ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2010; 6(3):199-212. doi:10.7150/ijbs.6.199 This issue Cite
Research Paper
Uncoupling oxidative/energy metabolism with low sub chronic doses of 3-nitropropionic acid or iodoacetate in vivo produces striatal cell damage
E Rodríguez1, I Rivera1, S Astorga1, E Mendoza1, F García2, E Hernández-Echeagaray1 ![]()
1. Unidad de Biomedicina, FES-I, Universidad Nacional Autónoma de México. Av. de los Barrios # 1, Los Reyes Iztacala, C.P. 54090, Tlalnepantla, México.
2. Instituto Nacional de Pediatría, Insurgentes Sur 3700, C.P. 04530, México, D.F.
Abstract
A variety of evidence suggests that the failure of cellular metabolism is one of the underlying causes of neurodegenerative diseases. For example, the inhibition of mitochondrial function produces a pattern of cellular pathology in the striatum that resembles that seen in Huntington's disease. However, neurons can also generate ATP through the glycolytic pathway. Recent work has suggested a direct interaction between mutated huntingtin and a key enzyme in the glycolytic pathway, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Yet little work has been gone into examination of the cellular pathology that results from the inhibition of this alternative energy source. Therefore, the aim of the present study is to characterize the cellular pathology that results in the striatum of mice after treatment with a toxin (iodoacete, IOA) that compromises anaerobic metabolism. This striatal pathology is compared to that produced by a widely studied blocker of mitochondrial function (3-nitropropionic acid, 3-NP).
We found that low doses of either toxin resulted in significant pathology in the mouse striatum. Signs of apoptosis were observed in both experimental groups, although apoptosis triggered by IOA treatment was independent from caspase-3 activation. Importantly, each toxin appears to produce cellular damage through distinct mechanisms; only 3-NP generated clear evidence of oxidative stress as well as inhibition of endogenous antioxidants. Understanding the distinct pathological fingerprints of cell loss produced by blockade of oxidative and anaerobic metabolisms may give us insights into neurodegenerative diseases.
Keywords: Huntington's disease, neurotoxins, 3-NP, IOA, apoptosis, neurodegeneration, lipoperoxidation.
Introduction
Failure in energy metabolism has been suggested as one of the most important mechanisms underlying neural degeneration. Indeed, neurons are exposed to oxidative damage due to their high O2 consumption to generate the ATP needed to maintain all neuronal functions [1]. Disruption of mitochondrial function in striatal tissue can produce cellular damage resembling that observed in postmortem tissue obtained from patients afflicted by HD [2-4]. GAPDH, an enzyme related to anaerobic metabolism, has also been implicated in neuronal degeneration [5, 6]. A reduction in GAPDH activity has been demonstrated in HD patients, in transgenic mouse model of HD [7] and intrastriatal inhibition of GAPDH produces cell death [8]. In addition to its glycolytic activity, GAPDH is implicated in other cellular functions, for example its inhibition induces the activation of apoptotic pathways and changes in transcription regulation [9-11].
Effects of glycolysis inhibition on striatal cell death have not been well explored or compared with damage produced by the inhibition of oxidative phosphorylation. In addition, it is unclear if metabolic uncoupling is associated with only necrosis or apoptosis, if necrotic and apoptotic death are both present, or if other types of cell death such as autophagy are happening. In fact, one of the unsolved problems in striatal degeneration observed in disorders like HD is the precise identification of the cellular process that produces neuronal death. Some authors have proposed that cells die through a necrotic process [12, 13]; still others have suggested that neuronal death is apoptotic [14].
Irreversible or reversible inhibition of mitochondria by 3-NP and malonate respectively, has been used to uncover cellular mechanisms underlying striatal degeneration. These compounds reproduce many of the histopathological and neurochemical distinctiveness of HD in animal models [15-17]. In fact, the use of 3-NP has provided some insights related to impairment in the oxidative metabolism in rodents, although experimental data regarding the process that conduces to neuronal death are still contradictory [18]. Neuronal damage produced by 3-NP administration can be the consequence of excitotoxicity [19], and calpain activation leading to necrotic death [20, 21]. However, several studies show that the blockade of oxidative metabolism induces apoptosis [22, 23].
Both oxidative phosphorylation and glycolysis can be involved in the pathogenesis of neural degeneration in the striatum. However, the type of cellular damage induced will depend on several experimental factors, namely, if the study was carried out in vitro or in vivo, whether rats or mice were used, the experimental procedure involving intrastriatal, or intraperitoneal injections, acute or chronic drug administration, and if low, sub toxic or toxic concentrations were used [4].
The aim of this study was to evaluate and compare neuronal damage induced in C57BL/6 mice, after blocking energy metabolism with sub toxic doses of 3-NP (which blocks oxidative phosphorylation) or IOA (which inhibits glycolysis). We selected mice of ~1 month of age at the beginning of the experimental treatment to be able to compare with other animal models where early striatal dysfunction starts at this age.
Materials and Methods
All experiments were carried out following the international regulations of Animal Care. C57BL/6 mice of 30 days old (Harlan, México) were housed in plastic cages, in groups of 2, kept at 20°C with a 12/12 light/dark cycle with free access to food and water. Two days after their arrival, animals were assigned to one of the following 3 groups: Experimental groups receive 3-NP (15 mg/kg), Iodoacetate (10 mg/kg) or vehicle (Phosphate Buffer; PB 0.01M; pH 7.4) for 5 days using intraperitoneal injections. After experimental treatments, mice were assigned at random, to perform IHC, histochemical, electron microscopy, or biochemical analysis.
Evaluation of motor disturbances: Motor disturbances were evaluated everyday during the treatment period and 24 hours after the last treatment, they were classified in terms of the presence of dystonia and gait abnormalities. Dystonia classification consisted of 0 = none, 1 = intermittent dystonia of one limb, 2 = intermittent dystonia of two limbs or permanent dystonia of one limb, 3 = permanent dystonia. Gait anormalities; 0= none, 1= uncoordinated and wobbling gait.
Haematoxylin and Eosin (H&E): To evaluate cellular damage, 3 mice per group were euthanized with halothane and perfused through the heart with saline solution (NaCl 0.15 M) followed by paraformaldehyde (4%) in phosphate buffer (PB, 0.1M, pH 7.4), brains were dissected and post-fixed in the same fixative for two hours at room temperature, and then transferred to a sucrose solution (20%). Coronal slices (20 μm) were obtained with a microtome (Leica, Germany) and mounted in glass slides for staining with conventional technique for H&E (H&E kit 75290, BIOCARE Medical; Concord, CA). Then tissue was covered with Cytoseal®.
Immunohistochemistry (IHC): Caspase-3 and caspase-9 expression was documented by IHC. The IHC procedure was carried out as previously described [24]. Briefly, coronal tissue sections (20 μm), adjacent to those used for H&E were incubated at 4ºC in blocking serum overnight. Thereafter, tissue sections were incubated for 24 hr in primary antibodies (Chemicon, International, Inc, Temecula CA, USA) raised against caspase-3 (1:100, AB3623 Polyclonal Antibody), caspase-9 (1:500, MAB4709 monoclonal antibody) or GAPDH (1:100, MAB374 Monoclonal Antibody), all diluted in blocking buffer. After rinse, tissue slices were incubated with the corresponding secondary antibodies (1:500) coupled to biotin and diluted in blocking solution. The AB Vector kit (Vector Laboratories, Inc. Burlingame, CA, USA) was used to detect biotinylated antibodies following the manufacturer's instructions. Tissue slices were mounted and cover slipped with mounting medium (Fisher Scientific, Pittsburgh, PA, USA). Control experiments were carried out by pre-absorbing the primary antibodies with their corresponding antigens omitting the primary antibody or the primary and secondary antibodies.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay: Tissue slices adjacent to those used for evaluating Caspases immunoreactivity were used to detect DNA fragmentation by TUNEL assay, following supplier instructions (Apoptosis detection kit, TA300, R&D Systems, Minneapolis, MN, USA). To finish, tissue was rinsed, dried and coverslipped with Permount (Fisher Scientific, Pittsburgh, PA, USA). TUNEL-labeled cells which displayed densely labeled, small particles in the cytoplasm (apoptotic bodies), and different types of chromatin condensation around the margin of the nucleus forming either crescent caps or rings, were quantified per field by light microscopy.
Cell counting: To count damaged cells, positive cells to IHC procedures and TUNEL assay, images were obtained and digitized using an image analysis system (Metamorph 4.0, Universal Imaging) connected to a microscope equipped with a video camera coupled to a computer monitor. The analysis consisted of obtaining squared probes of 1mm2 in 40X magnification to determine damaged or immunostained cells. The number of cells was obtained as an average of 3 fields per section, 10 sections per brain and 3 mice per experimental group.
Electron microscopy: To evaluate ultrastructural changes 3 animals of each experimental group were perfused with paraformaldehyde 4% and glutaraldehyde 2.5 % in phosphate buffer (0.1M, pH 7.4). Neostriatum was dissected, post fixed for 3 hours and cut in small segments. Later on, tissue slabs were rinsed and included in epon resin to obtain ultrathin section (60 nm). Slices were visualized with the aid of electron microscopy and micrographs were taken for further analysis. Sections were subject to blind examination regarding the experimental protocol and scored as follows: Vacuolization: 0 = none, 1 = mild, 2 = moderate and 3 = severe.
Ribonucleic acid (RNA) extraction and Reverse Transcription-Polymerase Chain Reaction (RT-PCR): Total RNA was isolated from 0.5 g samples of frozen striatal tissue (3 mice brains from each experimental condition), and the extraction was carried out with the TRIzol reagent method using the Total RNA Isolation Reagent (Invitrogen, Life Technologies, Inc.). RNA concentration was calculated from the optical density at 260 nm, and purity was determined by 260nm /280nm absorbance.
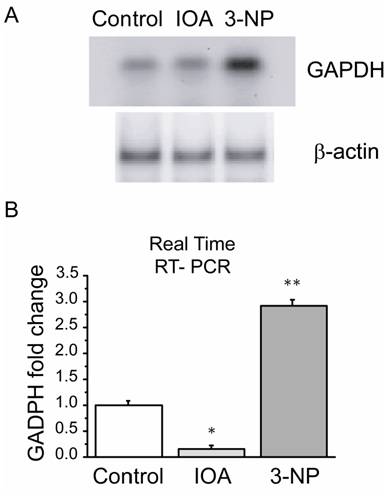
Quantitation of gene transcripts by real-time RT-PCR analysis: Total RNA (~300ng) from each sample was reverse transcribed (RT) using oligo(dT)12-18 primer and SuperScript I RT kit (Life Technologies), and 1 μg/μl RNA for 50 min at 42°C. Quantitative real-time PCR was performed using SYBR Green 1 μl, PCR buffer 1X 2.5 μl, 50 mM MgCl2 1 μl, 100 μM dNTPs 1 μl, 1 unit platinum Taq DNA polymerase, 50 μmol/μl gene-specific forward and reverse primers, and 50 ng cDNA. The reaction conditions were as follows: 4 min at 94°C (denaturalization), 1 min at 94°C, 1 min annealing temperature, and 1 min at 72°C (30 or 35 cycles). Gene-specific PCR products for GAPDH were continuously measured by a Rotor Gene RG 3000 detection system (Corbett Research, Australia). Three replicates for each experimental sample were performed, and differences were assessed with the 2_ΔΔCT method [25]. Using the 2_ΔΔCT method, the data are presented as the fold change in gene transcripts normalized to the housekeeping gene as one by definition [26]. Then computed analysis was used to reflect the relative mRNA transcript levels to demonstrate the fold change of GAPDH gene transcripts in C57BL /6 mice that received either toxin compared with the expression of β-actin gene. The sequences of the specific primers and alignment conditions used are listed in Table 1. Data are presented as the fold change.
Primer sequences Real Time PCR
| Gene | Primer (5'-3') | Alignment (°C) | Product size (pb) |
|---|---|---|---|
| β -actin | F-gTg gC CgC TCT Agg CAC CAA | 56°C | 536 pb |
| R-CTC TTT gAT TgC ACg CAC gAT TTC | |||
| GAPDH | F-CTC ATg ACC ACA gTC CAT gC | 54°C | 201 pb |
| R-CAC ATT ggg ggT Agg AAC AC |
Evaluation of nitric oxide (NO) levels
NO: NO production was evaluated indirectly by measuring nitrates in serum. Blood samples from treated (3-NP, n=5; IOA, n=5) and control (n=5) mice were incubated for about 15 minutes at 37°C, afterward the blood was centrifuged (12000 rpm) for 15 minutes. To quantify nitrates, 0.1% of napftiletilendiamine dihydrochloride was mixed in a plate of 96 wheels, with serum samples (100 µl, different concentrations) and 75µl of Griess reagent, incubated at room temperature for 10 min, and measured in 550 nm-620 nm in an ELISA lector. Absorbance spectrum of the colored azo compounds was quantified using serial dilutions of sodium nitrite as standard reference curve. Data are expressed as absorbance units.
Evaluation of lipid peroxidation
LPO: Lipid peroxidation was measured by the fluorescence of lipid peroxidation products, briefly striatal tissue from control (n=5), 3-NP (n=5) and IOA (n=5) treated mice was weighed and homogenized in 3 ml of saline solution (0.9% NaCl). Aliquots of 1 ml were added to 4 ml of a chloroform-methanol mixture (2:1 v/v). After stirring, the mixture was kept on ice (30 min) to permit phase separation and then, the fluorescence of the chloroform layer was measured in a Perkin-Elmer LS50B luminescence spectrometer (excitation 370 nm and emission 430 nm wavelengths). Results are expressed as fluorescent units/g of wet tissue.
Evaluation of antioxidant enzymes activation
Five mice per group were used to evaluate the activity of antioxidant enzymes
CAT activity: Catalase activity was determined at 25ºC analyzing the decomposition of H2O2 used as the substrate of the enzyme contained in the homogenized striatal sample [27]. Kinetics was analyzed by the equation, k= 2.3/t log Ao/A where k was the first point and t was the time when H2O2 was reduced by catalase activity. Results are expressed in k/mg of protein.
GPx activity: 30 μl of homogenized striatal tissue (control n=5, 3-NP, n=5; IOA, n=5) was incubated in the following solution: 0.5ml glutathione (GSH; 2.0 mM), 0.5 ml Na phosphate buffer (0.40 M, pH 7) containing EDTA (4 x 10-4 M), 0.25 ml NaN2 (0.1M) in water. After 5 min at 37 ºC, 0.1 ml H2O2 (0.2 mM) was added. Then, 1 ml of metaphosphoric acid was added and centrifuged (3500 rpm, 15 min). Free GSH was determined in 1ml of the centrifuged solution in 1ml of PB, 0.5 ml of 5, 5-di-tiobis-2-nitrobenzoic (DTNB). Absorbance was measured at 412 nm in a U.V. spectrophotometer (Lamda 20, Perkin-Elmer). The decrease in absorbance was directly proportional to the GPx concentration.
GSR activity: Glutathione reductase activity was measured by the method of Carlberg and Mannervik [28]. The enzyme activity was quantified at 25 °C by measuring the disappearance of NADPH at 340 nm. Optical density was recorded at 4 minutes and data expressed in mU/mg of protein.
Total SOD activity: The method used for determining SOD activity was based on nitroblue tetrazolium (NBT) reduction [29]. Xanthine-xanthine oxidase was utilized to generate a superoxide flux in the striatal sample (control, 3NP or IOA). The absorbance obtained from NBT reduction to blue formazan by superoxide was determined spectrophotometrically (560 nm, BEKMAN DU-64) at room temperature. One unit (U) of SOD activity was defined as the amount that reduced the absorbance change by 50%, and results were normalized on the basis of total protein content (U/mg protein) in each sample.
Copper-zinc-superoxide dismutase (CuZn-SOD) activity: It was differentiated from manganese superoxide dismutase (Mn-SOD) by the addition of 2 mM sodium cyanide, to inhibit the activity of CuZn-SOD. CuZn-SOD activity was calculated as the difference between total SOD and Mn-SOD activity. Data were normalized on the basis of total protein content and expressed as U/mg protein.
Statistical Analysis: Damage induced by 3-NP treatment in mice varies from mouse to mouse; also there are no studies carried out in vivo analyzing striatal damage in the presence of low doses of IOA [30], then to visualize all data distribution with our experimental conditions, data are presented in box plots unless otherwise indicated. Experiments on data normally distributed were compared using ANOVA followed by Bonferroni post hoc tests. Kruskall-Wallis One Way Analysis Of Variance followed by Dunnet post hoc comparisons were used if the normality test failed. The statistical analysis was conducted using SigmaStat statistical software for Windows, version 2.03 (Systat Software, Inc.) and p<0.05 was considered to be statistically significant.
Results
Motor disturbances in 3-NP and IOA treated animals
Treating animals with 15 mg/kg of 3-NP or 10 mg/kg of IOA for 5 days did not produce gait abnormalities or dystonia during the treatment period. In pilot experiments higher doses of 3-NP and IOA (20 mg/kg and 15 mg/kg, respectively), were utilized and motor disturbances were observed, but high animal mortality was also obtained (~60 %, data not shown). Lower doses of 3-NP (10mg/kg) or IOA (5mg/kg) did not produce tissue damage in comparison to control group. A combination of 3-NP and IOA together was given in some mice, but they died the following day. For those reasons, selected drug concentration were 15 (3-NP) and 10 (IOA) mg/kg administrated for 5 days, and only one toxin was administered per animal group or the drug vehicle to control mice.
Cellular damage in 3-NP and IOA treated animals
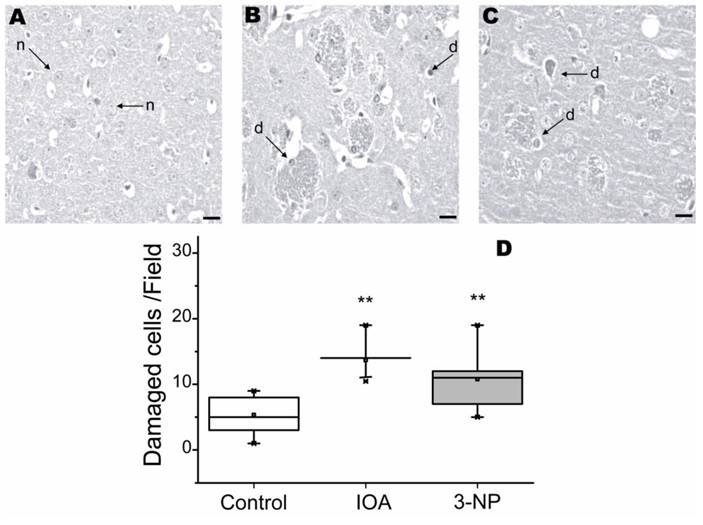
To evaluate striatal damage, tissue slices were stained with H&E histochemical reaction. Figure 1, shows cellular damage visualized with H&E. Tissue from control animals exhibited striatal neurons with normal (n) appearance (Fig. 1A). IOA and 3-NP treated mice, displayed significantly more damaged cells (Fig. 1A-D) than control tissue (Kruskal-Wallis, One Way Analysis of Variance on Ranks, H2=14.232, p<0.001). Multiple comparisons carried out with Dennett's method, revealed significant differences between each experimental group and the control group (IOA vs. control q=3.72, p<0.05; 3-NP vs. control q=2.287, p<0.05).
TUNEL assay in 3-NP and IOA treated animals
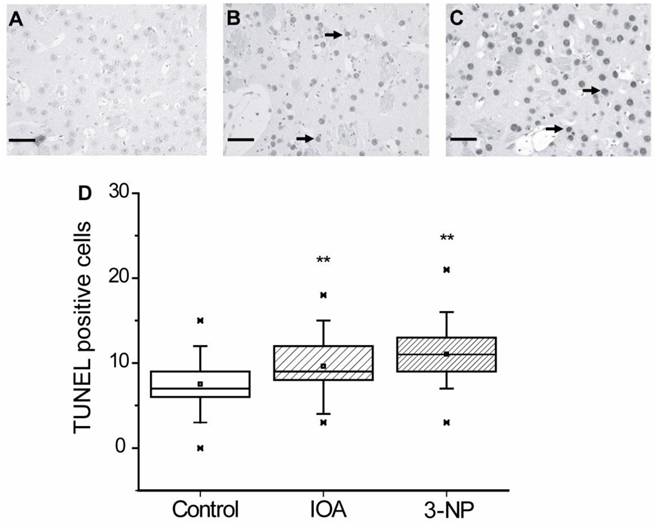
Nuclei alterations may be related to apoptotic damage, therefore TUNEL assay was carried out to evaluate if apoptosis was induced in our experimental manipulations. Figure 3 illustrates representative images obtained from brain slices of control (Fig. 3A), IOA (Fig. 3B) and 3-NP (Fig. 3C) treated mice. Figure 3D shows that both, IOA (28 %) and 3-NP (57 %) treatments significantly increased TUNEL positive cells, in comparison with control tissue (Kruskal-Wallis One Way Analysis of Variance on Rank, H2= 53.010, p<0.001, Multiple Comparisons versus Control Group Dennett's method, q'=4.283 (IOA) and q'=7.206 (3-NP), p<0.05).
Light micrographs of H&E stained striatal tissue of control (A), IOA (B), or 3-NP (C) treated mice. No histological abnormalities were present in the tissue from control brains and neurons exhibited normal (n) appearance, arrows point to normal cells (A). Striatal tissue from IOA treated mice exhibited some healthy cells, but there are several damaged cell (d) with condensed and pyknotic nuclei (B). Striatal tissue from mice treated with 3-NP exhibited damaged cells (d) with condensed and pyknotic nuclei and different degrees of neurodegeneration (C). Box plots represent Median, 25% and 75% of the damaged cells in each group (D) and demonstrated that IOA and 3-NP treated animals showed more damaged cells than control group (**p<0.001). Scale bar is 20 μm and magnification was 40X.
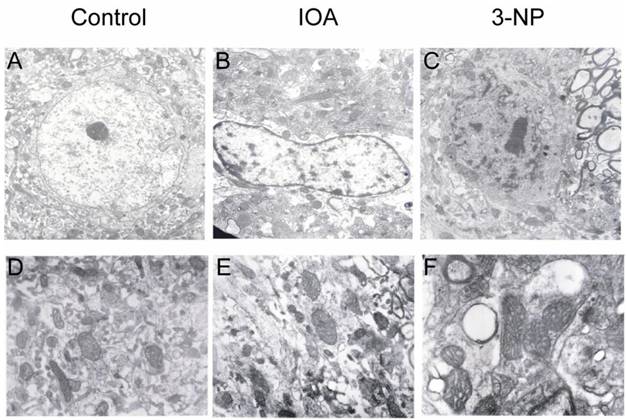
Electron microscopy analysis of cellular damage. A and D exhibit electron micrographs of control tissue. Cells show well defined nuclei with normal appearance (A) and mitochondria with normal membrane and clear crest (D). Tissue from IOA treated mice (B) shows altered nuclei; chromatin is oriented towards nuclear membrane, mitochondria are swollen and intracellular edema can be observed (E). Tissue of 3-NP treated mice (C) was characterized by altered nuclei and chromatin oriented to nuclear membrane. Nuclear membrane is also damaged. Prominent vacuoles are observed (F), as well as swollen mitochondria. Magnification in A, B and C is 7000X, and in D, E, and F is 12000X.
Light micrographs of TUNEL positive cells in the striatal tissue of control (A), IOA (B), or 3-NP treated mice (C). Tissue of IOA treated mice (B) as well as 3-NP treated tissue, exhibited more apoptotic cells (arrows) than control tissue (**p<0.001). Box plots expressed data in Medians, 25% and 75% of stained cells. Pictures were taken at 40X magnification. Scale bar ix 40 μm.
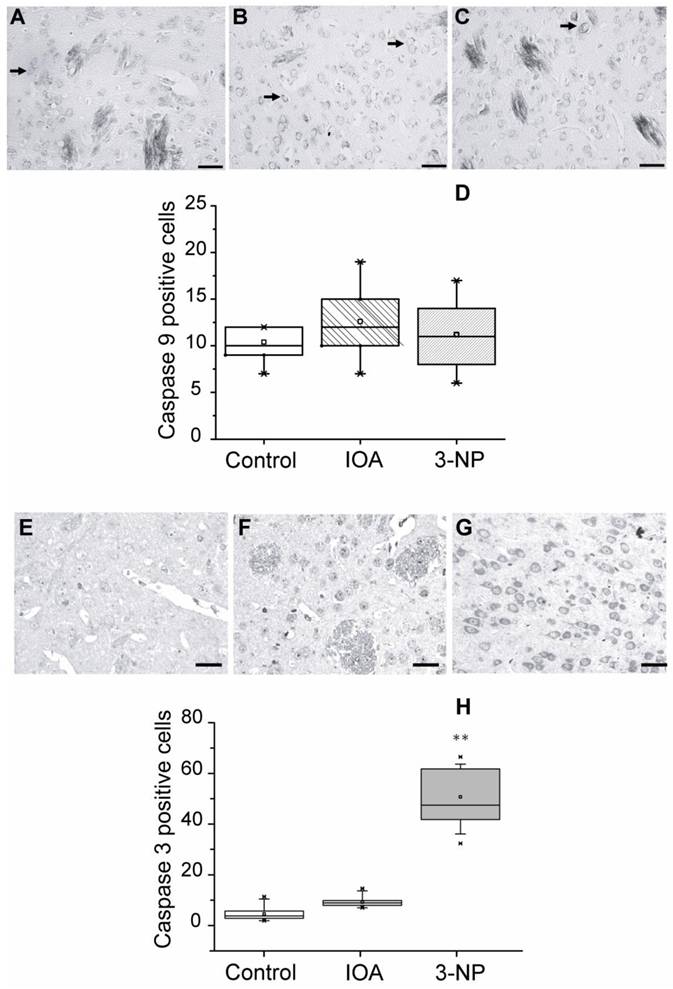
Light micrographs of Caspases 9 and 3 immunohistochemical localization in the striatal slices of control (A), IOA (B), or 3-NP treated mice (C). (Top) Analysis of the number of positive cells for caspase-9 showed no statistically significant changes among groups (D). (Bottom) Caspase-3 immunohistochemical localization in the striatal tissue of control (E), IOA (F), or 3-NP treated mice (G). Analysis of the number of positive cells for caspase-3 increased significantly in the 3-NP treated group (**p<0.001, H). Box plots expressed data in Medians, 25% and 75% of stained cells. Pictures were taken at 40X magnification. Scale bar is 30 μm.
Caspases activation in IOA and 3-NP treated animals
Apoptosis can be triggered by caspases activation or by other intracellular mechanisms; we estimated caspase-9 and caspase-3 involvement in the apoptotic damage produced by 3-NP and IOA, by means of IHC procedures in striatal slices adjacent to those used for TUNEL assay. Figure 4 (top) shows representative images of immunostained tissue for Caspase-9 from control (Fig. 4A), IOA (Fig. 4B), and 3-NP (Fig. 4C) treated animals. There were no significant differences in the number of positive cells for caspase-9 between groups (Kruskal-Wallis One Way Analysis of Variance on Rank, H 2= 2.499, P=0.287, Fig. 4 D). Figure 4 (bottom) illustrates representative images obtained from slices of control (Fig. 4E), IOA (Fig. 4F), and 3-NP (Fig. 4G) treated animals stained with caspase-3 antibody. Figure 4H shows that the number of positive cells for caspase-3 significantly increased in the 3-NP group (Kruskal-Wallis One Way Analysis of Variance on Rank, H2= 54.275, P <0.001, Multiple Comparisons versus Control Group, Dennett's Method, q' =6.661, P<0.05), but not in IOA treated animals (q' =0.6671, p>0.05).
Expression of mRNA GAPDH by Polymerase Chain Reaction (PCR) in IOA and 3-NP treated animals
GAPDH enzyme has been suggested as a multifunctional protein whose role is wider than that related to glycolysis, its deregulation has been linked to apoptosis induction and its expression may also be modified by altering energy and/or oxidative metabolism. Consequently, mRNAGAPDH expression levels were evaluated and compared in both treatments. Real time RT-PCR showed differences in the expression levels of mRNAGAPDH (F2= 85.894, p=0.002, Fig. 5B) between groups. There was a significant increase in mRNAGAPDH levels in the 3-NP treated group when compared to control (t= 8.773, p=0.009; Bonferroni`s t-test) and IOA treated groups (t=12.820, P=0.003, Bonferroni`s t-test). IOA treated mice striatal tissue also exhibited differences in comparison to control group (t=5.033; p=0.045; Bonferroni`s t-test).
Expression levels of striatal mRNAGAPDH by PCR. (A) PCR products of GAPDH in control, IOA and 3-NP treated groups stained with SYBR green on a 2 % agarose electrophoresis gel. (B) Quantitative PCR analysis of mRNAGAPDH levels from mice striatum of control, IOA and 3-NP treated groups displayed significant differences in the gene expression among groups (**p<0.002).
Estimation of NO levels in IOA and 3-NP treated animals
Inhibition of energy metabolism and oxidative phosphorylation may trigger an increase in NO levels, which in turn induces neuronal damage due to oxidative stress. Subsequently, to investigate if NO was involved in neuronal damage in Fig.1 and Fig. 2 NO presence was evaluated indirectly by measuring nitrite production. Levels of NO changed in IOA (48%) and in 3-NP treated animals (34 %) when compared with control group (Fig. 6A); however, those changes were not statistically significant (One Way Analysis of Variance, F2= 3.117, p=0.074).
Evaluation of NO and LPO activity. (A) Displays NO levels in control, IOA and 3-NP treated groups. No statistical differences were obtained among groups (p>0.074). (B) Displays LPO in experimental groups, 3NP treated group exhibited a significant increase in LPO levels (**p<0.001).
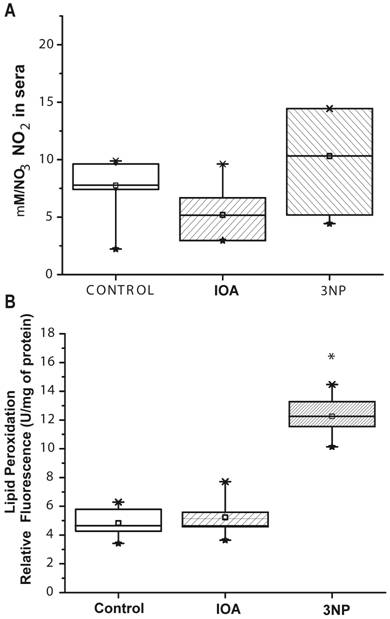
Estimation of LPO in IOA and 3-NP treated animals
Increase of reactive oxygen species (ROS) in cells produces lipid peroxidation (LPO); LPO presence is associated with an increase in NO, for that reason LPO levels were evaluated. Our evaluation showed significant changes in LPO among groups (One Way Analysis of Variance, F2=58.507, p<0.001). Striatal tissue taken from IOA treated mice did not differ in LPO levels from control mice (t=0.521, p=1.00; Multiple comparisons, Bonferroni´s t-test), however striatal tissue from 3-NP treated animal (Fig. 6B) displayed significant differences between control (t=9.618, p<0.001; Multiple Comparisons, Bonferroni´s t-test) and IOA treated group (t=9.097, p<0.001, Multiple Comparisons, Bonferroni´s t-test).
Estimation of antioxidant enzymes activities
Mechanisms of cellular protection were evaluated by measuring enzymatic activities of Catalase (CAT), Glutathione Peroxidase (GPx), Glutathione Reductase (GSR), total SOD, CuZn-SOD, and Mn-SOD after toxins treatment period to better understand the process underlying metabolic uncoupling.
Catalase
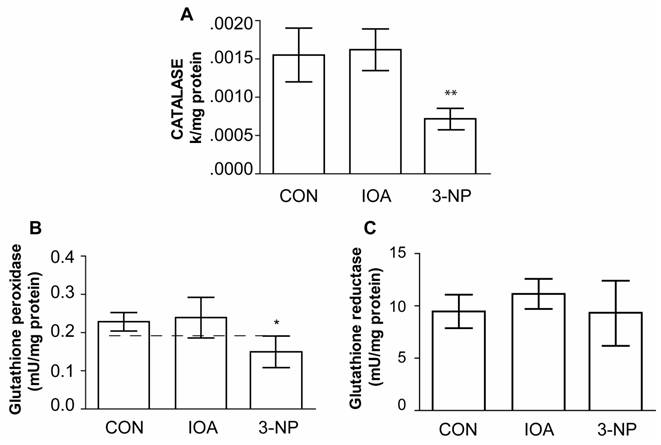
Figure 7 A, shows that Catalase activity was significantly different (One Way Analysis of Variance, F2=29.778, P<0.001) in the striatum of 3-NP treated animals in comparison to control mice (control vs. 3 NP, t=5.689, p<0.001); while IOA treated mice did not exhibit statistical changes in comparison to control group (control vs. IOA, t=0.441, p=1.000; Bonferroni`s t-test).
Glutathione peroxidase
GPx activity was statistically different among evaluated groups (F2=8.361, P<0.004, Fig. 7B), and the difference was observed between 3-NP vs control (t=2.922, p=0.021, Bonferroni`s t-test) but not between IOA and control groups (t=0.368; P=1.000; Bonferroni`s t-test).
Glutathione reductase
As GPx decreased in the striatal tissue of 3-NP treated mice, we hypothesized that GSR activity may also be diminished. However, there were no statistical differences in the GSR activity (F2=1.196, p=0.330) among evaluated groups (Fig. 7C).
Evaluation of CAT, GPx, and GSR activity. Activity of CAT (A), GPx (B) and GSR (C) is illustrated, IOA did not change any of the enzymes activity, while 3-NP significantly reduced CAT (** p<0.001, A) and GPx (*p< 0.004, B) activity levels.
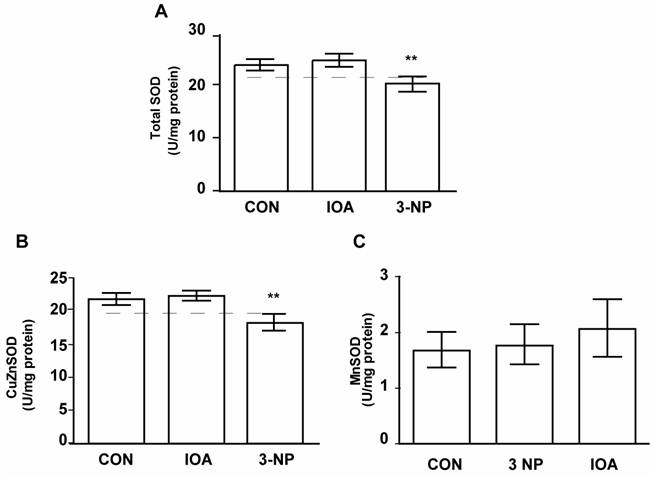
Evaluation of total SOD, CuZn-SOD, and MnSOD activity. Activity of Total SOD (A), CuZn-SOD (B) and MnSOD (C) is displayed, IOA did not change any of SOD enzymes activity, while 3-NP significantly reduced Total SOD(** p<0.001, A) and CuZn-SOD (** p< 0.001, B) activity levels.
Superoxide dismutase (SOD)
The other antioxidant enzyme which catalyzes the dismutation of superoxide into oxygen and hydrogen peroxide is the SOD. Subsequently, enzymatic activities of total SOD, CuZn-SOD, and Mn-SOD were measured.
Total SOD
Total SOD activity changed significantly among evaluated groups (Fig. 8A, One Way Analysis of Variance, F2= 28.20, p<0.001). There was a decrease in total SOD activity in 3-NP treated group in comparison to control group (Control vs 3-NP, t=4.997, p<0.001; Bonferroni`s t-test), but there were no differences between control and IOA treated group (Control vs. IOA, t=1.183, p=0.511; Bonferroni`s t-test).
Copper-zinc and Mn isoform of SOD
Similarly as total SOD, changes (F2=30.154, p<0.001) in the activity of the CuZn-SOD was found. Multiple Comparisons analysis demonstrated that CuZn-SOD activity was significantly different in 3-NP treated mice when compared to Control group (t=5.537, p<0.001; Bonferroni`s t-test; Fig. 8 B), but there were no differences between Control and IOA treated groups (t=716, p=0.970, Bonferroni`s t-test). Mn-SOD activity was not different among evaluated groups (One Way Analysis of Variance; F2=1.313, P=0.298, Fig. 8C).
Discussion
The present work compares in detail cellular damage and the activity of antioxidant enzymes in response to the inhibition of oxidative phosphorylation or glycolysis using low sub chronic doses of 3-NP or IOA respectively. Our main finding is that each toxin produces striatal apoptotic damage through different mechanisms.
Motor disturbances
Motor disturbances evaluation carried out with our experimental protocol did not display major motor disturbances, like dystonia and gait abnormalities. These results support the idea that the development of overt clinical signs, including weight loss and motor dysfunction, occurs when massive degeneration has taken place [4]. Evaluations of longer times after the last injection are currently carrying on, analyzing motor symptoms.
Cellular damage
Oxidative phosphorylation or glycolysis inhibition elicited cellular damage documented with H&E staining (Fig. 1B and 1C), and electron microscopy (see below). Several experimental reports describe that cell's alteration in neurodegenerative diseases induce neurons to a necrotic or apoptotic death [1, 2]. Thus, interrupting mitochondrial function with repetitive doses of 3-NP produced cellular damage like that earlier reported [3, 31]. Neuronal damage produced by intraperitoneal administration of 3-NP in low doses, has been described at the onset of the neurodegenerative process [4], is restricted to striatal calbinding positive cells (unpublished data from our lab), may initiate necrotic death by excitotoxicity [19], and triggers apoptotic pathways in mice like it does in rats [32].
Regarding mechanisms responsible for striatal cellular alteration triggered by glycolysis inhibition, our data demonstrates that IOA induces apoptosis (Fig.1B, 2B and 3B). Those results suggest that changes in energy metabolism may also underlie cellular dysfunction at the striatum [5].
TUNEL assay in our experiments revealed that inhibition of both, oxidative phosphorylation and glycolysis trigger apoptotic pathways (Fig. 3B and 3C). To analyze if caspases activation was involved in the apoptotic damage, immunehistochemical expression of caspase-3 and caspase-9 were carried out. Caspase-9, an initiator caspase [33], did not display significant differences in its expression after inhibition of oxidative phosphorylation or glycolysis. It is possible that by the time its expression was evaluated, it had already been inactivated. Conversely, a significant increase in the number of immunostained cells for caspase-3 was exhibited in tissue from 3-NP treated group but not in the IOA-treated group.
In previous experiments 3-NP did not show caspase-9 or caspase-3 expression in striatal cells in culture [34], nonetheless in vivo, caspase-9 activation was induced with two intraperitoneal injections (acute model) of 3-NP, or with constant infusion for 5 days (chronic model) through osmotic pumps. In the same study, caspase-3 was induced in the acute but not in the chronic model [20]. 3-NP administration scheme in the present study was neither acute nor constant, instead 3-NP was given daily in low doses (15 mg/kg), once a day, for 5 days; we considered our treatment as a sub-chronic administration, thus discrepancies between our results and those earlier reported, must be due to differences in experimental design.
In the present study, each toxin systemically administered, produced apoptosis as previously reported [32, 35]; however, they activate different cellular apoptotic pathways. It was recently demonstrated that IOA produces neuronal death by ROS formation in vitro [36]; in our conditions there were no changes in NO, LPO or in antioxidant enzymes activity after IOA administration in vivo.
Besides apoptotic damage, some signs of necrotic damage were manifested at the ultra structural level in 3-NP treated animals, for example, cellular edema and altered nuclei with chromatin towards nuclear membrane might be produced by water accumulation inside the cell, resulting from an alteration in mitochondrial function [37]. 3-NP treatment indirectly alters ion gradients due to energy depletion in neurons, leading to a prolonged increase in intracellular Ca2+ and Na+, producing cell edema [38]. Nuclear changes observed in the tissue treated with 3-NP may be induced by the release of pro-apoptotic molecules that participate in nuclear condensation and its fragmentation [39].
Inhibition of GAPDH with IOA did not induce necrotic damage; nevertheless, it was interesting to observe the presence of vacuolization in response to IOA or 3-NP treatments, since it suggests that some type of autophagy is activated after each toxin treatment. In normal conditions, damaged organelles, cell membranes and proteins are degraded by autophagy; however, a failure in autophagy could be responsible for protein accumulation in damaged cells [40]. Indeed autophagy has been related to cell destruction in several types of programmed cell death [41].
GAPDH role in cellular damage
The study of glycolysis inhibition at the brain with IOA has been carried out previously in vitro [30, 42, 43]; but there was a lack of information about glycolysis inhibition in vivo, and its effects on striatal tissue. Also, there were no previous studies evaluating changes of the glycolytic enzyme GAPDH levels when oxidative phosphorylation was blocked or inactivated. We found significant differences in GAPDH gene expression between IOA and 3-NP treatment. While IOA reduced mRNAGAPDH expression, 3-NP treated group increased GAPDH gene expression. Although an increase in GAPDH transcription would be expected to compensate the reduced activity of the enzyme after IOA treatment, the truth is that IOA is an irreversible inhibitor of GAPDH [43], then reduced mRNAGAPDH expression with chronic administration of IOA is also understandable. On the other hand, there are at least two explanations for the increase of mRNAGAPDH expression levels in the presence of 3-NP. Inhibition of mitochondrial respiration activates other metabolic pathways such as glycolysis in glial cells [44]; therefore it is possible that increased level of mRNAGAPDH in the 3-NP treated group was due to glycolysis activation in glial cells, because for RT-PCR procedures the whole striatal tissue was processed. An alternative explanation is related to other GAPDH functions; as known, besides its role in glycolysis, GAPDH is involved in DNA transcription, replication, reparation and apoptotic induction [9-11], in fact GAPDH increase is found in apoptotic cell death [45]; finally an increase of mRNAGAPDH levels have also been observed in non-neuronal tissue after oxidative stress induction [46]. Therefore, mRNAGAPDH increase in striatal tissue of 3-NP treated mice may indicate neuronal damage. Further experiments should be carried out to probe whether mRNAGAPDH increase in the presence of 3-NP is related to neuroprotection or to neurodegeneration.
Effects on NO and LPO production
In previous experiments, administration of both 3-NP and IOA increases free radicals production [46-47]. Superoxide anion and hydrogen peroxide, combined with other reactive species as nitric oxide (NO) produce peroxynitrite [48], and are capable of attacking cell membrane lipids and proteins. 3-NP increases oxidative stress because interferes with electron transport cascade, which results in a cellular energy deficit [49].
In the present study, 3-NP and IOA did not produce significant augmentation of NO, however 3-NP increased LPO. Previous reports looking at the involvement of NO in the toxicity of 3-NP have not helped in reaching clear conclusions [50], but it has been suggested that 3-NP acts as NO donor, increasing nitro anions (NO-) levels [47, 49]. 3-NP also induces Ca2+ increase, and interferes with mitochondrial function, increasing mitochondrial O2·- which in turn interacts with NO to form more NO- which interacting with arachidonic acid induces lipid peroxidation, which may explain LPO increase in the 3-NP treated group (Fig. 6B).
IOA inhibits glycolysis, decreases ATP production [30] and increases free radicals in vitro [36], we did not evaluate directly changes in free radicals formation, but at the doses evaluated and administration route, IOA did not produce changes in NO or LPO levels.
Effects on antioxidant enzymes activation
To better understand the process underlying energy uncoupling we searched for mechanisms of cellular protection, evaluating the activity of the antioxidant enzymes CAT, GPx, GSR, Total SOD, CuZn-SOD and Mn-SOD, which counteract the deleterious effects of reactive oxygen species and oxidative stress. In our experimental conditions, IOA-treated animals did not exhibit changes in any of the enzymes evaluated. This finding supports the conclusion that IOA effects on striatal tissue result from the activation of different cellular pathways than those activated in 3-NP treated animals [43].
On the contrary 3-NP treated group exhibited a reduction in CAT, GPx, Total SOD and CuZn-SOD enzymatic activities while Mn-SOD, did not change. We do not have an answer to explain it, but Superoxide Dismutases convert superoxide radicals to hydrogen peroxide, after that, glutathione peroxidase and catalase converts H2O2 to water. If one of these enzymatic reactions fails, antioxidant mechanisms fail too, leading to neuronal damage [51]. Decrement in CuZn-SOD activity, as the one we observed in the present study, has been reported in animal models of Parkinson's disease [52] and Huntington's disease [53]. Then, 3-NP treated mice possibly were subject to oxidative stress. These results are consistent with the hypothesis which suggests that in cell degeneration there is an imbalance between ROS production and the antioxidant defense system function.
Conclusions
Several studies have revealed that energy deficiency causes neurodegenerative diseases; however, energy sources may come from two different routes, glycolysis and oxidative phosphorylation. In our experimental conditions the inhibition of glycolysis or oxidative phosphorylation induced cellular damage; but, glycolysis inhibition did not produce striatal alterations to the extent that oxidative phosphorylation inhibition did. This information should be taking in count when designing experimental protocols to induced striatal damage where a failure in bioenergetics has been presumed.
Acknowledgements
We thank Dr. C. S. Colwell and S. Espíndola for their comments to the manuscript and P. Maldonado for processing tissue samples for enzymatic assay. This work was supported in part by CONACyT 81062, DGAPA-PAPPIT (UNAM) IN201307 and UC-Mexus Grant.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging. 2001;18:685-716
2. Hamilton BF, Gould DH. Nature and distribution of brain lesions in rats intoxicated with 3-NP a type of hypoxic (energy deficient) brain damage. Acta Neuropathol. 1987;72:286-297
3. Beal MF, Brouilliet E, Jenkins BG, Ferrante RJ, Kowal NW, Millar JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histological characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181-4192
4. Brouillet E, Conde F, Beal MF, & Hantraye P. Replicating Huntington's disease phenotype in experimental animals. Prog Neurobiol. 1999;59:427-468
5. Browne SE, Beal MF. The energetics on Huntington´s Disease. Neurochem Res. 2004;29:531-546
6. Burke JR, Enghild JJ, Martin ME, Jou YS, Roses AD, Vance JM, Strittmater WJ. Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat Med. 1996;2:347-350
7. Senatorov VV, Charles V, Reddy PH, Tagle DA, Chuang DM. Overexpression and nuclear accumulation of glyceraldehyde-3-phosphate dehydrogenase in a transgenic mouse model of Huntington's disease. Mol Cell Neurosci. 2003;22:285-97
8. Matthews RT, Ferrante RJ, Jenkins BG, Browne SE, Goetz K, Berger S, Chen IY, Beal MF. Iodoacetate produces striatal excitotoxic lesions. J Neurochem. 1997;69:285-289
9. Kragten E, Lalande I, Zimmermann K, Roggo S, Schindler P, Muller D, Van Oostrum J, Waldmeier P, Furst P. Glyceraldehyde-3-phosphate dehydrogenase, the putative target of the antiapoptotic compounds CGP 3466 and R-(-)-deprenyl. J Biol Chem. 1998;273:5821-5828
10. Sheline CT, Choi DW. Neuronal death in cultured murine cortical cells is induced by inhibition GAPDH and triosephosphate isomerase. Neurobiol Dis. 1998;5:47-54
11. Morgenegg G, Winkler GC, Hubscher U, Heizmann CW, Mous J, Kuenzle CC. Glyceraldehyde-3-phosphate dehydrogenase is a nonhistone protein and a possible activator of transcription in neurons. J Neurochem. 1986;47:54-62
12. Novelli A, Reilly AJ, Lysko PG, Henneberry RC. Glutamate becomes neurotoxic via N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205-212
13. Fink SL, Ho DY, Sapolsky RM. Energy and glutamate dependency of 3-nitropropionic acid neurotoxicity in culture. Exp Neurol. 1996;138:298-304
14. Hickey MA, Chesselet M-F. Apoptosis in Huntington's disease. Prog Neuro-Psycopharmacol Biol Psych. 2003;27:255-265
15. Alston T, Mela L, Brig HJ. 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivation of succinate deshidrogenase. PNAS. 1977;74:3767-3771
16. Coles CJ, Edmondson DE, Singer YP. Inactivation of succinate deshydrogenase by 3-Nitropropionate. J Biol Chem. 1979;254:5161-5167
17. Andreassen OA, Ferrante RJ, Hughes DB, Klivenyi P, Dedeoglu A, Ona VO, Friedlander RM, Beal MF. Malonate and 3-nitropropionic acid neurotoxicity are reduced in transgenic mice expressing a caspase-1 dominant-negative mutant. J Neurochem. 2000;75:847-52
18. Nasr P, Gursahani HI, Pang Z, Bondada V, Lee J, Hadley RW, Geddes JW. Influence of cytosolic and mitochondrial Ca2+, ATP, mitochondrial membrane potential, and calpain activity on the mechanism of neuron death induced by 3-nitropropionic acid. Neurochem Int. 2003;43:89-99
19. Schulz JB, Matthews RT, Jenkins BG, Ferrante RJ, Siwek D, Henshaw DR, Cipolloni PB, Mecocci P, Kowall NW, Rosen BR, Beal MF. Blockade of neuronal nitric oxide synthase protects against excitotoxicity in vivo. J Neurosci. 1995;15:8419-8429
20. Bizat N, Hermel JM, Boyer F, Jacquard C, Créminon C, Ouary S, Escartin C, Hantraye P, Kajewski S, Brouillet E. Calpain is a major cell death effector in selective striatal degeneration induced in vivo by 3-nitropropionate: implications for Huntington's disease. J Neurosci. 2003;23:5020-30
21. Lee ST, Chu K, Park JE, Kang L, Ko SY, Jung KH, Kim M. Memantine reduces striatal cell death with decreasing calpain level in 3-nitropropionic model of Huntington's disease. Brain Res. 2006;1118:199-207
22. Kim GW, Chan PH. Oxidative stress and neuronal DNA fragmentation mediate age-dependent vulnerability to the mitochondrial toxin 3-nitropropionic acid, in the mouse striatum. Neurobiol Dis. 2001;8:114-126
23. Rosenstock TR, Carvalho AC, Jurkiewicz A, Frussa-Filho R, Smaili SS. Mitochondrial calcium, oxidative stress and apoptosis in a neurodegenerative disease model induced by 3-nitropropionic acid. J Neurochem. 2004;88:1220-1228
24. Zermeño V, Espíndola S, Mendoza E, Hernández-Echeagaray E. Differential expression of neurotrophins in postnatal C57BL/6 mice striatum. Int J Biol Sci. 2009;5(2):118-127
25. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C(T)) Method. Methods. 2001;25:402-408
26. Ponchel F, Toomes C, Bransfield K, Leong FT, Douglas SH, Field SL, Bell SM, Combaret V, Puisieux A, Mighell AJ, Robinson PA, Inglehearn CF, Isaacs JD, Markham AF. Real-time PCR based on SYBR-Green I fluorescence: an alternative to the TaqMan assay for a relative quantification of gene rearrangements, gene amplifications and micro gene deletions. BMC Biotechnol. 2003;13:3-18
27. Goth LA. A simple method for determination of serum catalase activity, and revision of reference range. Clin Chim Acta. 1991;196:143-152
28. Carlberg I, Mannervik B. Purification and characterization of the flavoenzime gluthatione reductase from rat liver. J Biol Chem. 1975;250:5475-5480
29. Sun Y, Oberley LW, Li Y. A simple method for clinical assay of superoxide dismutase. Clin Chem. 1988;34:497-500
30. Schmidt MM, Dringen R. Differential effects of iodoacetamide and iodoacetate on glycolysis and gluthathione metabolism of cultured astrocytes. Frontiers Neuroenergetics. 2009;1:1-10
31. Alexi T, Hughes PE, Knusel B, Tobin AJ. Metabolic compromise with systemic 3-nitropropionic acid produces striatal apoptosis in Sprague-Dawley rats but not in BALB/c ByJ mice. Exp Neurol. 1998;153:74-93
32. Sato S, Gobbel GT, Honkaniemi J. Apoptosis in the striatum of rats following intraperitoneal injection of 3-nitropropionic acid. Brain Res. 1997;745:343-347
33. Cohen GM. "Caspases: the executioners of apoptosis". Biochem J. 1997;326:1-16
34. Galas MC, Bizat N, Cuvelier L, Bantubungi K, Brouillet E, Schiffmann SN, Blum D. Death of cortical and striatal neurons induced by mitocondrial defect involves differential molecular mechanisms. Neurobiol Dis. 2004;15:152-159
35. Otsuki S, Morshed SR, Chowdhury SA, Takayama F, Satoh T, Hashimoto K, Sugiyama K, Amano O, Yasui T, Yokote Y, Akahane K, Sakagami H. Possible link between glycolysis and apoptosis induced by sodium fluoride. J Dent Res. 2005;84:919-23
36. Hernández-Fonseca K, Cárdenas-Rodriguez N, Pedraza-Chaverri J, Massieu L. Calcium production of reactive oxygen species is involved in neuronal damage induced during glycolysis inhibition in cultured hippocampal neurons. J Neurosci Res. 2008;86:1768-1780
37. Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 2001;21:2-14
38. Choi DW. Calcium and excitotoxic neuronal injury. Ann NY Acad Sci. 1994;747:162-71
39. van Gurp M, Festjens N, van Loo G, Saelens X, Vandenabeele P. Mitochondrial intermembrane proteins in cell death. Biochem Biophys Res Commun. 2003;304:487-97
40. Keller JN, Dimayuga E, Chen Q, Thorpe J, Gee J, Ding Q. Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain". Int J Biochem Cell Biol. 2004;36:2376-91
41. Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Differen. 2005;12:1528-1533
42. Zeewalk GD, Nicklas WJ. Chemically induced hypoglycemia and anoxia: relationship to glutamate receptor-mediated toxicity in retina. J Pharmachol Exp Ther. 1990;253:1285-1292
43. Sabri MI, Ochs S. Inhibition of glyceraldehyde-3-phosphate dehydrogenase in mammalian nerve by iodoacetic acid. J Neurochem. 1971;18:1509-1514
44. Almeida A, Almeida J, Bolaños JP, Moncada S. Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically-generated ATP in astrocyte protection. PNAS. 2001;98:294-299
45. Ishitani R, Sunaka K, Hirano A, Saunders P, Katsube N, Chuang DM. Evidence that glyceraldehyde-3-phosphate dehydrogenase is involved in age-induced apoptosis in mature cerebellar neurons in culture. J Neurochem. 1996;66:928-935
46. Ito Y, Pagano PJ, Tornheim K, Brecher P, Cohen RA. Oxidative stress increases glyceraldehyde-3-phosphate dehydrogenase mRNA levels in isolated rabbit aorta. Am J Physiol. 1996;270:81-87
47. Kim GW, Chan PH. Involvement of superoxide in excitotoxicity and DNA fragmentation in striatal vulnerability in mice after treatment with the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab. 2002;22:798-809
48. Brorson JR, Schumaker PT, Zhang H. Nitric oxide acutely inhibits neuronal energy production. J Neurosci. 1999;19:147-158
49. Deshpande SB, Hida H, Takei Io N, Masuda T, Baba H, Nishino H. Involvement of nitric oxide in 3-nitropropionic acid-induced striatal toxicity in rats. Brain Res. 2006;1108:205-215
50. Hellweg R, von Armin CA, Buchner M, Huber R, Reipe MW. Neuroprotection and neuronal dysfunction upon repetitive inhibition of oxidative phosphorylation. Exp Neurol. 2003;183:346-354
51. Michiels C, Reas M, Toussaint O, Remacle J. Importance of Se-glutathione peroxidase, catalase, and Cu/Zn-SOD for cell survival against oxidative stress. Free Radic Biol Med. 1994;17:235- 248
52. Fu YT, He FS, Zhang SL, Zhang JS. Lipid peroxidation in rats intoxicated with 3-nitropropionic acid. Toxicon. 1994;33:327-331
53. Przedborski S, Kostic V, Jackson -Lewis V, Naini AB, Fahn S, Carlson E, Epstein CJ, Cadet JL. Transgenic mice with increased Cu/Zn-superoxide dismutase activity are resistant t N-methyl-4-phenyl-1,2,3,6-tetrahydropyri-dine induced neurotoxicity. J Neurosci. 1992;12:1658-1667
Author contact
![]() Corresponding author: Laboratorio de Neurofisiología del Desarrollo y la Neurodegeneración. Unidad de Biomedicina, FES-I, Universidad Nacional Autónoma de México. Av. de los Barrios # 1, Los Reyes Iztacala, C.P. 54090, Tlalnepantla, México. elihernandeziztacala.unam.mx. Phone + (52) (55) 5623 1333 Ext 39787. FAX + (52) (55) 5623-1138.
Corresponding author: Laboratorio de Neurofisiología del Desarrollo y la Neurodegeneración. Unidad de Biomedicina, FES-I, Universidad Nacional Autónoma de México. Av. de los Barrios # 1, Los Reyes Iztacala, C.P. 54090, Tlalnepantla, México. elihernandeziztacala.unam.mx. Phone + (52) (55) 5623 1333 Ext 39787. FAX + (52) (55) 5623-1138.
Received 2009-12-7
Accepted 2010-4-15
Published 2010-4-22