Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(7):649-654. doi:10.7150/ijbs.6.649 This issue Cite
Short Research Communication
Mechanical Forces Used for Cell Fractionation Can Create Hybrid Membrane Vesicles
Izhar Salomon, Hans Janssen, Jacques Neefjes ![]()
The Netherlands Cancer Institute, Division of Cell Biology. Amsterdam, the Netherlands.
Received 2010-5-19; Accepted 2010-10-17; Published 2010-10-20
Abstract
The ability to understand the inner works of the cell requires methods for separation of intracellular membrane-enclosed compartments. Disruption of the plasma membrane (PM) by mechanical forces to investigate the content of the cell is common practice. Whether vesicles or membranes of different sources can fuse as a result is unclear. If such contamination occurs, conclusions based on these techniques should consider these. Utilizing an endoplasmic reticulum (ER) membrane marker and a PM marker, we were able to detect the source of membranes following the breakup of cells using flow cytometry and immuno Electron Microscopy (immuno EM). Fractionation processes produced a small fraction of new membrane entities from two distinctively different origins generated during the initial disruption steps in a temperature independent manner, stressing that defining organelles or intrinsic fusion events based on such procedures and markers are valid when exceeding the small number of vesciles fused during the fractionation process.
Keywords: membrane fusion, fractionation, trafficking, signalling, proteomics.
1. Introduction
Cell biology uses a plethora of methods to understand the complexity of an individual cell. Breaking the cell's membranes to retrieve smaller organelles, particles, proteins and nucleic acids is common practice. The study of processes such as pathogen invasion (1), antigen presentation (2-4), intracellular trafficking (3), cellular signalling and dynamics, maturation and fusion of different compartments (4, 5) and the analysis of protein complexes (6) demand organelle separation, determination of the organelle-specific markers and detection of foreign proteins or peptides on non-related membranes (7). Imperative for these studies is that the technique applied does not influence the data retrieved. We tested whether fusion of membranes could artificially be achieved by mechanical force or sonication. Although cell fractionation is a common technique, such a 'control' experiment has not been performed before and it is unclear whether a large, small or non-existing fraction fused during the fractionation procedure. We selected the endoplasmic reticulum (ER) and the plasma membrane (PM) as two separate compartments that under non-disrupted conditions would not mix. Tap1-GFP (Peptide Transporter subunit 1 tagged with Green Fluorescent Protein) was used as an ER marker. MHC-II (major histocompatibility complex class II) detected by an antibody: L243-directly conjugated with Alexa 647, was used to mark the PM.
Using common techniques for PM disruption and organelle fractionation such as homogenization and sonication (8), we were able to trace particles exhibiting those markers by flow cytometry and electron microscope (EM). These vesicles showed a continuous lipid bilayer containing both the ER and the PM markers, indicating fusion events of these separate compartments as a response to mechanical force. Varying the experimental conditions showed that fusion of a small fraction of vesicles occurred during the first steps of fractionation, with no direct relationship to sheering forces and temperature.
2. Materials and Methods
Fluorescent cell sort staining
MelJuSo (MJS) cells stably transfected with Tap1-GFP were trypsinized, washed and stained with mouse anti human L243-Alexa 647 or L243-Cy5 for 15 minute at 4oC to exclude endocytosis of the MHC-II surface molecules, then washed 3 times to remove traces of antibody.
Membrane disruption
The integrity of the cells was compromised by an EMBL 8.020mm “cell cracker” homogenizer with ball sizes 8.010, 8.008 or 8.004nm (gap of 0.005, 0.006 or 0.008nm between ball and wall of chamber, respectively) either 2, 10 or 30 times. Alternatively, cell disintegration was achieved by sonication pulses of approximately 0.5 second by a Branson sonifier 250 (Duty cycle 50%, output control 5) or Diagenode Bioruptor (High level 0.5 seconds on/off interval), for either 4 (short), 8 (intermediate) or 12 (long) pulses. For detergent-based cell disintegration, 0.1(v/v)% Triton X-100 in PBS was used for 5 or 10 minutes on ice, spun down (5 min at 10.000xg) and both supernatant and PBS resuspended pellet were analyzed.
Reading was done on a Beckman-Coulter MoFlo fluorescent cell sorter.
EM preparation
Particles exhibiting both GFP and Alexa 647 were sorted using flow cytometry and spun at 40,000 rpm onto a nitrocellulose membrane, fixed in 2(v/v)% paraformaldehyde + 0.2(v/v)% glutaraldehyde in 0.1 M PHEM buffer, (60 mM PIPES, 25 mM HEPES, 2 mM MgCl2, 10 mM EGTA, pH 6.9), blocked sequentially with 0.15 M glycine/PBS and 1(w/v)% BSA/PBS. The filter was cut up in 4 pieces and each piece was used for immunolabelling with either mouse anti-GFP (αGFP - gift by J.Bonifacino), rabbit anti-human MHC-II serum (9) or control antibody rabbit anti-human Lactoferrin (αLactoferrin - Cappel antibodies, West Chester, PA) for 60 minutes at room temperature (RT), washed and stained with protein A/gold-10nm (EMlab, University of Utrecht) for 30 minutes. Fixed with 1(v/v)% glutaraldehyde/PBS for 10 minutes, blocked again and incubated with a second antibody, αGFP, anti-MHC class II or αLactoferrin (depending on the first antibody) for 60 minutes, washed and stained at RT with protein A/gold 15nm (EMlab, University of Utrecht). The labeled filters were then prepared for plastic embedding by Karnovsky fixation (4% paraformaldehyde + 2.5% glutaraldehyde in 0.1 M cacodylate buffer pH7.2), followed by staining with 1(w/v)% osmiumtetroxide for 1 hour, dehydration by isopropanol series, toluene and embedded in embed 812 (Electron Microscopy Sciences), sectioned and viewed with a Philips CM10 Electron Microscope.
3. Results
Hybrid vesicles are formed after mechanical membrane disruption
To monitor possible effects on fusion by sheer force-based disruption methods, MelJuSo (MJS) cells stably transfected with Tap1-GFP were stained with anti-MHC-II antibody L243-Alexa 647nm at low temperature to prevent endocytosis thus effectively labelling only the plasma membrane pool of MHC-II. The cells' membranes were then disrupted by an EMBL “cell cracker” homogenizer, sonication or by a mild detergent step (0.1% Triton X-100) as a control (Figure 1A) and read by MoFlo cell sorter to detect and collect particles exhibiting fluorescent signals from both GFP (Tap1, the ER protein) and Alexa 647 (MHC-II, the PM protein). We gated on small material defined by FACS on the basis of Forward (FSC) and Side (SSC) scatter and determined the fluorescence on this population only (Figure 1B). This fraction did not alter under the different conditions tested in this study.
Hybrid membranes created by sheer forces. (A) Outline of the experimental procedure: MelJuSo cells stably transfected with Tap1-GFP (17) were Alexa647-L243 Ab stained for 15min at 4oC before the unbound antibody was removed. Cells were fractionated by sonication or douncing under controlled conditions with the 'EMBL Cell Cracker' before analysis by two-color flow cytometry. (B) Representative flow cytometer plots i. and ii. control sample (WT MJS); iii. and iv. a representative sample (ball 8.004 - 30 strokes). i. and iii. side (SSC) and forward (FSC) scatter showing whole sample population, ii. and iv. Two color fluorescence analysis of the R1 region in plot I and III, representing the small particles containing ER marker TAP1-GFP (FL1) and PM marker L243-Alexa647 (FL6). R1: cell particles, R2: whole cells, R3: double positive gated population. (C) ER-PM positive samples isolated by FACS were processed for immuno-EM and stained with anti-GFP (10 nm indicated by arrowheads in the insert) and anti-MHC II (15nm). A representative continuous membrane containing both markers representing a hybrid vesicle is shown.
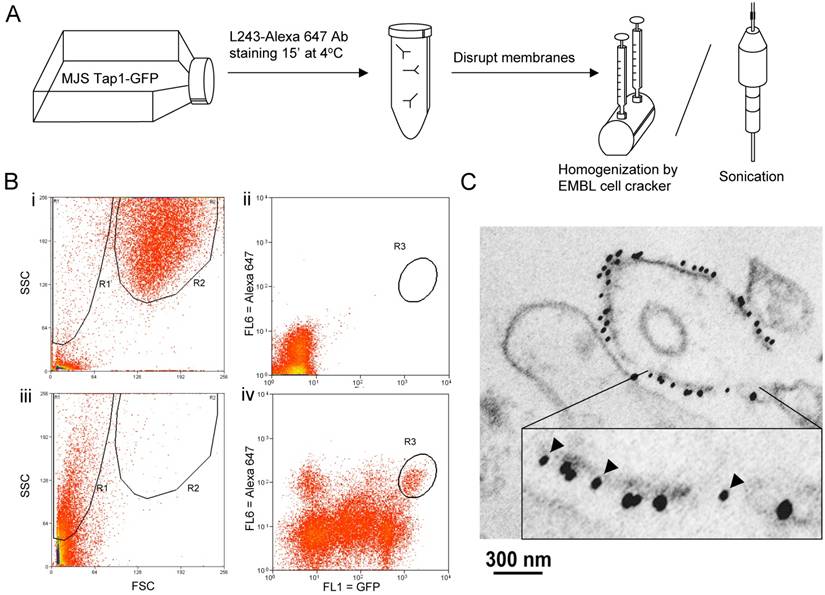
A small (around 0.5% of vesicles) but distinct population of particles presenting both the ER and the PM markers was detected under almost all conditions (Figure 1B and C). To further validate that vesicles labelling for TAP-GFP and L243-Alexa 647 were genuinely fused rather that associated vesicles, double positively stained particles were sorted by FACS and concentrated on a nitrocellulose filter. This procedure did not yield hybrid mitochondrial-late endosome fusions, as we had tested before (data not shown). The samples were fixed in formaldehyde/glutaraldehyde and stained with antibodies detecting GFP (10 nm gold) and detecting MHC-II (or lactoferrin as control) with 15 nm gold before analysis by cryo-immuno-electronmicroscopy. Of note, this procedure detects some 10% of antigens implying that the real numbers of antigens is considerably higher than detected. Vesicles not labeled by this procedure may in fact still be positive for the markers. Still, vesicles were observed staining either for ER and PM marker proteins (15% of vesicles) or both marker proteins (8% of vesicles; Figure 1C), indicating that new hybrid and fused membrane vesicles have been formed.
Conditions for fractionation-induced vesicle fusion
To control for antibody contamination of vesciles and fusion post-fractionation, we mixed vesicle fractions of MJS cells expressing Tap1-GFP as ER marker with vesicle fractions of control MJS cells stained MHC-II as PM marker and compared the recovery of double positive vesicles with those where the cells were first mixed before fractionation or where the MJS TAP1-GFP cells were labelled with L243-Alexa 647 before fractionation (Figure 2A). The double-positive fluorescent vesicles were detected by FACS. This experiment reveals that most double-positive vesicles are created during fractionation.
Quantifying hybrid particles presenting both ER and PM markers. The membrane integrity of MelJuSo cells was compromised by EMBL 8.020mm “cell cracker” homogenizer. For all experiments, the percentage of hybrid vesicles labeling for TAP1-GFP and MHC class II-L243 relative to single or non-labeled vesicles was determined by a Beckman-Coulter MoFlo fluorescent cell sorter and represented in the graphs. All experiments are performed in multiplo. Shown is mean + SD. (A) Discernable cell populations with either ER (Tap1-GFP) or PM (stained MHC-II) markers were broken in separate tubes and than mixed ('Stained: Separately, Fractionated: Separately') or broken in the same tube simultaneously ('Stained: Separately, Fractionated: Together'). As a control cells labeled with both markers were used ('Stained: Together, Fractionated: Together'). (B) Fractionation strokes were performed 2, 10 or 30 times (ball size 8.010mm) or (D) with ball sizes of 8.010, 8.008 or 8.004mm (30 strokes). As a soft detergent 0.1% Triton X-100 in PBS (Triton) was used for 5 or 10 minutes on ice. (C) Sonication pulses of approximately 0.5 second by a Branson sonifier 250 (Duty cycle 50%, output control 5) or Diagenode Bioruptor (High level 0.5 seconds on/off interval), for either 4 (short), 8 (intermediate) or 12 (long) pulses. The released cell fragments were spun down and both supernatant and PBS resuspended pellets were analyzed. (E) The effect of temperature variations on formation of hybrid vesicles test was performed by EMBL cell cracker with 8.010mm ball size. Fractionation was 30 times at 4, 10, 18, 25 and 37oC.
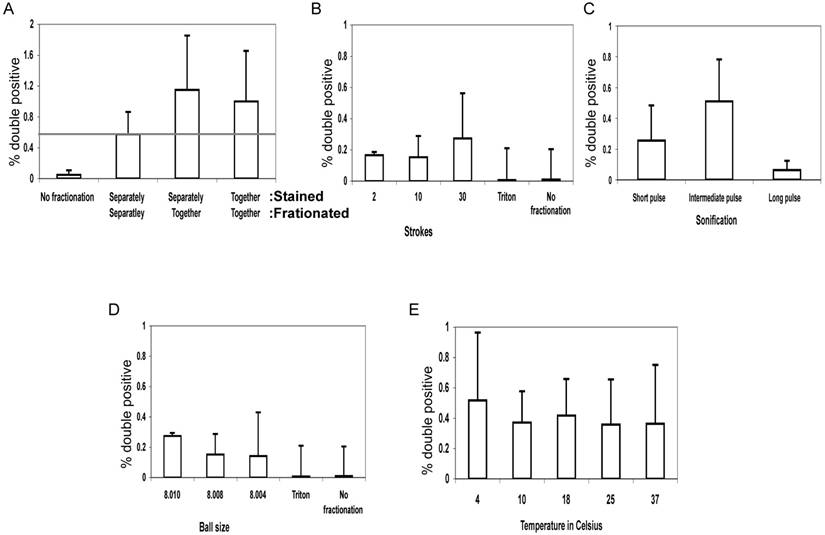
Several conditions for fractionation were explored using the EMBL “cell cracker” and sonication. No hybrid vesicles were observed in flow cytometry when cells were exposed to mild detergent (0.1% TX-100), implying that mechanical force was required for their formation. We varied the number of fractionation steps (the number of strokes in the EMBL cell cracker) or the length of sonication to test whether formation of hybrid ER-PM structures continued after the initial breaking apart steps of the cell or whether hybrid structures were formed ab initio. Almost all cases showed a detectable population of vesicles containing both ER and PM markers signal but this was not affected by the number of douncing steps or the length of sonication (Figure 2B,C).
We then varied the size of the ball in the EMBL cell cracker chamber where cells and vesicles were pushed along. The EMBL cell cracker has a chamber with a radius of 8.020 mm containing metal balls with a radius of 8.004 up to 8.010 mm. The distance between ball and chamber is thus varied between 0.005-0.008 mm representing different shearing forces on cells and vesicles. Although some variation in yield of vesicles labelling for TAP-GFP and L243 was detected (Figure 2D), no relationship was observed between ball size and yield of hybrid vesicles suggesting that shearing forces were not directly related to the rate of fusion of unrelated organelles.
Fluidic state of membranes do not alter hybrid vesicle formation
We then tested whether the fluidic state of the membranes had any effect on the formation of hybrid organelles. Lipid bilayers will convert from a fluidic to a solid state around 20oC, depending on lipid composition (10). Breaking the cells by EMBL “cell cracker” homogenizer at different temperatures (4oC, 10oC, 18oC, 25oC and 37oC) yielded similar numbers of hybrid ER-PM organelles, as detected by flow cytometry (Figure 2E). This suggests that hybrid organelles arise by processes that do not require a fluidic lipid membrane state (11).
4. Discussion
To progress and widen our understanding of molecular processes, we must break open the outer membrane and explore the inner works of the cell. A gamut of methods has evolved to answer such a need, especially cell fractionation by sheering forces (12) or following mild detergent lyses have been used. It was usually assumed -but never experimentally verified- that fusion of vesicles did not appear as a result of fractionation and that the end-result of fractionation reflected the original situation in cells. Here, we described that this assumption in not completely correct as novel vesciles are formed by sheering forces. These new particles are few, in our experiments typically around 0.5% with some variation between experiments. Still those instances occur and must come into the overall calculation and conclusion of experiments done in such a fashion. For example, the identification by mass spectrometry of ER proteins in highly purified phagosomal fractions may contain a contribution of hybrid ER-phagosome vesicles. As the ER is the largest intracellular compartment, contaminations will constitute predominantly this compartment, followed by mitochondrial and other membrane contaminations. These contaminations can be detected by novel highly sensitive methods like mass spectrometry (13). The conclusion that the ER can fuse to phagosomes (which could be relevant for an immunological process called cross-presentation (14)) could in part be based on analysis of hybrid organelles which can be excluded by considering the small contamination that we detect in our experiments.
Chemical agents pose an array of problems in breakage and separation of cellular components. The obvious advantages in harvesting specific proteins, DNA or RNA can be a hindrance when trying to study bigger composite structures with constituents that react differently to detergents or other chemicals that dissociate large protein and membrane structures like organelles (15). Thus even though chemical disturbance of the membrane may not produce new hybrid vesicles (Figure 2), it does not allow conclusions on intact membrane structures. Non-detergent based cellular fractionation therefore remains an essential technique in cell biology.
Considering that most of the cell's membranes are ER and a much smaller portion is the PM, (for example, 2% of membranes are PM, about 50% ER, 7% Golgi and about 40% mitochondria in an hepatocyte (16)), it seems logical to assume that the formation of these hybrid vesicles occurs more often than our detection resolution. We were only able to follow a small part of these fusion instances that involved the PM, whereas a substantial number of other events involving the reformation of a membrane entity from different origins may happen under the sheering stress.
Mechanical or chemical methods each have their advantages and disadvantages and those should be taken into consideration when results are analysed. A small but reproducible population of artefact hybrid particles does not necessarily devaluate the importance of a method nor the conclusion derived from the method. It does however require an attentive examination of data obtained in such a manner, subjecting it to statistical analysis and additional controls.
In order to investigate the mechanisms inside a cell, we have to manipulate the membranes and their contents before further analysis in isolation. Still we must consider that the lipid bilayers' basic physics, that nature has so elegantly harnessed might create artefacts when we, in our somewhat brutal way, break in the walls of the house to see how the kitchen stove light works.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Raupach B & Kaufmann S. Immune responses to intracellular bacteria. Curr Opin Immunol. 2001;13(4):417-28
2. van Niel G. et al. Dendritic cells regulate exposure of MHC class II at their plasma membrane by oligoubiquitination. Immunity. 2006;25(6):885-894
3. Behnia R & Munro S. Organelle identity and the signposts for membrane traffic. Nature. 2005;438(7068):597-604
4. Gagnon E. et al. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell. 2002;110(1):119-131
5. Bonifacino JS & Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116(2):153-166
6. Mathias RA & Simpson RJ. Towards understanding epithelial-mesenchymal transition: a proteomics perspective. Biochim Biophys Acta. 2009;1794(9):1325-1331
7. Schaub BE, Nair P, & Rohrer J. Analysis of protein transport to lysosomes. Current protocols in cell biology. 2005;15:15-8
8. Ramachandra L, Sramkoski RM, Canaday DH, Boom WH, & Harding CV. Flow analysis of MHC molecules and other membrane proteins in isolated phagosomes. J Immunol Methods. 1998;213(1):53-71
9. Neefjes JJ, Stollorz V, Peters PJ, Geuze HJ, & Ploegh HL. The biosynthetic pathway of MHC class II but not class I molecules intersects the endocytic route. Cell. 1990;61(1):171-183
10. Reits EA & Neefjes JJ. From fixed to FRAP: measuring protein mobility and activity in living cells. Nat Cell Biol. 2001;3(6):E145-147
11. Shibata Y, Hu J, Kozlov MM, & Rapoport TA. Mechanisms shaping the membranes of cellular organelles. Annu Rev Cell Dev Biol. 2009;25:329-354
12. Aubry L & Klein G. Purification techniques of subcellular compartments for analytical and preparative purposes. Methods Mol Biol. 2006;346:171-185
13. Brunner Y, Schvartz D, Couté Y, & Sanchez J-C. Proteomics of regulated secretory organelles. Mass spectrometry reviews. 2009;28(5):844-867
14. Guermonprez P. et al. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature. 2003;425(6956):397-402
15. Schubert D, Boss K, Dorst HJ, Flossdorf J, & Pappert G. The nature of the stable noncovalent dimers of band 3 protein from erythrocyte membranes in solutions of Triton X-100. FEBS Lett. 1983;163(1):81-84
16. Alberts B. et al. Molecular Biology of the Cell. New York: Garland. 2007
17. Reits EA, Vos JC, Grommé M, & Neefjes. The major substrates for TAP in vivo are derived from newly synthesized proteins. Nature. 2000;404(6779):774-778
Author contact
![]() Corresponding author: Dr. Jacques Neefjes, The Netherlands Cancer Institute, Division of Cell Biology, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands. Phone: +31 20 512 2017; E-mail: j.neefjesnl
Corresponding author: Dr. Jacques Neefjes, The Netherlands Cancer Institute, Division of Cell Biology, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands. Phone: +31 20 512 2017; E-mail: j.neefjesnl