International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Technical advances in iPSC...
Generation of Human iPSCs from...
Hepatic Differentiation of human...
Disease modeling with human iPSCs
Drug toxicity screening and drug...
Therapeutic Potentials of human...
Conclusion
References
Introduction
Technical advances in iPSC...
Generation of Human iPSCs from...
Hepatic Differentiation of human...
Disease modeling with human iPSCs
Drug toxicity screening and drug...
Therapeutic Potentials of human...
Conclusion
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(7):796-805. doi:10.7150/ijbs.6.796 This issue Cite
Review
Applications of Patient-Specific Induced Pluripotent Stem Cells; Focused on Disease Modeling, Drug Screening and Therapeutic Potentials for Liver Disease
Yong Soon Chun, Pooja Chaudhari, Yoon-Young Jang ![]()
Department of Oncology, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA
Received 2010-12-8; Accepted 2010-12-13; Published 2010-12-14
Citation:
Chun YS, Chaudhari P, Jang YY. Applications of Patient-Specific Induced Pluripotent Stem Cells; Focused on Disease Modeling, Drug Screening and Therapeutic Potentials for Liver Disease. Int J Biol Sci 2010; 6(7):796-805. doi:10.7150/ijbs.6.796. https://www.ijbs.com/v06p0796.htm
Other stylesAbstract
The recent advances in the induced pluripotent stem cell (iPSC) research have significantly changed our perspectives on regenerative medicine by providing researchers with a unique tool to derive disease-specific stem cells for study. In this review, we describe the human iPSC generation from developmentally diverse origins (i.e. endoderm-, mesoderm-, and ectoderm- tissue derived human iPSCs) and multistage hepatic differentiation protocols, and discuss both basic and clinical applications of these cells including disease modeling, drug toxicity screening/drug discovery, gene therapy and cell replacement therapy.
Keywords: Stem Cell, Disease Modeling, Drug Screening, Liver Disease
Introduction
In 2006, Yamanaka and co-workers first reported the generation of induced pluripotent stem cells (iPSCs) from mouse somatic fibroblasts by the retroviral transduction of Oct4, Sox2, Klf4 and c-Myc genes (1). Subsequently, human iPSCs have been generated from embryonic, neonatal and adult fibroblasts (2-4). In addition, derivation of patient-specific iPSCs for various diseases/disorders has also been reported (5-9). More recently several groups have explored the possibilities of disease modeling using patient derived iPSCs and directed differentiation technologies (7, 10-18). Since iPSCs resemble embryonic stem cells (ESCs) in their pluripotency, and offer potential solutions for histo-incompatibility issues that limit the use of ESC-based therapies, patient-specific iPSCs hold great potential as an unlimited cell source not only for generating disease models but also drug screening and cell replacement therapy for various diseases.
One of the main hurdles for achieving these goals is to develop efficient directed differentiation technologies that are also functional and safe. During recent years, in vitro hepatic differentiation from both human ESCs and iPSCs has been achieved and improved by several laboratories including ours (15, 19-22). For patients with end-stage liver disease, liver transplantation is the only curative method of treatment (23). However, limited availability of donor livers and immunological incompatibilities are major obstacles to liver or hepatocyte transplantation (24). The use of ex vivo adult human hepatocytes is a desirable option for cellular therapies or drug testing. However, these cells have limited proliferation potential, and lose function and viability upon isolation. Although there have been great advances in liver stem cell biology (25-27), adult liver stem cells are rare within tissue, making their isolation difficult and expansion unfavorable for large-scale applications (28). Therefore it is critical to develop alternative strategies such as iPSC derived functional hepatic cells as an unlimited hepatocyte source which can be utilized in drug screening, disease modeling and cell therapy. In this review, we summarize the current progress in the field of human iPSCs and hepatic differentiation, and discuss both basic and clinical applications of the iPSCs with an emphasis on liver disease.
Technical advances in iPSC derivation methods
Most patient-specific iPSCs have been established using retrovirus vectors. However, the retrovirally derived iPSCs carry the risk of numerous transgene integrations in the genome, which may result in leaky expression. This could disturb the endogenous transcription factor network and lead to failure of directed differentiation. Also, transgene integration has a risk of tumorigenesis after transplantation. In particular, c-Myc, one of the reprogramming factors, is a well-known oncogene, and its reactivation could give rise to transgene driven-tumor formation (29). There have been several reports of improvements of the transduction method for generating safe iPSCs. Removal of the c-Myc transgene from reprogramming cocktail is one important approach. Human and mouse iPSCs have been established from fibroblasts with only Oct4, Sox2, and Klf4, although the efficiency is significantly reduced (30). Other different approaches have also been devised to introduce reprogramming factors into somatic cells (Table 1). A transposon system encoding a reprogramming cassette has been used for iPSC induction (31, 32). The transduction of a plasmid-based transposon vector enables it to integrate into the host genome with the help of transposase, and induces iPSC colony formation. The re-expression of the transposase recognizes the terminal repeat of the integrated transposon vector, and excises it from the genome. The excision of the transposon does not leave a footprint in most cases, and hence maintains the original endogenous sequence. Several other methods accomplished iPSC induction by the transient expression of reprogramming factors. These methods include viral vectors (adenovirus and Sendai virus) (33, 34), DNA vectors (plasmid and episomal plasmid vector) (35, 36), small molecules (37), and direct protein delivery (38). However, the efficiency of most of these integration free iPSC induction systems is significantly lower than that with retrovirus vectors. More recently, the Rossi group used synthetic mRNA to reprogram human fibroblasts to pluripotency and induced differentiation terminally into myogenic cells (39). The use of synthetic mRNA overcomes the innate antiviral responses, and showed superior conversion efficiency and kinetics than the established viral protocols. Although relatively technically complex, synthetic mRNA completely eliminates the risk of genomic integration and thus insertional mutagenesis inherent to all DNA-based methodologies. Also, the potential of temporal control over the individual factor expression afforded by this system could help to elucidate the stage-specific roles of individual transcription factors during reprogramming.
Table 1
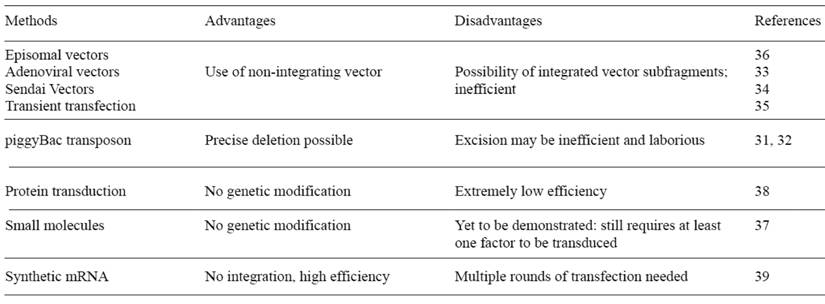
Integration-free human iPS induction methods
Generation of Human iPSCs from Different Somatic Cell Types
One of the most important issues to be addressed before human iPSCs can be used for clinical purposes is the generation of safe and functional cell types for therapy. Embryonic fibroblasts and tail-tip fibroblasts in the mouse and dermal fibroblasts in the human have been the most widely used cell types for reprogramming, largely due to their availability and relative ease of culture. A comprehensive study using various mouse iPSCs has demonstrated that the origin of the iPSCs has a profound influence on the tumor-forming propensities in a cell transplantation therapy model (40). Mouse tail-tip fibroblast iPSCs (mesoderm origin) have shown the highest tumorigenic propensity, whereas gastric epithelial and hepatocyte derived iPSCs (both with endodermal origin) have shown significantly lower tumorigenic propensities (40). The molecular mechanism underlying this phenomenon is not yet fully understood, but evidence suggests that epigenetic memory of the somatic cell of origin is retained in the iPSCs, and that the memory may influence their directed differentiation potential into blood cells (41, 42).
Based upon these mouse iPSC studies, it is reasonable to speculate that the somatic memory of human iPSCs might have an effect on their differentiation potential, which is critical for disease modeling and therapy. Although it has been demonstrated that human iPSCs retain certain gene expression patterns of the parent cells (43), it remains largely unclear if the cell origin could affect the safety and functionality of human iPSCs. It is therefore extremely important to establish human iPSC lines from different developmental origins and thoroughly examine the source that might affect both the safety aspects and their differentiation potentials. Human iPSCs have been derived mostly from the mesodermal (i.e. fibroblasts and blood cells) or the ectodermal origin cells (i.e. keratinocytes, and neural stem cells) (Table 2).
Table 2
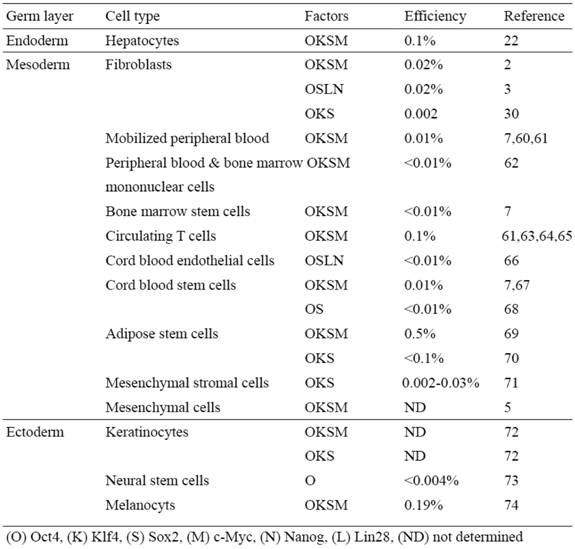
Human iPS cells derived from different somatic cell types
We have recently shown the reprogramming of human primary hepatocytes (i.e. endodermal origin) into iPSCs (22) (Table 2). The technology to develop human endoderm tissue-derived iPSC lines, together with other established human iPSC lines, provides a foundation to elucidate the mechanisms of cellular reprogramming and study the safety and efficacy of differentially originated human iPSCs for cell therapy. For studying pathogenesis of liver disease, this technology also provides a potentially more amenable system to generate liver disease-specific iPSCs. The ability to reprogram human hepatocytes is crucial for developing iPSC-based liver disease models, especially for certain liver disorders involving acquired somatic mutations that occur only in hepatocytes of patients and not in other cell types (44-48).
Recently, two groups have reported derivation of human iPSCs from malignant cell lines (Table 3) demonstrating that these iPSCs lose certain cancer cell characteristics after reprogramming (49, 50). However, it remains to be determined whether these cancer derived iPSCs still retain genetic/epigenetic memory of the original cancer tissue; and more importantly if these iPSCs can be utilized for disease modeling, in order to study complicated cancer pathogenesis and drug screening purposes. In this regard, it is valuable to establish multiple cancer-derived iPSC lines, preferably from primary tumors and comprehensively evaluate their disease modeling potentials.
Table 3
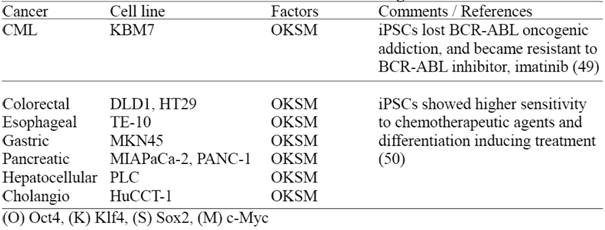
Human iPS cells derived from cultured human malignant cell lines
Hepatic Differentiation of human iPSCs
Directed differentiation of human iPSC lines into hepatic cells has been improved with more efficient and functional methods including our multistage differentiation protocol (19, 21, 22, 48, 51). Our stepwise hepatic differentiation protocol is composed of three differentiative stages resembling liver development, i.e. definitive endoderm, hepatic progenitors, and mature hepatocytes (22, 48). This iPSC based hepatic differentiation technology holds great promise as an ultimate hepatocyte source which can be utilized for drug screening, disease modeling and cell therapy, however, more research is required to improve their function. The functionality of human iPSC derived mature hepatocytes can be analyzed in vitro, by various methods, including analyses for cytochrome P-450 activity and glycogen storage ability with the Periodic Acid-Schiff assay (22). Although these in vitro methods are highly informative and convenient, the most definitive proof for the functionality of human iPSC derived hepatic cells would be the demonstration of hepatic engraftment in vivo using animal models (48) and detection of secreted human liver proteins in animal serum/plasma.
A recent study demonstrated the feasibility of using in vitro hepatic differentiation of human iPSCs to model several inherited liver diseases (15). Although the in vitro culture may recapitulate certain disease features and may be suitable for drug screening purposes, successful regenerative therapy will require hepatic cells that can functionally engraft the liver. To date, the in vivo functionality of human iPSC derived hepatic cells is largely unknown (47, 50). Therefore, it remains to be determined whether the human iPSC -derived multistage hepatic cells possess in vivo functional activities and which differentiative stage of hepatic cells is most suitable for efficient, functional and safe regeneration of the injured liver.
Disease modeling with human iPSCs
The concept of utilizing iPSCs to model a disease in a culture dish is based on the unique capacity of these cells to continuously self-renew and their potential to give rise to all cell types in human body. The greatest advantage of iPSC technology is that it allows for the generation of pluripotent cells from any individual in the context of his or her own particular genetic identity, including individuals with sporadic forms of disease; and those affected by complex multifactorial diseases of unknown genetic identity, such as type 1 diabetes (52) and liver cancers (44-48).
Recently, a number of studies have reported the successful generation of patient-specific iPSC lines from individuals with different diseases; however, effective disease modeling has been demonstrated on a few studies (Table 4). For example, Ebert et al. reported the differentiation of iPSC-derived motor neurons from a patient diagnosed with a genetic from of spinal muscular atrophy (SMA), a neurodegenerative disease that leads to loss of lower motor neurons (11). Although motor neurons derived from the patient-specific iPSCs were initially similar in morphology and number, to those derived from wild-type iPSCs, their numbers and size selectively declined after eight weeks in culture. Furthermore, these cells exhibited a deficiency in the survival of motor neuron (SMN) protein aggregates, which is a characteristic phenotype associated with SMA. Importantly, this deficiency in SMN levels could possibly be reversed with drug treatment, thus providing a basis for future drug screens.
Generation of liver disease patient iPSCs has recently been reported from patients who have inherited liver diseases that arise as a result of loss of function mutation (15, 16). By focusing on a representative disease for each of the different mechanisms in liver diseases such as protein misfolding, cell surface receptor dysfunction and obstruction of cytosolic metabolism, and generating iPSC lines that successfully recapitulate cellular pathological features in vitro; it is now plausible that iPSC technology can be used to model other diseases involving these mechanisms (Table 4). Importantly, this demonstrates that subtle intracellular processes can be studied in an in vitro cellular system, and that they can be accurately preserved despite the high stress exerted on cells by the reprogramming and differentiation protocols.
While the generation of disease-specific iPSC lines is a critical first step, ultimately, it will be necessary to derive a representative set of human iPSCs from different patients; as the phenotype and severity of many of these diseases can vary markedly within the population. Of note, even though some of these non-hepatic sourced iPSCs could be sufficient for regeneration therapy and drug screening for many liver disorders including inherited diseases, hepatic-sourced iPSCs will still be required for disease modeling of certain acquired liver diseases such as cirrhosis and hepatocellular carcinoma.
Drug toxicity screening and drug discovery using human iPSCs
The availability of human iPSCs offers exciting opportunities for reliable high throughput drug screening. The ability to use human cell types in toxicology studies has the potential to increase the efficiency of novel drug development, while reducing drug attrition in the final stages of development and costs. Additionally, the use of human iPSCs would also enable the study of a number of single nucleotide polymorphisms that influence the ability of an individual to effectively metabolize and clear drugs and toxins. Accurate prediction of human drug toxicity is a vital element of drug discovery process. In particular, hepatotoxicity and cardiotoxicity are two principal causes of drug failure during preclinical testing, while the variability in individual responses to potential therapeutic agents is also a major problem in effective drug development (53). However the safety evaluation process is hindered by the availability and quality of primary human liver models with which to study drug toxicity. Development of the scalable and high-fidelity human hepatocytes from fetal and adult progenitors has been hindered due to limited organ availability, and rapid loss of hepatocyte function in culture. The advantage of iPSC technology is that it allows the generation of a library of human cell lines that may represent the genetic and potentially epigenetic variation of a broad spectrum of the population. The use of this tool in high-throughput screening assays could allow better prediction of the toxicology and therapeutic responses induced by newly developed drugs, and offer insight into the underlying mechanisms. The net result of this approach would substantially decrease the risk and cost associated with early-stage clinical trials and could lead toward a more personalized approach in drug administration. Because iPSCs grow indefinitely in culture, they provide an unlimited resource of any desired specialized cells. Ultimately, the goal of this approach is to use patient iPSC based disease models to identify novel drugs to treat the disease. In order to determine individual drug effects on disease specific iPSC derived functional cell types (e.g hepatocytes differentiated from alpha 1 antitrypsin deficiency patient iPSCs), the disease phenotypes needs to be consistently and uniformly recapitulated. Although there have been many attempts to derive patient specific iPSCs, only a few reports have demonstrated some of the disease features with variable degrees (Table 4). It remains, however, to further develop disease modeling conditions and optimize differentiation conditions for more accurate disease modeling and drug screening.
Table 4
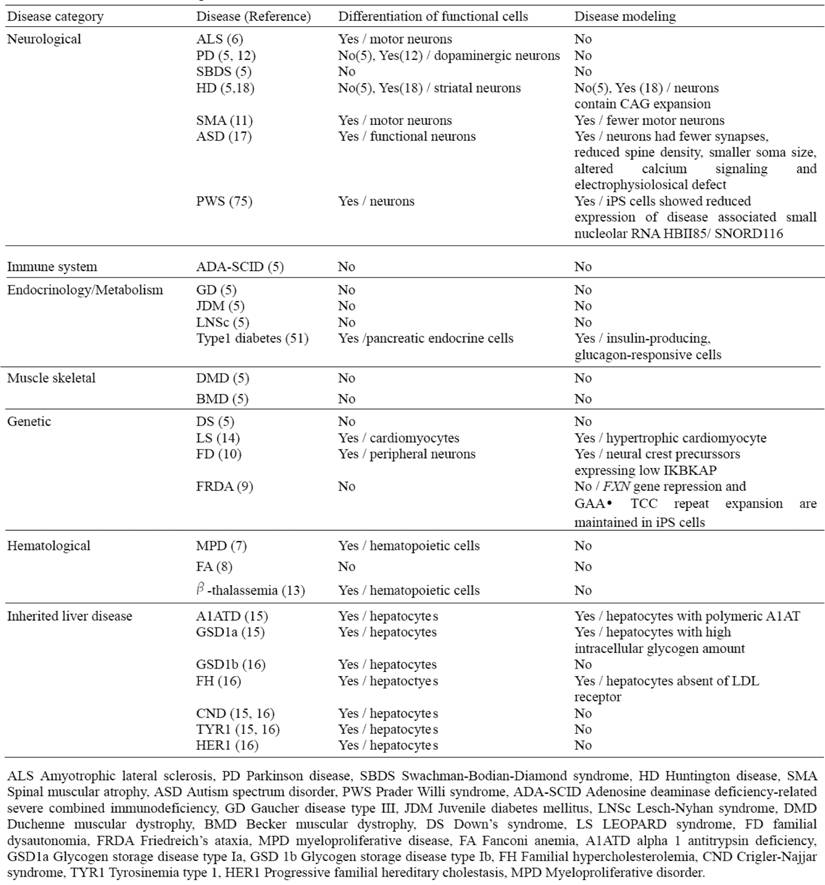
List of current disease-specific iPS cell lines
Therapeutic Potentials of human iPSCs
An ultimate goal for iPSC research will be to use the iPSC (generation and differentiation) technology for therapy. Because iPSCs can bypass the ethical concerns related to ESC derivation and potential issues of allogenic rejection, they may represent a more ideal source to produce patient-specific and disease-specific adult cells for future clinical applications and drug development. Organ transplantation among non-related individuals is also complicated by the limited availability of matched tissues and the requirement for life-long treatment with immunosuppressive drugs that can have serious side effects. Human iPSCs might potentially circumvent these problems, as they could be coaxed into the desired cell types that would already be genetically matched with the patient.
Another key advantage of iPSCs over current transplantation approaches is the possibility of repairing disease-causing mutations by gene targeting and correction technologies. A proof of principle that iPSCs can be used to treat disease by correction of the underlying genetic defect was demonstrated in a mouse model of sickle cell anemia (54). The wild-type β-globin gene was used to replace the defective gene by homologous recombination. Remarkably, transplantation with genetically corrected iPSC-derived hematopoietic progenitors was successful in ameliorating the symptoms of anemia and for restoring physiological function in the diseased animal. In principle, this approach could be applied to any disease in humans for which the underlying mutation is known, and that can be treated by cell transplantation. A similar approach was taken with human individuals with Fanconi anemia, a disease characterized by severe genetic instability (8). In this case, the mutant gene was replaced using lentiviral vectors prior to epigenetic reprogramming of the patient's fibroblasts and keratinocytes, as the genetic instability of the mutant fibroblasts made them non-permissive for iPSC generation. Importantly, these iPSCs could be differentiated into hematopoietic progenitors as efficiently as wild-type iPSCs, stably maintaining the disease-free phenotype in vitro.
Hepatocytes are the main functional cells of the liver and perform a variety of important endocrine and exocrine functions. Research employing human hepatocytes for cellular transplantation has been constrained by the difficulties in sourcing and maintaining viable hepatocytes. A number of innovative cell-based and animal model studies of human liver disorders have highlighted the remarkable regenerative capacity of hepatocytes in vivo, indicating the feasibility of hepatocyte transplantation as a means of replacing lost or diseased hepatic tissue (55-58). Transplantation of hepatocytes derived from human iPSCs could represent an alternative either to liver transplantation in acute liver failure or for the correction of genetic disorders resulting in metabolically deficient states (59).
Conclusion
Since the first description of iPSC generation, there has been remarkable progress toward basic and clinical implementation of the iPSC technologies (Figure 1). However, several key issues remain to be addressed for more accurate disease modeling and therapy; first, patient specific iPSCs needs to be derivated from diseased tissue portions (i.e. hepatocyte within liver cancer) rather than the tissues which do not carry any pathogenetic events (i.e. skin fibroblasts for liver cancer). Second, major challenges also remain in directed differentiation protocols and disease recapitulation technologies that efficiently lead to pure and functional populations of many disease-relevant cell types for the purpose of disease modeling and drug screening. Finally, extensive characterization of the functionality of iPSC-derived somatic cells and their functional equivalence with in vivo counterparts needs to be demonstrated along with the safety for the future regeneration therapy. Given the rapid pace of developments within the iPSC field, it is likely that the generation of safe and effective iPSCs for use in cell therapy as well as in disease modeling and drug screening will be achieved in the near future.
Figure 1
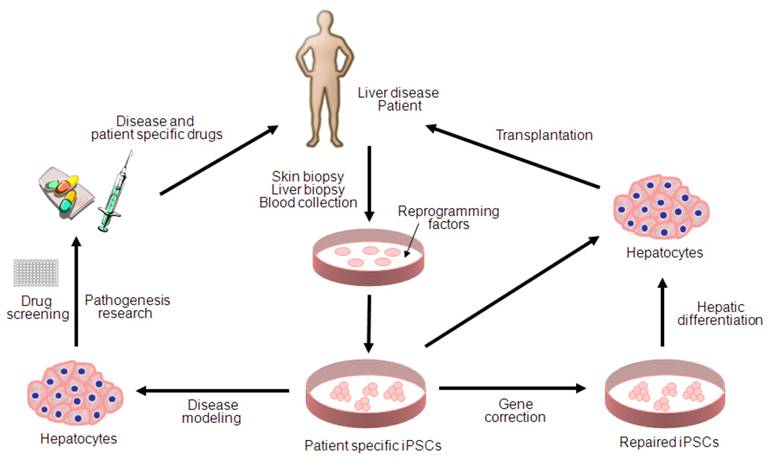
Potential applications of human iPSCs for liver diseases. iPSC technology can be potentially utilized in disease modeling, pathogenesis research, drug discovery, gene therapy and cell replacement therapy. Genetic mutations can be corrected by gene targeting approaches before or after reprogramming. Hepatic cells differentiated from patient specific iPSCs can be used for disease modeling and transplantation purposes.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-76
2. Takahashi K, Tanabe K, Ohnuki M el al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861-72
3. Yu J, Vodyanik MA, Smuga Otte-K. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917-20
4. Park IH, Zhao R, West JA. et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141-6
5. Park IH, Arora N, Huo H. et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877-86
6. Dimos JT, Rodolfa KT, Niakan KK. et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218-21
7. Ye Z, Zhan H, Mali P, Dowey S, Williams DM, Jang YY, Dang CV, Spivak JL, Moliterno AR, Cheng L. Human induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood. 2009;114:5473-5480
8. Raya A, Rodriguez-Piza I, Guenechea G. et al. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature. 2009;460:53-9
9. Ku S, Soraguni E, Campau E. et al. Friedreich's ataxia induced pluripotent stem cells model intergenerational GAA∙TTC triplet repeat instability. Cell Stem Cell. 2010;7:631-7
10. Lee G, Papapetrou EP, Kim H. et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461:402-6
11. Ebert AD, Yu J, Rose FF Jr. et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277-80
12. Soldner F, Hockemeyer D, Beard C. et al. Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964-77
13. Ye L, Chang JC, Lin C. et al. Induced pluripotent stem cells offer new approach to therapy in thalassemia and sickle cell anemia and option in prenatal diagnosis in genetic disease. Proc Natl Acad Sci USA. 2009;106:9826-30
14. Carvajal-Vergara X, Sevilla A, D'Souza SL. et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465:808-12
15. Rashid ST, Corbineau S, Hanna N. et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest. 2010;120:3127-36
16. Ghodsizadeh A, Taei A, Totonchi M. et al. Generation of liver disease-specific induced pluripotent stem cells along with efficient differentiation to functional hepatocyte-like cells. Stem Cell Rev and Rep. 2010;6:622-32
17. Marchetto MC, Carromeu C, Acab A. et al. A model for neural development and treatment of rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527-39
18. Zhang N, An MC, Montoro D. et al. Characterization of human Huntington's disease cell model from induced pluripotent stem cells. PLos Curr. 2010;2:RRN1193
19. Song Z, Cai J, Liu Y. et al. Efficient generation of hepatocyte-like cells from human induced pluripotent stem cells. Cell Res. 2009;19:1233-42
20. Agarwal S, Holton KL, Lanza R. Efficient differentiation of functional hepatocytes from human embryonic stem cells. Stem Cells. 2008;26:1117-27
21. Sullivan GJ, Hay DC, Park IH. et al. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology. 2010;51:329-35
22. Liu H, Ye Z, Kim Y. et al. Generation of endoderm-derived human induced pluripotent stem cells from primary hepatocytes. Hepatology. 2010;51:1810-19
23. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209-18
24. Haridass D, Narain N, Ott M. Hepatocyte transplantation: waiting for stem cells. Curr Opin Organ Transplant. 2008;13:627-32
25. Herrera MB, Bruno S, Buttiglieri S. et al. Isolation and characterization of a stem cell population from adult human liver. Stem Cells. 2006;24:2840-50
26. Lazaro CA, Rhim JA, Yamada Y. et al. Generation of hepatocytes from oval cell precursors in culture. Cancer Res. 1998;58:5514-22
27. Sahin MB, Schwartz RE, Buckley SM. et al. Isolation and characterization of a novel population of progenitor cells from unmanipulated rat liver. Liver Transpl. 2008;14:333-45
28. Czyz J, Wiese C, Rolletschek A. et al. Potential of embryonic and adult stem cells in vitro. Biol Chem. 2003;384:1391-409
29. Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:318-24
30. Nakagawa M, Koyanagi M, Tanabe K. et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101-6
31. Kaji K, Norrby K, Paca A. et al. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771-5
32. Woltjen K, Michael IP, Mohseni P. et al. piggyback transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766-70
33. Stadtfeld M, Nagaya M, Utikal J. et al. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945-9
34. Fusaki N, Ban H, Nishiyama A. et al. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348-62
35. Okita K, Nakagawa M, Hyenjong H. et al. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322:949-53
36. Yu J, Hu K, Smuga-Otto K. et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797-801
37. Huangfu D, Maehr R, Guo W. et al. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol. 2008;26:795-7
38. Zhou H, Wu S, Joo JY. et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009;4:381-4
39. Warren L, Manos PD, Ahfeldt T. et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cell with synthetic modified mRNA. Cell Stem Cell. 2010;7:618-30
40. Miura K, Okada Y, Aoi T. et al. Variation in the safety of induced pluripotent stem cell lines. Nat Biotechnol. 2009;27:743-45
41. Kim K, Doi A, Wen B. et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:280-1
42. Polo JM, Liu S, Figueroa ME. et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nature Biotechnol. 2010;28:848-55
43. Marchetto MCN, Yeo GW, Kainohana O. et al. Transcriptional signature and memory retention of human induced pluripotent stem cells. PLoS One. 2009;4:e7076
44. Laurent-Puig P, Zucman-Rossi J. Genetics of hepatocelluar tumors. Oncogenes. 2006;25:3778-86
45. Bluteau O, Jeannot E, Bioulac Sage P. et al. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat Genet. 2002;32:312-5
46. Wong CM, Fan ST, Ng IO. Beta-catenin mutation and overexpression in hepatocellular carcinoma: clinicopathologic and prognostic significance. Cancer. 2001;92:136-45
47. Tanaka S, Toh Y, Adachi E. et al. Tumor progression in hepatocellular carcinoma may be mediated by p53 mutation. Cancer Res. 1993;53:2884-7
48. Choi SM, Liu H, Kim Y. et al. Multistage hepatic differentiation from human induced pluripotent stem cells. Methods and Protocols. 2010 in press
49. Carette JE, Pruszak J, Varadarjan M. et al. Generation of iPSCs from cultured human malignant cells. Blood. 2010;115:4039-42
50. Miyoshi N, Ishii H, Nagai K. et al. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc Natl Acad Sci USA. 2010;107:40-5
51. Si-Tayeb K, Noto FK, Nagaoka M. et al. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297-305
52. Maehr R, Chen S, Snitow M. et al. Generation of pluripotent stem cells from patients with type 1 diabetes. Proc Natl Acad Sci USA. 2009;106:15768-73
53. Rubin LL. Stem cells and drug discovery: the beginning of a new era? Cell. 2008;132:549-52
54. Hanna J, Wernig M, Markoulaki S. et al. Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science. 2007;318:1920-3
55. Overturf K, al-Dhalimy M, Ou CN. et al. Serial transplantation reveals the stem-cell- like regenerative potential of adult mouse hepatocytes. Am J Pathol. 1997;151:1273-80
56. Rhim JA, Sandgren EP, Degen JL. et al. Replacement of diseased mouse liver by hepatic cell transplantation. Science. 1994;263:1149-52
57. Sandgren EP, Palmiter RD, Heckel JL. et al. Complete hepatic regeneration after somatic deletion of an albumin-plasminogen activator transgene. Cell. 1991;66:245-56
58. Jang YY, Collector MI, Baylin SB, Diehl AM, Sharkis SJ. Hematopoietic stem cells convert into liver cells within days without fusion. Nature Cell Biol. 2004;6:532-9
59. Seifinejad A, Tabebordbar M, Baharvand H. et al. Progress and promise towards safe induced pluripotent stem cells for therapy. Stem Cell Rev and Rep. 2010;6:297-306
60. Loh YH, Agarwal S, Park IH. et al. Generation of induced pluripotent stem cells from human blood. Blood. 2009;113:5476-9
61. Staerk J, Dawlaty MM, Gao Q. et al. Reprogramming of human peripheral blood cells to induced pluripotent stem cell. Cell Stem Cell. 2010;7:20-4
62. Kunisato A, Wakatsuki M, Shinba H. et al. Direct generation of induced pluripotent stem cells from human nonmobilized blood. Stem Cells Dev. 2010 [Epub ahead of print]
63. Seki T, Yuasa S, Oda M. et al. Generation of induced pluripotent stem cells from human terminally differentiated circulation T cells. Cell Stem Cell. 2010;7:11-4
64. Brown ME, Rondon E, Rajesh D. et al. Derivation of induced pluripotent stem cells from human peripheral blood T lymphocytes. PLos One. 2010;5:e11373
65. Loh YH, Hartung O, Li H. et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell. 2010;7:15-9
66. Haase A, Olmer R, Schwanke K. et al. Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell. 2009;5:434-41
67. Eminli S, Foudi A, Stadtfeld M. et al. Differentiation stage determines potential of hematopoietic cells fro reprogramming into induced pluripotent stem cells. Nat Genet. 2009;41:968-76
68. Giorgetti A, Montserrat N, Aasen T. et al. Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2. Cell Stem Cell. 2009;5:353-57
69. Sugii S, Kida Y, Kawamura T. et al. Human and mouse adipose-derived cells support feeder-independent induction of pluripotent stem cells. Proc Natl Acad Sci USA. 2010;107:3558-63
70. Aoki T, Ohnishi H, Oda Y. et al. Generation of induced pluripotent stem cells from human adipose-derived stem cells without c-MYC. Tissue Eng Part A. 2010;16:2197-206
71. Oda Y, Yoshimura Y, Ohnishi H. et al. Induction of pluripotent stem cells from human third molar mesenchymal stromal cells. J Biol Chem. 2010;285:29270-8
72. Aasen T, Raya A, Barrero MJ. et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276-84
73. Kim JB, Greber B, Arauzo-Bravo MJ. et al. Direct reprogramming of human neural stem cells by OCT4. Nature. 2009;461:649-53
74. Utikal J, Maherali N, Kulalert W. et al. Sox2 is dispensible for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J Cell Sci. 2009;122:3502-10
75. Yang J, Cai J, Zhang Y. et al. Induced pluripotent stem cells can be used to model the genomic imprinting disorder Prader Willi syndrom. J Biol Chem. 2010 [Epub ahead of print]
Author contact
![]() Corresponding author: Yoon-Young Jang, M.D., Ph.D., Stem Cell Biology laboratory, Department of Oncology, Johns Hopkins University School of Medicine, 1550 Orleans Street, CRB2 Rm552, Baltimore, MD 21231. Office (410) 502-8195; Email: yjang3edu
Corresponding author: Yoon-Young Jang, M.D., Ph.D., Stem Cell Biology laboratory, Department of Oncology, Johns Hopkins University School of Medicine, 1550 Orleans Street, CRB2 Rm552, Baltimore, MD 21231. Office (410) 502-8195; Email: yjang3edu
Citation styles
APA
Chun, Y.S., Chaudhari, P., Jang, Y.Y. (2010). Applications of Patient-Specific Induced Pluripotent Stem Cells; Focused on Disease Modeling, Drug Screening and Therapeutic Potentials for Liver Disease. International Journal of Biological Sciences, 6(7), 796-805. https://doi.org/10.7150/ijbs.6.796.
ACS
Chun, Y.S.; Chaudhari, P.; Jang, Y.Y. Applications of Patient-Specific Induced Pluripotent Stem Cells; Focused on Disease Modeling, Drug Screening and Therapeutic Potentials for Liver Disease. Int. J. Biol. Sci. 2010, 6 (7), 796-805. DOI: 10.7150/ijbs.6.796.
NLM
Chun YS, Chaudhari P, Jang YY. Applications of Patient-Specific Induced Pluripotent Stem Cells; Focused on Disease Modeling, Drug Screening and Therapeutic Potentials for Liver Disease. Int J Biol Sci 2010; 6(7):796-805. doi:10.7150/ijbs.6.796. https://www.ijbs.com/v06p0796.htm
CSE
Chun YS, Chaudhari P, Jang YY. 2010. Applications of Patient-Specific Induced Pluripotent Stem Cells; Focused on Disease Modeling, Drug Screening and Therapeutic Potentials for Liver Disease. Int J Biol Sci. 6(7):796-805.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.