ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2010; 6(2):151-162. doi:10.7150/ijbs.6.151 This issue Cite
Research Paper
Tumors Induced in Mice by Direct Inoculation of Plasmid DNA Expressing Both Activated H-ras and c-myc
Li Sheng-Fowler1, Fang Cai1,4, Haiqing Fu1,5, Yong Zhu1,6, Brian Orrison1, Gideon Foseh1, Don G. Blair2,7, Stephen H. Hughes2, John M. Coffin2,3, Andrew M. Lewis Jr1 ![]() , Keith Peden1
, Keith Peden1 ![]()
1. Division of Viral Products, OVRR, CBER, FDA, Bethesda, MD 20892, USA
2. Frederick Cancer Research Facility, National Cancer Institute, Frederick, MD 21702, USA
3. Present address: Center for Drug Evaluation and Research, White Oak, MD, USA
4. Present address: National Cancer Institute, Bethesda, MD, USA
5. Present address: University of Texas, Galveston, TX, USA
6. Present address: National Institutes of Health, Rockville, MD, USA
7. Tufts University, Boston, MA 02111, USA
Abstract
Vaccines contain residual DNA derived from the cells used to produce them. As part of our investigation to assess the risk of this cellular DNA, we are developing a quantitative in vivo assay to assess the oncogenicity of DNA. In an earlier study, we had generated expression plasmids for two oncogenes - human activated T24-H-ras and murine c-myc - and had shown that these two plasmids, pMSV-T24-H-ras and pMSV-c-myc, could act in concert to induce tumors in mice, although the efficiency was low. In this study, we took two approaches to increase the oncogenic efficiency: 1) both oncogene-expression cassettes were placed on the same plasmid; 2) transfection facilitators, which increase DNA uptake and expression in vitro, were tested. The dual-expression plasmid, pMSV-T24-H-ras/MSV-c-myc, is about 20-fold more efficient at tumor induction in newborn NIH Swiss mice than the separate expression plasmids, with tumors being induced with 1 µg of the dual-expression plasmid DNA. However, none of the transfection facilitators tested increased the efficiency of tumor induction. Based on these data, the dual-expression plasmid pMSV-T24-H-ras/MSV-c-myc will be used as the positive control to develop a sensitive and quantitative animal assay that can be used to assess the oncogenic activity of DNA.
Keywords: H-ras, c-myc, oncogenes
Introduction
Because all viral vaccines contain some residual DNA from the cell substrate used to produce them, it is important to establish whether this DNA could pose a risk to vaccine recipients and, if so, to quantify the risk. The issue of the potential oncogenic risk posed by DNA was one of the reasons that the Armed Forces Epidemiological Board recommendation in 1954 against the use of tumorigenic cells, or cells derived from human tumors, for the manufacture of vaccines for human use [1, 2]. This recommendation remained in effect for over 40 years. However, opinions on the risk posed by residual cellular DNA have varied from there being no risk to it being considered to be an important risk factor [3-5], particularly for vaccines manufactured in cells that are tumorigenic or are derived from tumors. The potential risk from cellular DNA arises both from its oncogenic activity and from the possibility that the DNA could encode infectious agents [6]. Studies on DNA infectivity has been presented elsewhere (Sheng-Fowler et al., in press).
The possibility that vaccines produced in neoplastic cells might have the capacity to induce a neoplastic phenotype in the cells of vaccine recipients through the injection and uptake of residual DNA from the neoplastic cell substrate is supported by the presence of oncogenes in the genomes of eukaryotic cells [7] and the demonstration that these cellular oncogenes can transform primary cells in culture [8-10] into cells that can become tumorigenic in animals [8, 11]. Another mechanism by which residual cellular DNA could induce oncogenesis would be through the integration of the DNA into the genome of cells of vaccine recipients. Integration of residual cellular DNA could potentially activate a dominant cellular oncogene or inactivate a tumor suppressor gene. However, oncogenesis through integration of DNA has been considered to be unlikely, because integration of exogenous DNA per se is a low probability event [12] and the probability of integration at a particular site is even lower [13-18] (Sheng-Fowler et al., in press). Oncogenesis through the inactivation of a cellular tumor-suppressor gene would be even less likely, because both alleles of the suppressor gene would have to be inactivated. Therefore, the major oncogenic risk of DNA in vaccines would be through the introduction of a dominant activated oncogene whose expression either could directly transform a cell in a vaccinated individual into a cell that could form a tumor [8-10], or could cause the “initiation” of the tumorigenic process that, with the acquisition of additional mutations over time, could lead to the development of a tumor [19-22].
It has been estimated that the genomes of normal mammalian cells have more than 200 proto-oncogenes [23], although, in normal cells, these proto-oncogenes have not been activated to become dominant transforming genes. However, for several reasons, this number is likely an underestimate. For example, ordinary housekeeping genes such as phosphoglycerate mutase [24] and certain cellular receptors such as the constant gamma chain of the IL-2 receptor [25] have been shown to be capable of becoming transforming oncogenes. In addition, it has recently been demonstrated that certain microRNA (miRNA) genes can induce a polyclonal B-cell malignancy in transgenic mice [26], and some of these miRNAs have been implicated in human cancer [27-31], thus expanding the number of genes in cellular DNA that have the potential to become dominant oncogenes.
Because few studies had attempted to measure DNA oncogenicity in vivo, we undertook to develop an animal model that could be used to estimate the oncogenic potential of various DNAs. In a previous study, we generated expression plasmids for the activated human T24 H-ras gene and for the murine c-myc gene [32]. These oncogenes were initially chosen, because they had been shown to transform rodent cells in vitro into cells that were tumorigenic in vivo [8, 33, 34]. Our initial studies demonstrated that these plasmids were oncogenic in vivo in two mouse strains. We found that NIH Swiss mice were more sensitive to oncogenic DNA than C57BL/6, and newborn mice were more sensitive than adult mice. Both oncogenes were required for tumor induction, and tumors were only induced by the highest amounts of DNA used (12.5 µg of each plasmid). In this assay, a ten-fold dilution of the DNA showed no activity [32]; this was likely due to a requirement that both oncogenes be taken up by the same cell.
In the current study, we took two approaches to increase the efficiency of tumor induction by oncogenic DNA. In the first, we generated dual-expression plasmids that contained both the human T24-H-ras and mouse c-myc oncogenes and expressed each gene from its own promoter. In the second, we evaluated whether transfection facilitators, reagents that increase DNA uptake and expression in vitro, were able to enhance the efficiency of tumor induction by DNA in vivo.
The results showed that having both oncogenes on the same molecule increased the efficiency of tumor induction. As little as 1 µg of the dual-expression plasmid (pMSV-T24-H-ras/MSV-c-myc) DNA was able to induce tumors in newborn mice. However, none of the transfection facilitators increased the oncogenic efficiency of the dual-expression plasmid in mice. The dual-expression plasmid pMSV-T24-H-ras/MSV-c-myc will be the positive control oncogenic DNA that will be used to establish a sensitive and quantitative in vivo assay for DNA oncogenesis. Once this assay is established, it will be used to estimate the oncogenic risk of cellular DNA in vaccines and other biological products. In addition, an assay that can detect the oncogenic activity of DNA introduced directly into mice could also be used to evaluate the in vivo oncogenic potential of cellular or viral oncogenes, thus circumventing the indirect approach of generating cell lines expressing these oncogenes and then assessing the tumorigenicity of these cells in vivo.
Materials and Methods
Construction of oncogene expression plasmids
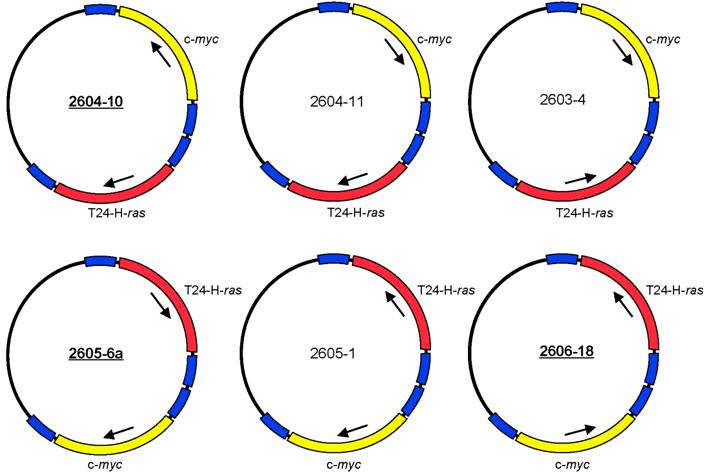
The expression plasmids pMSV-T24-H-ras and pMSV-c-myc [32] were digested with NruI, and the oncogene-containing fragments were purified by gel electrophoresis. The vector pLS3 [32], which has single sites for EcoRI, NruI, SacI, KpnI, SalI, PstI, PvuII, NotI, XbaI, AvrII BamHI, SpeI, NdeI, ClaI, BglII, HpaI, XhoI, SphI, BstBI, MluI, NcoI, NheI, and HindIII, was digested with NruI and separately with HpaI. After dephosphorylation with Antarctic phosphatase (New England BioLabs, Ipswich, MA), the vectors were purified by gel electrophoresis. The oncogene-containing NruI fragments were ligated both to NruI-digested pLS3 vector and to HpaI-digested pLS3. Those plasmids that contained either T24-H-ras or c-myc insertions at the HpaI site were digested with NruI for insertion of the other oncogene, and those plasmids that contained either T24-H-ras or c-myc insertions at the NruI site were digested with HpaI for insertion of the other oncogene. Of the eight possible dual-oncogene plasmid configurations, six were obtained (Fig. 1). Despite each plasmid having four long terminal repeat (LTR) elements in various orientations, all were stable to propagation in bacteria. Plasmid DNA was prepared as described [32].
Structures of six configurations of the dual expression plasmid pMSV-T24-H-ras/MSV-c-myc.
Cell lines and cell culture
The NIH 3T3 cells (CRL-1658) were obtained from the American Type Culture Collection (Manassas, VA) and Rat-1 cells were originally obtained from Bill Topp (Cold Spring Harbor Laboratory). They were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 1% glutamine and either 5% fetal bovine serum (FBS; DMEM-5) for NIH-3T3 cells or 10% FBS (DMEM-10) for Rat-1 cells.
Transfection reagents
Transfection facilitators were: PolyFect, SuperFect and Effectene (Qiagen Inc., Valencia, CA); Lipofectamine and DMRIE-C (Invitrogen, Inc., Carlsbad, CA); DOTAP, DOSPER, and FuGene HD (Roche Applied Sciences, Indianapolis, IN); Transfectol (GeneChoice, Frederick, MD); aurintricarboxylic acid (Sigma, St. Louis, MO); and in vivo-JetPEI (PolyPlus Transfection Inc., New York, NY).
Animals and procedures
NIH Swiss mice were purchased from the National Cancer Institute, Division of Cancer Treatment, Frederick, MD. Three of the dual-expression plasmids were selected for testing in animals - 2604-10, 2605-6a, and 2606-18 (Fig. 1).
Plasmid DNA in PBS (total volume 50 µL) was inoculated via the subcutaneous route above the scapulae in both adult (two to four months of age) and newborn (one to two days of age) NIH Swiss mice using a 26-gauge needle and a 0.5 mL syringe. DNA amounts are indicated in the text; the total amount of DNA was made up to a constant amount by the addition of appropriate quantities of the empty expression vector pMSV/LS [32]. Animals were housed in cages with food and water ad libitum and a 12-hour light/dark cycle. Protocols were approved by the CBER Animal Use and Care Committees. Processing of tumors was done as described [32].
Establishment of cell lines from tumors
Cell lines were established from tumor tissue as described using trypsin dispersal of the cells [32]. Cell lines were expanded in DMEM-10 and frozen as described [32].
Extraction of DNA from cell lines
Cells from the tumor-cell lines were harvested from T150 tissue-culture flasks by trypsinization and were washed in PBS. Between 2 x 107 and 1 x 108 cells (1 to 4 flasks depending on the cell line) were used to extract genomic DNA by the Qiagen Blood and Cell Culture DNA kit (Qiagen Inc., Valencia, CA).
PCR analysis
Genomic DNA was subjected to PCR using primers that were designed to detect the plasmid-encoded oncogenes while not reacting with the endogenous genes [32]. PCR analysis was done with 300 ng of genomic DNA (cell line or tumor) or 200 pg of plasmid DNA. A negative control was performed by omitting DNA (genomic DNA or plasmid). PCR products (2 µL of the 50 µL reaction) were resolved by electrophoresis on a 1.8% agarose gel containing 0.025% ethidium bromide. Gels were photographed using an AlphaImager 3400 (Alpha Innotech, San Leandro, CA).
Southern DNA analysis
Genomic DNA (50 µg) from the tumor-cell lines was digested with restriction enzymes and analyzed for complete digestion by analytical agarose-gel electrophoresis. For Southern analysis, 8 µg of each digested DNA were fractionated on a 1% agarose gel. After photographing, the gel was gently shaken at room temperature for 15 min in 0.25 N HCl to partially depurinate the DNA. The gel was then neutralized in 0.4 N NaOH for 30 min, and the DNA was transferred for 3 h by downward capillary action to Amersham Hybond-N+ membrane (GE Healthcare, Piscataway, NJ) in the same buffer using the Schleicher and Schuell TurboBlotter (Whatman Inc., Florham Park, NJ 07932). The DNA was cross-linked to the wet membrane using a UV Stratalinker 1800 (Stratagene, La Jolla, CA) with 70 mJoules/cm2 of energy. The dried membranes were stored between two filter papers in plastic bags at 23-25°C.
Prehybridization and hybridization were done in glass roller bottles using a Hybaid Maxi 14 incubator. Membranes were prehybridized in 10 mL of PERFECTHYB PLUS (Sigma, St. Louis, MO) at 68°C for 1 - 2 h. Probes were the gel-purified ras or myc genes (excised from the expression plasmids using NotI plus BamHI digestion) labeled using the North2South Biotin Random Prime Kit (Pierce, Rockford, IL). Probes (50 ng/mL) were added to the prehybridization solution, and the membranes were hybridized overnight at 68°C in glass cylinders. Membranes were washed once in 2 x SSC, pH 7.2, 0.1% SDS for 30 min at room temperature, twice in 0.5 x SSC, pH 7.2, 0.1% SDS for 30 min each time at 68°C, and once in 0.1 x SSC, pH 7.2, 0.1% SDS for 30 min at 68°C.
Probe detection was done according to the manufacturer's protocol (Pierce, Rockford, IL). Briefly, the membrane was incubated in blocking buffer with shaking for 15 min at room temperature and then in fresh blocking buffer with the Stabilized Streptavidin-Horseradish Peroxidase Conjugate for another 15 min at room temperature with shaking. The membrane was washed with 1 x wash solution 5 times for 5 min each time and incubated in substrate equilibration buffer for 5 min with gentle shaking followed by addition of the chemiluminescent substrate working solution buffer for 5 min without shaking. The membranes were exposed to X-ray film for times varying for a few seconds to several minutes.
Preparation of cell lysates and western blot analysis
This was done as described by Sheng et al. [32].
Results
Development of the dual-expression plasmids
Our previous studies had demonstrated that activated human H-ras and murine c-myc could cooperate to induce tumors in immune-competent adult and newborn mice. Newborn mice were more sensitive than adults and NIH Swiss mice were more susceptible than C57BL/6 mice [32]. However, the efficiency of tumor induction was low: 12.5 µg of each of the plasmids pMSV-T24-H-ras and pMSV-c-myc were required for tumor induction. Because tumor induction required that a single cell take up both plasmids, it was reasonable to expect that having both oncogenes on the same plasmid would increase the efficiency of tumor induction.
The two oncogene-expression cassettes were combined in the plasmid pLS3 (the steps are outlined in Materials and Methods). Of the eight possible arrangements of the two oncogenes, six were obtained (Fig. 1). These plasmids differed in the orientation of the oncogene-expression cassette and the position of the ras and the myc genes relative to each other. Because each gene is expressed from its own murine sarcoma virus (MSV) long terminal repeat (LTR) promoter and has its own termination signal, we expected that the arrangement of the oncogenes in the plasmid would not affect the oncogenic activity of the plasmids. Despite the presence of four MSV LTR elements in various orientations, all of the plasmids were stable when propagated in bacteria.
Each of the dual-expression plasmids was tested in vitro for its capacity to induce loss of contact inhibition in a focus-formation assay in both NIH 3T3 cells and Rat-1 cells. All six versions of pMSV-T24-H-ras/pMSV-c-myc induced foci in both cell lines and had indistinguishable activities (data not shown). Three were chosen - 2604-10, 2605-6a, and 2606-18 - to assess their ability to induce tumors in vivo.
Activity in newborn and adult NIH Swiss mice
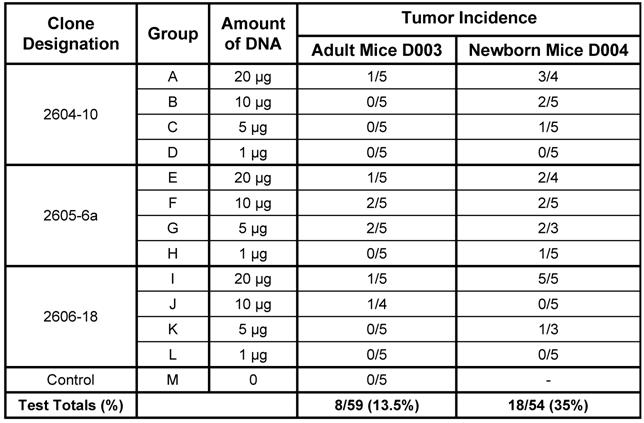
Four doses (20 µg, 10 µg, 5 µg, and 1 µg) of the three versions of the dual-expression plasmid pMSV-T24-H-ras/MSV-c-myc were inoculated subcutaneously into both adult and newborn NIH Swiss mice. Animals were monitored daily for general health and twice weekly for tumor formation. Tumors began to appear after about 4 weeks, and no additional tumors were found after about nine weeks. The newborn mouse was more sensitive than the adult mouse to tumor induction by DNA. This is shown by the combined number of mice with tumors at all doses of DNA. For example, tumors were induced in 8 out of 59 (13.5%) adult mice and in 18 out of 54 (35%) newborn mice (Table 1). The newborn mouse was also more sensitive to tumor induction by lower doses of DNA: 1 µg of DNA caused tumors in newborn mice, whereas no tumors were induced in adult mice with less than 5 µg of DNA.
From the results of Table 1, there was no clear dose-response relationship with any one group. It is likely that this is a consequence of the low numbers of animals used and of the variation commonly seen is animal experiments. Additional experiments are planned to investigate this variability.
Tumor induction in adult and newborn NIH Swiss Mice by three variants of the dual expression plasmid.
PCR analysis of the DNA from tumor-cell lines
Cell lines were established from five of the tumors induced in adult mice (D003-A, D003-F, D003-I, D003-J, D003-E) and from four of the tumors induced in newborn mice (D004-B. D004-F, D004-I, D004-K). To determine whether the tumors were induced by the inoculated oncogenes, DNA was extracted from the cell lines, and PCR was carried out using primers that were selective for the exogenous H-ras and c-myc genes and would not amplify the endogenous genes [32].
PCR analysis of DNA isolated from cell lines established from tumors induced by the dual-expression plasmids (pMSV-T24-H-ras/MSV-c-myc) in adult NIH Swiss mice. DNA from the tumor cell lines was amplified by PCR using Oligos610/511 for the 5' myc gene and Oligos513/515 for the 3' myc gene and Oligos509/512 for the 5' ras gene and Oligos514/515 for the 3' ras gene. The PCR products were resolved by electrophoresis in a 1.8% agarose gel. A. The 5' myc and 3' myc gene. B. The 5' ras and 3' ras gene. Lanes M; marker; lanes a: no DNA control; lanes b: positive controls pMSV-T24-H-ras or pMSV-c-myc; lanes c: D003-A; lanes d: D003-F; lanes e: D003-I; lanes f: D003-J; lanes g: D003-E.
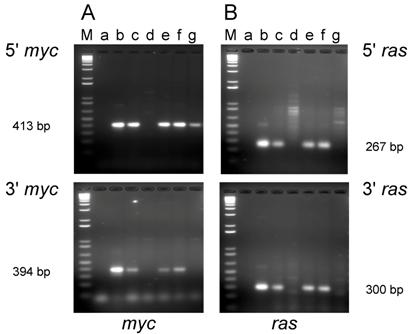
PCR analysis of the cell lines established from tumors induced in adult mice revealed that DNA from three out of the five lines (D003-A, D003-I, D003-J) produced PCR products that co-migrated with the products amplified from the control plasmids with both the 5' and 3' primer sets for both the c-myc and H-ras genes (Fig. 2A and B; lanes c, e, and f). DNA from the other two lines (D003-F, D003-E) either failed to yield the expected PCR product with both of the myc primer sets (D003-F)(Fig. 2A; lane d) or only yielded the expected product with the 5' myc primer set (D003-E)(Fig. 2A; lane g). DNA from these same two lines did not yield the expected ras gene PCR product with either the 5' or the 3' ras primers (Fig. 2B; lanes d and g).
The most likely reason for the absence of oncogene DNA in these two cell lines was that the tumor cells were selectively lost during the establishment of these cell lines from the tumors, possibly as a consequence of over digestion with the trypsin used to disperse the tumor tissue. If this explanation were correct, then DNA isolated directly from the tumor would be expected to carry the pMSV-T24-H-ras/pMSV-c-myc sequences. As shown in Fig. 3, PCR analysis of DNA isolated from tumors D003-E and D003-F were positive with both the 5' and 3' myc primers (Fig. 3A, lanes d and f, respectively) and with both the 5' and 3' ras primers (Fig. 3B, lanes d and f, respectively).
PCR analysis of DNA isolated from tumors and from cell lines established from the tumors induced by the dual-expression plasmids (pMSV-T24-H-ras/MSV-c-myc) in adult NIH Swiss mice. DNA was amplified by PCR using Oligos610/511 for the 5' myc gene and Oligos513/515 for the 3' myc gene and Oligos509/512 for the 5' ras gene and Oligos514/515 for the 3' ras gene. The PCR products were resolved by electrophoresis on a 1.8% agarose gel. A. The 5' myc and 3' myc gene. B. The 5' ras and 3' ras gene. Lanes M; marker; lanes a: no DNA control; lanes b: positive controls pMSV-T24-H-ras or pMSV-c-myc; lanes c: D003-E cell line; lanes d: D003-E tumor; lanes e: D003-F cell line; lanes f: D003-F tumor.
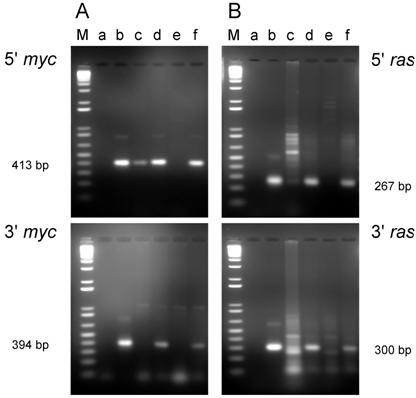
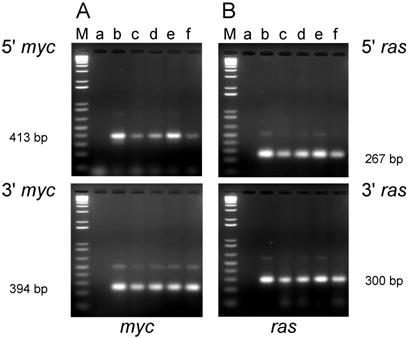
PCR analysis of DNA isolated from cell lines from tumors induced by the dual-expression plasmids (pMSV-T24-H-ras/MSV-c-myc) in newborn NIH Swiss mice. DNA was amplified by PCR using Oligos610/511 for the 5' myc gene and Oligos513/515 for the 3' myc gene and Oligos509/512 for the 5' ras gene and Oligos514/515 for the 3' ras gene. The PCR products were resolved by electrophoresis in a 1.8% agarose gel. A. The 5' myc and 3' myc gene. B. The 5' ras and 3' ras gene. Lanes M; marker; lanes a: no DNA control; lanes b: positive controls pMSV-T24-H-ras or pMSV-c-myc; lanes c: D004-B; lanes d: D004-F; lanes e: D004-I; lanes f: D004-K.
Expression of c-Myc and H-Ras proteins
If the c-myc and activated H-ras genes were inducing the tumorigenic phenotype in mouse cells, then the oncoproteins from these genes should be expressed in the tumor-cell lines. The expression of the c-Myc and H-Ras proteins was assessed using Western analysis of lysates prepared from the tumor-cell lines discussed above.
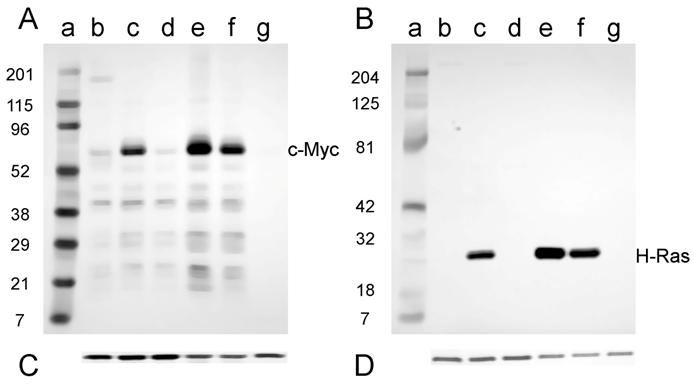
Expression of the c-Myc and H-Ras oncoproteins in cell lines established from tumors induced in adult NIH Swiss mice is shown in Fig. 5. The antibody to mouse c-Myc detected a protein of approximately 60 kD (D003-A, D003-I, and D003-J; Fig. 5A, lanes c, e, and f); this is the expected size for the c-Myc protein. Conversely, those cell lines that did not express c-Myc (D003-F and D003-E; Fig. 5A, lanes d and g) were the cell lines whose DNA also did not have amplifiable c-myc sequences (Fig. 2A). The less intense band visible in the samples prepared from line D003-F (Fig. 5A, lane d) is likely due to the endogenous c-Myc protein, as samples prepared from NIH 3T3 cells had a band of similar intensity and size (Fig. 5A, lane b). A similar correlation was found for H-Ras expression, viz., the cell lines that expressed the approximately 21 kD protein (D003-A, D003-I, and D003-J; Fig. 5B, lanes c, e, and f) and those that did not (D003-F and D003-E; Fig. 5B, lanes d and g) were those cell lines whose DNA either did, or did not, contain H-ras DNA as measured by PCR amplification (Fig. 2B). The absence of c-Myc or H-Ras bands was not a consequence of protein not being loaded, since the same membranes were probed with anti-actin antibody and similar amounts of that protein were detected in all lanes (Fig. 5C and D).
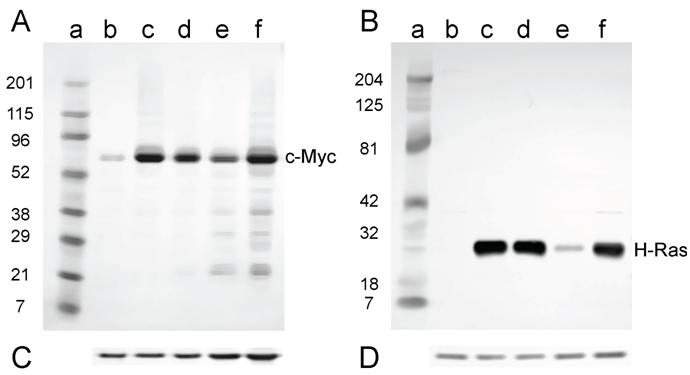
For the cell lines that were derived from tumors induced in newborn mice, all lines expressed both c-Myc and H-Ras proteins (Fig. 6A and B) as expected, since these lines contained both the c-myc and H-ras genes (Fig. 4A and B).
Integration patterns of the plasmid DNA
The integration pattern of the oncogene-expression plasmid pMSV-T24-H-ras/MSV-c-myc provides information on the number of copies and the sites of integration; this information can be used to determine whether the tumors were clonal. If the tumors were monoclonal, then a single cell would have been transformed, which would explain the low efficiency of tumor induction by DNA. In contrast, if the tumor were oligoclonal or polyclonal, then the efficiency of tumor induction would be correspondingly higher.
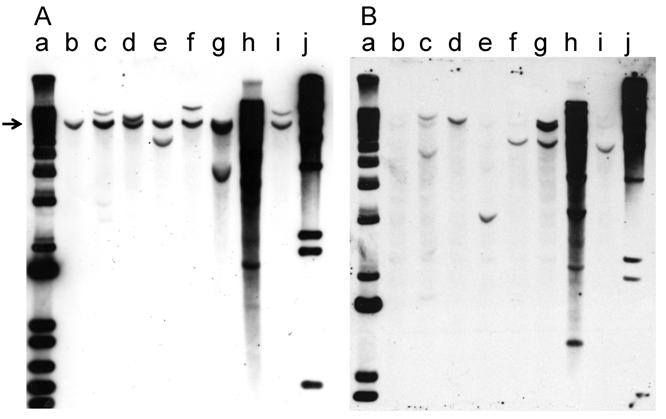
Southern hybridization was used to analyze the integration pattern of the oncogene-expression plasmid pMSV-T24-H-ras/MSV-c-myc. DNA prepared from the three PCR-positive cell lines derived from tumors induced in adult mice and the four cell lines derived from tumors induced in newborn mice was digested with BglII, an enzyme that cleaves pMSV-T24-H-ras/MSV-c-myc once. The digested DNAs were fractionated by electrophoresis on 1% agarose gels and the DNA was transferred to Hybond-N+ membranes. One membrane was hybridized with a c-myc probe (Fig. 7A), while the other was hybridized with an H-ras probe (Fig. 7B). The endogenous mouse c-myc gene was the only hybridizing band seen in NIH 3T3 DNA (Fig. 7A, lane b), and a corresponding band was seen in all lanes (indicated by the arrow in Fig. 7A). Most of the DNAs prepared from tumor-cell lines from tumors induced in adult mice (Fig. 7A, lanes c, d, e) or in newborn mice (lanes f, g, i) had one or two additional c-myc-hybridizing bands, demonstrating that each of these tumors was likely derived from a single oncogenic event and that the tumors were clonal. The exception was tumor-cell line D004-I (Fig. 7, lane h), which was derived from a tumor induced in newborn mice. This line had multiple copies of the c-myc plasmid sequences, and thus it is not possible to determine whether the original tumor was clonal.
With the H-ras probe, the endogenous mouse H-ras gene was less apparent in NIH 3T3 DNA (Fig. 7B, lane b), presumably because the probe was prepared from the human gene [32]. As seen with the analysis with the c-myc probe, in the analysis of the DNA from all but one line, the patterns of integration were simple, with one or two bands present. The exception was D004-I, whose DNA had multiple H-ras-hybridizing bands (Fig. 7B, lane h); DNA from this line also had multiple c-myc-hybridizing bands.
Because each of the tumor-cell lines had a different pattern of hybridization, the tumors were derived from different oncogenic events. Because the patterns of hybridizing bands in the Southern blots were simple in all but one case, most of the tumors were likely to be clonal, a conclusion also reached in our earlier study [32].
Western blot analysis of the expression of c-Myc and H-Ras in lysates of tumor cell lines derived from tumors induced in adult NIH Swiss mice. Lysates (2.5 µg for H-Ras and 30 µg for c-Myc) prepared from cell lines established from tumors were fractionated by SDS-PAGE (4-20% gel) and transferred to a PVD membrane. A. Antibody 9402 to c-Myc. B. Antibody C-20 to H-Ras. Lanes a: marker; lanes b: NIH 3T3; lanes c: D003-A; lanes d: D003-F; lanes e: D003-I; lanes f: D003-J; lanes g: D003-E. C. and D. The PVD membranes were reacted with the anti-actin antibody SC-1616; a band migrating at approximately 43 kD is seen in all lanes.
Western analysis for the expression of c-Myc and H-Ras in lysates of tumor cell lines derived from tumors induced in newborn NIH Swiss mice. Lysates (2.5 µg for H-Ras and 30 µg for c-Myc) prepared from cell lines established from tumors were fractionated by SDS-PAGE (4-20% gel) and transferred to a PVD membrane. A. Antibody 9402 to c-Myc. B. Antibody C-20 to H-Ras. Lanes a: marker; lanes b: NIH 3T3; lanes c: D004-B; lanes d: D004-F; lanes e: D004-I; lanes f: D004-K. C. and D. The PVD membranes were reacted with the anti-actin antibody SC-1616; a band migrating at approximately 43 kD is seen in all lanes.
Southern blot analysis of the integration pattern of the oncogenes in tumor cell lines. Tumor-cell-line DNA (8 µg) was digested with BglII, fractionated by electrophoresis on a 1% agarose gel, and analyzed by Southern hybridization. A. myc probe; B. ras probe. Lanes a: DNA size marker 1 kb ladder; lanes b: NIH 3T3; lanes c: D003-A; lanes d: D003-I; lanes e: D003-J; lanes f: D004-B; lanes g: D004-F; lanes h: D004-I; lanes i: D004-K; lanes j: DNA size maker lambda HindIII.
Assessment of transfection facilitators
If DNA uptake were a limiting step for the conversion of a normal mouse cell into a tumor cell, then increasing the efficiency of DNA uptake might increase the overall efficiency of tumor induction. We tested eleven commercially available reagents that have been widely used to increase transfection efficiency in vitro to determine if any of these reagents would increase the efficiency of tumor formation in vivo. These compounds represent most of the different types of transfection facilitators: liposomal (Lipofectamine, DMRIE-C, DOTAP, DOSPER, FuGene HD, and Transfectol); cationic liposomal (Effectene); cationic polymer (in vivo-JetPEI); polyamidoamine dendrimer (SuperFect and PolyFect); and a DNase inhibitor (aurintricarboxylic acid). Several of these facilitators had been used in animals to enhance the immunogenicity of DNA vaccines.
Because the total volume that we could safely inoculate into newborn mice was 50 µL, and because we wanted to use the ratio of facilitator to DNA recommended by the manufacturers to optimize the in vitro activity for their products, we were restricted to 2 µg of the plasmid pMSV-T24-H-ras/MSV-c-myc. DNA and facilitator were mixed, and each complex was inoculated via the subcutaneous route into newborn NIH Swiss mice. No tumors were found with any of the inoculations that included a transfection facilitator; one tumor was observed when DNA was inoculated with the DNase inhibitor aurintricarboxylic acid.
One possibility for this result was that the transfection facilitators stimulated the cellular immune system resulting in rejection of the tumor. In this case, then transfection facilitators might be expected to have activity in mice defective in cellular immunity. We assessed this possibility by testing a number of the facilitators in newborn athymic nude mice, because these animals have a defect in T-cell function. Two micrograms of pMSV-T24-H-ras/MSV-c-myc, either alone or complexed with DMRIE-C, in vivo-JetPEI, or SuperFect were inoculated into newborn athymic nude mice between 1 and 3 days after birth. Because each litter of the athymic nude mouse has pups that can be nude (T-cell defective) or haired (immune competent), the experiment had an internal control for whether the immune system played a role. The results showed that none of the facilitators increased the tumor incidence and that the nude mice and their haired littermates had comparable tumor incidences, indicating that tumor suppression by an active T-cell system did not explain the lack of activity of the transfection facilitators.
Discussion
Our previous experiments had shown that two cooperating oncogenes are needed to induce tumors in vivo. The results we present here show that, as expected, having both oncogenes on the same plasmid induces tumors more efficiently than having the two cooperating oncogenes on separate plasmids. The lowest amount of the dual-expression plasmid pMSV-T24-H-ras/MSV-c-myc that was able to induce tumors in newborn NIH Swiss mice was 1 µg. With adult mice, the lowest amount was 5 µg. This result agrees with our earlier finding with the separate expression plasmids [32], where newborn mice were found to be more sensitive to DNA-mediated tumor induction than adult mice.
Although the incidence of tumors induced by the three versions of the dual-expression plasmid pMSV-T24-H-ras/MSV-c-myc generally reflected the amount of DNA inoculated, there was no strict dose-response relationship, a result that was probably due to the relatively low number of animals used per group. This conclusion is supported if all three versions of the pMSV-T24-H-ras/MSV-c-myc plasmid are considered to be functionally indistinguishable (their in vitro transformation activities are the same) and the results for each DNA dose in Table 1 are combined. In this case, the incidence becomes: 20% for 20 µg, 21% for 10 µg, 13% for 5 µg, and 0% for 1 µg in the adult mouse, and 77% for 20 µg, 27% for 10 µg, 36% for 5 µg, and 7% for 1 µg in the newborn mouse. While there is still some variability, there is evidence of a dose-response relationship. Additional studies with larger numbers of mice are planned to address this experimental variability.
Establishing the clonality of the tumors was considered to be important. If the tumors were polyclonal, then they would be derived from the transformation of multiple normal mouse cells, and thus the efficiency of tumor induction by DNA would be higher than if the tumors were monoclonal and derived from a single cell transformed by DNA. With one possible exception, the integration patterns of the plasmid DNA in the tumors were consistent with a single cell being converted into a tumor cell. The exception was the D004-I (Fig. 7A and B, lanes h), where the Southern analysis showed multiple BglII fragments containing T24-H-ras or c-myc sequences, indicating that multiple plasmid sequences were taken up and integrated in cells in the tumor. Whether the oncogenes in D004-I were taken up by a single cell, in which case the tumor would be clonal, or by multiple cells, in which case the tumor would not be clonal, remains to be determined. In our earlier study [32], a tumor-cell line whose DNA gave rise to multiple restriction fragments that hybridized to the ras and myc probes was shown, by single-cell cloning, to be clonal.
This conclusion, that the tumors were mostly, if not always, clonal would only be true if there were no bias in which of the cells in the tumor gave rise to the established cell lines in culture. While the tumors were not analyzed in this study, we have shown in other studies that oncogene-containing cells in tumor-cell lines accurately reflect the population of oncogene-containing cells in the original tumors (unpublished observations).
One of the important practical findings from this study was that there could have been a selective loss of tumor cells when the tumor tissue was dispersed by trypsin digestion, thus resulting in the outgrowth of normal cells and the loss of tumor cells, as seen with two lines established from tumors induced in adult mice (Fig. 2). Whether tumor cells in these tumors are more sensitive to trypsin digestion than normal untransformed cells is not known, but we have recently found that cutting up the tumors into small pieces and allowing tumor cells to grow out from these explants in culture has resulted in a higher success rate in establishing tumor-cell lines.
An unexpected finding was that reagents that increase DNA uptake in vitro did not increase the efficiency of tumor induction DNA in vivo. In fact, the transfection facilitators tested appeared to have a negative effect. One possible explanation for this finding was that the facilitators stimulated the immune system and this immune activation caused a rejection of the tumor in the mouse. Because the cellular arm of the immune system is believed to be involved in tumor rejection, we assessed whether transfection facilitators would be effective in a mouse strain that is defective in T-cell function, the nude mouse. Although the numbers were small, no differences were found in the efficiency of DNA-induced tumor formation between the nude mice and their immune-competent littermates. This could indicate either that some part of the immune system other than the cellular arm is being stimulated or that DNA uptake is not limiting. Additional experiments will be necessary to explain the lack of activity of the transfection reagents in DNA-induced tumor formation.
The DNA oncogenicity assay described in this paper might provide a new way of measuring the activity of cellular or viral oncogenes in vivo without having to create cell lines expressing the oncogenes and then testing the tumorigenicity of these cells in either syngeneic animals or immune-deficient animals. For example, transfection of both activated H-ras and c-myc into normal rodent fibroblasts cells can convert these cells into cells that can form tumors in vivo [35-37]. However, direct inoculation of oncogenes into animals would circumvent the in vitro step, potentially revealing hitherto unknown activities of these oncogenes. Direct inoculation of oncogenes might also be used to evaluate potential differences in the induction of tumors by DNA in vivo compared with the formation of tumors by the injection of cells transformed in vitro.
This study confirms results of our earlier experiments showing that activated oncogenes from neoplastic cells are capable of inducing normal cells to become tumorigenic after direct injection into an animal [32], a finding that argues that the potential risk from the residual DNA in vaccines produced in neoplastic cells can no longer be considered hypothetical. However, because of the relative sizes of the oncogene expression plasmid and the mammalian genome, if 1 µg of the plasmid induces tumors, then 100 µg to 1 mg of mammalian DNA would be required. We are currently evaluating different strains of mice, such as mice with various degrees of immune deficiencies and mice predisposed to tumor development in response to carcinogens, for their sensitivity to DNA oncogenic activity in order to optimize assay sensitivity and more clearly define levels of risk posed by residual DNA in vaccines.
Acknowledgements
This work was initiated following support from a grant from the Office of the Commissioner, FDA. Additional support came from a grant from the National Vaccine Program Office; current work is supported by a contract from the Division of Microbiology and Infectious Diseases of the National Institute of Allergy and Infectious Diseases through an Interagency Agreement with CBER/FDA. F.C., H.F. and Y.Z. were supported by the NIAID contract. This work was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research (to S.H.H). JMC was a research Professor of the American Cancer Society, with support from the George Kirby Foundation. We thank Hana Golding, Arifa Khan, Jerry Weir, Phil Krause, and Robin Levis for discussions and/or comments on the manuscript.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Hilleman MR. Cells, vaccines, and the pursuit of precedent. Natl Cancer Inst Monogr. 1968;29:463-469
2. Hilleman MR. Line cell saga - an argument in favor of production of biologics in cancer cells. Adv Exp Med Biol. 1979;118:47-58
3. Petricciani JC, Horaud FN. DNA, dragons and sanity. Biologicals. 1995;23:233-238
4. Petricciani JC, Regan PJ. Risk of neoplastic transformation from cellular DNA: calculations using the oncogene model. Dev Biol Stand. 1987;68:43-49
5. Petricciani J, Loewer J. An overview of cell DNA issues. Dev Biol. 2001;106:275-282
6. Lewis AMJr, Krause P, Peden K. A defined-risks approach to the regulatory assessment of the use of neoplastic cells as substrates for viral vaccine manufacture. Dev Biol. 2001;106:513-535
7. Bishop JM, Baker B, Fujita D. et al. Genesis of a virus-transforming gene. Natl Cancer Inst Monogr. 1978;48:219-223
8. Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature. 1983;304:596-602
9. Santos E, Pulciani S, Barbacid M. Characterization of a human transforming gene isolated from T24 bladder carcinoma cells. Fed Proc. 1984;43:2280-2286
10. Bos TJ. Oncogenes and cell growth. Adv Exp Med Biol. 1992;321:45-99
11. Thompson TC, Southgate J, Kitchener G, Land H. Multistage carcinogenesis induced by ras and myc oncogenes in a reconstituted organ. Cell. 1989;56:917-930
12. Ledwith BJ, Manam S, Troilo PJ. et al. Plasmid DNA Vaccines: Investigation of Integration into Host Cellular DNA following Intramuscular Injection in Mice. Intervirology. 2000;43:258-272
13. Kurth R. Risk potential of the chromosomal insertion of foreign DNA. Ann N Y Acad Sci. 1995;772:140-151
14. Kurth R. Risk potential of the chromosomal insertion of foreign DNA. Dev Biol Stand. 1998;93:45-56
15. Doehmer J. Residual cellular DNA as a potential transforming factor. Dev Biol Stand. 1987;68:33-41
16. Löwer J. Risk of tumor induction in vivo by residual cellular DNA: quantitative considerations. J Med Virol. 1990;31:50-53
17. Temin HM. Overview of biological effects of addition of DNA molecules to cells. Dev Biol Stand. 1998;93:37-44
18. Griffiths E. Major issues associated with the use of cell substrates for the production of vaccines. Dev Biol. 2001;106:25-35
19. Barrett JC. Mechanisms of multistep carcinogenesis and carcinogen risk assessment. Environ Health Perspect. 1993;100:9-20
20. Fearon ER. Genetic alterations underlying colorectal tumorigenesis. Cancer Surv. 1992;12:119-136
21. Hennings H, Glick AB, Greenhalgh DA. et al. Critical aspects of initiation, promotion, and progression in multistage epidermal carcinogenesis. Proc Soc Exp Biol Med. 1993;202:1-8
22. Pitot HC. The molecular biology of carcinogenesis. Cancer. 1993;72:962-970
23. Hesketh R. The Oncogene and Tumour Suppressor Gene FactsBook. Second Edition. London: Academic Press. 1997
24. Kondoh H, Lleonart ME, Gil J. et al. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177-185
25. Davé UP, Jenkins NA, Copeland NG. Gene therapy insertional mutagenesis insights. Science. 2004;303:333
26. Costinean S, Zanesi N, Pekarsky Y. et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A. 2006;103:7024-7029
27. Dalmay T. MicroRNAs and cancer. J Intern Med. 2008;263:366-375
28. Fabbri M, Croce CM, Calin GA. MicroRNAs. Cancer J. 2008;14:1-6
29. Voorhoeve PM, Agami R. Classifying microRNAs in cancer: the good, the bad and the ugly. Biochim Biophys Acta. 2007;1775:274-282
30. Wiemer EA. The role of microRNAs in cancer: no small matter. Eur J Cancer. 2007;43:1529-1544
31. Cowland JB, Hother C, Gronbaek K. MicroRNAs and cancer. Apmis. 2007;115:1090-1106
32. Sheng L, Cai F, Zhu Y. et al. Oncogenicity of DNA in vivo: Tumor induction with expression plasmids for activated H-ras and c-myc. Biologicals. 2008;36:184-197
33. Kohl NE, Ruley HE. Role of c-myc in the transformation of REF52 cells by viral and cellular oncogenes. Oncogene. 1987;2:41-48
34. Ruley HE. Transforming collaborations between ras and nuclear oncogenes. Cancer Cells. 1990;2:258-268
35. Land H, Chen AC, Morgenstern JP, Parada LF, Weinberg RA. Behavior of myc and ras oncogenes in transformation of rat embryo fibroblasts. Mol Cell Biol. 1986;6:1917-1925
36. Small MB, Hay N, Schwab M, Bishop JM. Neoplastic transformation by the human gene N-myc. Mol Cell Biol. 1987;7:1638-1645
37. Zerlin M, Julius MA, Cerni C, Marcu KB. Elevated expression of an exogenous c-myc gene is insufficient for transformation and tumorigenic conversion of established fibroblasts. Oncogene. 1987;1:19-27
Author contact
![]() Corresponding authors: Keith Peden, Building 29A, Room 3D08, or Andrew Lewis, Building 29A, Room 1B21, CBER, FDA, 29 Lincoln Drive, Bethesda, MD 20892. Telephone: 301 827 1708 (KP); 301 827 0650 (AL); FAX: 301 496 1810; E-mail: keith.pedenhhs.gov; andrew.lewishhs.gov
Corresponding authors: Keith Peden, Building 29A, Room 3D08, or Andrew Lewis, Building 29A, Room 1B21, CBER, FDA, 29 Lincoln Drive, Bethesda, MD 20892. Telephone: 301 827 1708 (KP); 301 827 0650 (AL); FAX: 301 496 1810; E-mail: keith.pedenhhs.gov; andrew.lewishhs.gov
Received 2010-2-18
Accepted 2010-3-22
Published 2010-3-29