappa B, and Glucose Regulation. Cancer Res. 2008;68(16):6643- 6651
30. Ladu S, Calvisi D.F, Conner E.A, Farina M, Factor V.M, Thorgeirsson SS. E2F1 Inhibits c-MYC Driven Apoptosis via PIK3CA/Akt/mTOR and Cox-2 on a Mouse Model of Human Liver Cancer. Gastroenterology. 2008;135(4):1322- 1332
31. Liang J, Slingerland JM. Multiple Roles of the PI3K/PKB (Akt) Pathway in Cell Cycle Progression. Cell Cycle. 2003;2(4):339- 345
32. Kops G.J.P.L, Medema R.H, Glassford J, Essers M.A.G, Dijkers P.F, Coffer P.J, Lam E.W.F, Burgering BMT. Control of Cell Cycle Exit and Entry by Protein Kinase B-Regulated Forkhead Transcription Factors. Mol Cell Biol. 2002;22(7):2025- 2036
33. Suzuki A, Kusakai G, Kishimoto A, Lu J, Ogura T, Lavin M.F, Esumi H. Identification of a Novel Protein Kinase Mediating Akt Survival Signaling to the ATM protein. J Biol Chem. 2003;278(1):48- 53
34. Gems D, Sutton A.J, Sundermeyer M.L, Albert P.S, King K.V, Edgley M.L, Larsen P.L, Riddle DL. Two Pleiotropic Classes of Daf-2 Mutation Affect Larval Arrest, Adult Behavior, Reproduction and Longeivity in Caenorhabditis elegans. Genetics. 1998;150(1):129- 155
35. Paradis S, Ailion M, Toker A, Thomas J.H, Ruvkun G. A PDK1 homolog is Necessary and Sufficient to Transduce AGE-1 PI3 Kinase Signals that Regulate Diapause in Caenorhabditis elegans. Genes Dev. 2009;13:1438- 1452
36. Gami M.S, Iser W.B, Hanselman K.B, Wolkow CA. Activated AKT/PKB Signaling in C. elegans Uncouples Temporally Distinct Outputs of DAF-2/Insulin-Like Signaling. BMC Dev Biol. 2006;6:45
37. Lee S.S, Kennedy S, Tolonen A.C, Ruvkun G. Daf-16 Target Genes that Control C. elegans Life-Span and Metabolism. Science. 2003;300(5619):644- 647
38. Fukuyama M, Rougvie A.E, Rothman JH. C. elegans Daf-18/PTEN Mediates Nutrient-Dependent Arrest of Cell Cycle Growth in the Germline. Curr Biol. 2006;16(8):773- 779
39. Shaw R, Dillin A. PPTR-1 CounterAkts Insulin Signaling. Cell. 2009;136:816- 818
40. Oh S.W, Mukhopadhyay A, Svrzikapa N, Jiang F, Davis R.J, Tissenbaum HA. JNK Regulates Lifespan in Caenorhabditis elegans by Modulating Nuclear Translocation of Forkhead Transcription Factor/ DAF-16. Proc Natl Acad Sci USA. 2005;102(12):4494- 4999
41. Kim A.H, Sasaki T, Chao MV. JNK-Interacting Protein 1 Promotes Akt1 Activation. J Biol Chem. 2003;278(32):29830- 29836
42. Wolkow C.A, Munoz M.J, Riddle DL, Ruvkun G. Insulin Receptor Substrate an P55 Orthologous Adaptor Proteins Function in the Caenorhabditis elegans Daf-2/Insulin-like Signaling Pathway. J Biol Chem. 2002;277(51):49591- 49597
43. An J.H, Vranas K, Lucke M, Inoue H, Hisamoto N, Matsumoto K, Blackwell TK. Regulation of the Caenorhabditis elegans Oxidative Stress Defense Protein SKN-1 by Glycogen Synthase Kinase-3. Proc Natl Acad Sci USA. 2005;102(45):16275- 16280
44. Kell A, Ventura N, Kahn N, Johnson TE. Activation of SKN-2 by Novel Kinases in Caenorhabditis elegans. Free Rad Biol Med. 2007;43:1560- 1566
45. Hart G.W, Housley M.P, Slawson C. Cycling of O-linked β -N-acetylglucosamine on Nucleocytoplasmic Proteins. Nature. 2007;446(7139):1017- 1022
46. Kang E.S, Han D, Park J, Kwak T.K, Oh M.A, Lee S.A, Choi S, Park Z.Y, Kim Y, Lee JW. O-GlcNAc Modulation at Akt1 Ser473 Correlates with Apoptosis of Murine Pancreatic Beta Cells. Exp Cell Res. 2008;314(11-12):2238- 2248
47. Wu M.Y, Hill CS. TGF-β Superfamily Signaling in Embryonic Development and Homeostasis. Dev Cell. 2009;16:329- 343
48. Wrighton K.H, Lin X, Feng XH. Phospho-Control of TGF- β Superfamily Signaling. Cell Res. 2009;19(1):8- 20
49. Inoue T, Thomas JH. Targets of TGF-β Signaling in Caenorhabditis elegans Dauer Formation. Dev Biol. 2000;217:192- 204
50. Wang J, Mohler W.A. and C. Savage-Dunn. C-terminal Mutants of C. elegans Smads Reveal Tissue Specific Requirements for Protein Activation by TGF-β Signaling. Development. 2005;132:3505- 3513
51. Daniels S.A, Ailion M, Thomas J.H, Sengupta P. Egl-4 Acts Through a Transforming Growth Factor β/SMAD Pathway in Caenorhabditis elegans to Regulate Multiple Neuronal Circuits in Response to Sensory Cues. Genetics. 2000;156:123- 141
52. Swiech L, Perycz M, Malik A, Jaworski J. Role of mTOR and Pathology of the Nervous System. Biochim Biophys Acta. 2008;1784:116-132
53. Proud C.G. mTOR Unleashed. Science. 2007;318(9):926- 927
54. Hong F, Larrea M.D, Doughty C, Kwiatkowski D.J, Squillace R, Slingerland JM. mTOR-Raptor Binds and Activates SGK1 to Regulate p27 Phosphorylation. Mol Cell. 2008;30:701- 711
55. Jia K, Chen D, Riddle DL. The TOR Pathway Interacts with the Insulin Signaling Pathway to Regulate C. elegans Larval Development, Metabolism and Life Span. Development. 2004;131:3897-3906
56. Padmanabhan S, Mukhopadhyay A, Narasimhan S.D, Tesz G, Czech M.P, Tissenbaum HA. A PP2A Regulatory Subunit Regulates C. elegans Insulin/IGF-1 Signaling by Modulating AKT-1 Phosphorylation. Cell. 2009;136:939- 951
57. Jones K.T, Greer E.R, Pearce D, Ashrafi K. Rictor/TORC2 Regulates Caenorhabditis elegans Fat Storage, Body Size, and Development through sgk-1. PLoS Biol. 2009;7(3):604- 615
58. Narbonne P, Roy R. Caenorhabditis elegans Dauers Need LKB1/AMPK to Ration Lipid Reserves and Ensure Long-Term Survival. Nature. 2009;457(8):210- 215
59. Wang Y, Levy DE. C. elegans STAT: Evolution of a Regulatory Switch. FASEB J. 2006;20(10):1641- 1652
60. Stephanou A, Latchman DS. STAT-1: A Novel Regulator of Apoptosis. Intl J Exp Path. 2003;84:239- 244
61. Stephanou A. Role of STAT-1 and STAT-3 in Ischaemia/Reperfusion Injury. J Cell Mol Med. 2004;8(4):519- 525
62. Lin J, Tang H, Jin X, Jia G, Hsieh JT. P53 Regulates Stat3 Phosphorylation and DNA Binding Activity in Human Prostrate Cancer Cells Expressing Constitutively Activated Stat3. Oncogene. 2002;21:3082- 3088
63. Munnarriz E, Barcaroli D, Stephanou A, Townsend P.A, Maisse C, Terrinoni A, Neale M.H, Martin S.J, Latchman D.S, Knight R.A, Melino G, DeLaurenzi V. PIAS-1 is a Checkpoint Regulator Which Affects Exit from G1 and G2 by Sumoylation of P73. Mol Cell Biol. 2004;24(24):10593- 10610
64. Bode J.G, Gatsios P, Ludwig S, Rapp U.R, Haussinger D, Heinrich P.C, Graeve L. The Mitogen-Activated Protein (MAP) Kinase P38 and Its Upstream Activator MAP Kinase Kinase 6 are Invovled in the Activation of Signal Transducer and Activator of Transcription by Hyperosmolarity. J Biol Chem. 1999;274(42):302222- 30227
65. Inaba M, Saito H, Fujimoto M, Sumitani S, Ohkawara T, Tanaka T, Kouhara H, Kasayama S, Kawase I, Kishimoto T, Naka T. Suppressor of Cytokine Signalling 1 Suppresses Muscle Differentiation through Modulation of IGF-1 Receptor Signal Transduction. Biochem Biophys Res Commun. 2005;328(4):953- 961
66. Himpe E, Kooijman R. Insulin Like Growth Factor-I Receptor Signal Transduction and the Janus Kinase/Signal Transducer and Activator of Transcription (JAK-STAT) Pathway. BioFactors. 2009;35(1):76- 81
67. Wang Y, Levy DE. C. elegans STAT Co-operates with DAF-7/TGF-β Signaling to Repress Dauer Formation. Curr Biol. 2006;16:89- 94
68. Ferraro E, Cecconi F. Autophagic and apoptotic response to stress signals in mammalian cells. Arch Biochem Biophys. 2007;462:210- 219
69. Smith D.J, Ng H, Kluck R.M, Nagley P. The Mitochondrial Gateway to Cell Death. Life. 2008;60(6):383- 389
70. Samara C. and N.Tavernarakis. Autophagy and Cell Death in Caenorhabditis elegans. Curr Pharm Des. 2008;14(2):97- 115
71. Brady G.F, Duckett CS. A Caspase Homolog Keeps CED-3 in Check. Trends Bioc Sci. 2009;34(3):104- 107
72. Tan F.J, Husain M, Manlandro C.M, Koppenol M, Fire A.Z, Hill RB. CED-9 and Mitochondrial Homeostasis in C. elegans Muscle. J Cell Sci. 2008;121(20):3373- 3782
73. Breckenridge D.G, Kang B.H, Kokel D, Mitani S, Staehelin A, Xue D. Caenorhabditis elegans drp-1 and fis-2 Regulate Distinct Cell-Death Execution Pathways Downstream of ced-3 and Independent of ced-9. Mol Cell. 2008;31:586- 597
74. Jagasia R, Grote P, Westermann B, Conradt B. DRP-1 Mediated Mitochondrial Fragmentation During EGL-1 Induced Cell Death in C. elegans. Nature. 2005;433(7027):754- 760
75. Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1- Mediated Phosphorylation of Bcl-2 Regulates Starvation Induced Autophagy. Mol Cell. 2008;30(6):678- 688
76. Wei Y, Sinha S, Levine B. Dual Role of JNK1-Mediated Phosphorylation of Bcl-2 in Autophagy and Apoptosis Regulation. Autophagy. 2008;4(7):949- 951
77. Pimkina J, Humbey O, Zilfou J.T, Jarnik M, Murphy ME. ARF Induced Autophagy by Virtue of Interaction with Bcl-xl. J Biol Chem. 2009;284(5):2803- 2810
78. Zong W.X, Moll U. P53 in Autophagy Control. Cell Cycle. 2008;7(19):2947- 2948
79. Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of Macroautophagy by mTOR and Beclin 1 Complexes. Biochimie. 2008;90:313- 323
80. Morck C, Pilon M. Caloric restriction in autophagy in Caenorhabditis elegans. Autophagy. 2007;3(1):51-53
81. Melendez A, Talloczy Z, Seaman M, Eskelinen E.L, Hall D.H, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301:1387-1391
82. Kourtis N, Tavernarakis N. Autophagy and Cell Death in Model Organisms. Cell Death and Diff. 2009;16:21- 30
83. Lavin M.F, Gueven N. The Complexity of p53 Stabilization and Activation. Cell Death Diff. 2006;13:941- 950
84. Bos R, Van Diest P.J, Van der Groep P, Shvarts A, Greijer A. and E. Van der Wall. Expression of Hypoxia-Inducible Factor 1 α and Cell Cycle Proteins in Invasive Breast Cancer are Estrogen Receptor Related. Breast Cancer Res. 2004;6:450- 459
85. Rosenbluth J.M, Pietenpol JA. mTOR Regulates Autophagy Associated Genes Downstream of p73. Autophagy. 2009;5(1):114- 116
86. Derry W.B, Putzke A.P, Rothman JH. Caenorhabditis elegans p53: Role in Apoptosis, Meiosis, and Stress Resistance. Science. 2001;294(5542):591- 595
87. Arum O, Johnson TE. Reduced Expression of the Caenorhabditis elegans p53 Ortholog cep-1 Results in Increased Longevity. J Gerontol. 2007;62(9):951- 959
88. Schumacher B, Schertel C, Wittenburg N, Tuck S, Mitani S, Gartner A, Conradt B, Shaham S. C.elegans ced-13 Can Promote Apoptosis and is Induced in Response to DNA Damage. Cell Death Diff. 2005;12:153- 161
89. Gao M.X, Liao E.H, Yu B, Wang Y, Zhen M, Derry WB. The SCFFsn-1 Ubiquitin Ligase Controls Germline Apoptosis Through CEP-1/p53 in C. elegans. Cell Death Differ. 2008;15:1054- 1062
90. Greiss S, Hall J, Ahmed S, Gartner A. C. elegans SIR-2.1 Translocation is Linked to a Proapoptotic Pathway Parallel to cep-1/p53 during DNA damage-Induced Apoptosis. 2009. Genes Dev. 2009;22:2831- 2842
91. Tavernarakis N, Pasparaki A, Tasedemir E, Maiuri M.C, Kroemer G. The Effects of p53 on Whole Organism Longevity are Mediated by Autophagy. Autophagy. 2008;4(7):870- 873
92. Ohtsubo M, Theodoras A.M, Schumacher J, Roberts J.M, Pagano M. Human Cyclin E, a Nuclear Protein Essential for the G1-to-S Phase Transition. Mol Cell Biol. 1995;15(5):2612- 2624
93. Diehl J.A, Cheng M, Roussel M.F, Sherr CJ. Glycogen Synthase Kinase-3 Beta Regulates Cyclin D1 Proteolysis and Subcellular Localization. Genes Dev. 1998;12(22):3499- 3511
94. Sherr C.J, Roberts JM. CDK Inhibitors: Positive and Negative Regulators of the G1-Phase Progression. Genes Dev. 1999;13(12):1501- 1512
95. Van den Heuvel S, Dyson NJ. Conserved Functions of the pRB and E2F Families. Nat Rev Mol Cell Biol. 2008;9(9):713- 724
96. Berthet C, Kaldis P. Cell-Specific Responses to Loss of Cyclin-Dependent Kinases. Oncogene. 2007;26:4469- 4477
97. Booher R.N, Holman P.S, Fattaey A. Human Myt1 is a Cell Cycle-Regulated Kinase That Inhibits Cdc2 but not Cdk2 Activity. J Biol Chem. 1997;272(35):22300- 22306
98. Rudolph J. Cdc25 Phosphatases: Structure, Specificity, and Mechanism. Biochemistry. 2007;46(12):3595- 3604
99. Uto K, Inoue D, Shimuta K, Nakajo N, Sagata N. Chk1, but not Chk2, Inhibits Cdc Phosphatases by a Novel Common Mechanism. EMBO J. 2004;23(16):3386- 3396
100. Mateyak M.K, Obaya A.J, Sedivy JM. C-Myc Regulates Cyclin D-CDK4 and- CDK6 Activity but Affects Cell Cycle Progression at Multiple Independent Points. Mol Cell Biol. 1999;19(7):4672- 4683
101. Ladu S, Calvisi D.F, Conner E.A, Farina M, Factor V.M, Thorgeirsson SS. E2F1 Inhibits c-MYC Driven Apoptosis via PIK3CA/Akt/mTOR and Cox-2 on a Mouse Model of Human Liver Cancer. Gastroenterology. 2008;135(4):1322- 1332
102. Van den Heuvel S. Cell-cycle regulation. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook 1.28.1. 2005
103. Clayton JE, Van den Heuvel S.J, Saito RM. Transcriptional Control of Cell-Cycle Quiescence during C. elegans Development. Dev Biol. 2008;313(2):603- 613
104. Koreth J. and S. Van den Heuvel. Cell-Cycle Control in Caenorhabditis elegans: How the Worm Moves from G1 to S. Oncogene. 2005;24:2756- 2764
105. Derry W.B, Bierings R, Van Iersel M, Satkunendran T, Reinke V, Rothman JH. Regulation of Developmental Rate and Germ Cell Proliferation in Caenorhabditis elegans by the p53 gene network. Cell Death Differ. 2007;14(4):662- 670
106. Gartner A, Boag P.R, Blackwell TK. Germline Survival and Apoptosis. Wormbook ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook 1.145.1. 2008
107. Kato M, de Lencastre A, Pincus Z, Slack FJ. Dynamic Expression of Small Non-Coding RNAs, Including Novel MicroRNAs and piRNAs/21U-RNAs, During C. elegans development. Genome Biology. 2009;10(5):R54
108. Chivukula R.R, Mendell JT. Circular Reasoning: MicroRNAs and Cell-Cycle Control. Trends Biochem Sci. 2008;33(10):474- 481
Author contact
![]() Correspondence to: Kenneth B. Storey, Institute of Biochemistry, Carleton University, 1125 Colonel By DriveOttawa, Ont. K1S 5B6, Canada. Tel. +1 613 520 3678, Fax +1 613 520 2569, E-Mail kenneth_storeyca
Correspondence to: Kenneth B. Storey, Institute of Biochemistry, Carleton University, 1125 Colonel By DriveOttawa, Ont. K1S 5B6, Canada. Tel. +1 613 520 3678, Fax +1 613 520 2569, E-Mail kenneth_storeyca
Citation styles
APA
Lant, B., Storey, K.B. (2010). An Overview of Stress Response and Hypometabolic Strategies in Caenorhabditis elegans: Conserved and Contrasting Signals with the Mammalian System. International Journal of Biological Sciences, 6(1), 9-50. https://doi.org/10.7150/ijbs.6.9.
ACS
Lant, B.; Storey, K.B. An Overview of Stress Response and Hypometabolic Strategies in Caenorhabditis elegans: Conserved and Contrasting Signals with the Mammalian System. Int. J. Biol. Sci. 2010, 6 (1), 9-50. DOI: 10.7150/ijbs.6.9.
NLM
Lant B, Storey KB. An Overview of Stress Response and Hypometabolic Strategies in Caenorhabditis elegans: Conserved and Contrasting Signals with the Mammalian System. Int J Biol Sci 2010; 6(1):9-50. doi:10.7150/ijbs.6.9. https://www.ijbs.com/v06p0009.htm
CSE
Lant B, Storey KB. 2010. An Overview of Stress Response and Hypometabolic Strategies in Caenorhabditis elegans: Conserved and Contrasting Signals with the Mammalian System. Int J Biol Sci. 6(1):9-50.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.