Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(1):74-86. doi:10.7150/ijbs.7.74 This issue Cite
Research Paper
Lipocalin-2-induced cytokine production enhances endometrial carcinoma cell survival and migration
Hsiu-Hsia Lin1,3, Chi-Jr Liao1, Ying-Chu Lee2, Keng-Hsun Hu2, Hsien-Wei Meng2, Sin-Tak Chu1,2 ![]()
1. Institute of Biochemical Science, College of Life Science, National Taiwan University, Taiwan;
2. Institute of Biological Chemistry, Academia Sinica, Taipei, Taiwan;
3. Department of Internal Medicine, National Taiwan University Hospital, Taiwan.
Received 2010-9-22; Accepted 2011-1-11; Published 2011-1-18
Abstract
Lipocalin-2 (Lcn-2) is an acute-phase protein that has been implicated in diverse physiological processes in mice, including: apoptosis, ion transport, inflammation, cell survival, and tumorigenesis. This study characterized the biological activity of Lcn-2 in human endometrial carcinoma cells (RL95-2). Exposure of RL95-2 cells to Lcn-2 for >24 h reduced Lcn-2-induced cell apoptosis, changed the cell proliferation and up-regulated cytokine secretions, including: interleukin-8 (IL-8), inteleukin-6 (IL-6), monocyte chemotatic protein-1 (MCP-1) and growth-related oncogene (GRO). However, IL-8 mRNA and protein levels were dramatically increased in Lcn-2-treated RL95-2 cells. To determine the IL-8 effect on Lcn-2-treated RL95-2 cells was our major focus. Adding recombinant IL-8 (rIL-8) resulted in decreased caspase-3 activity in Lcn-2-treated cells, whereas the addition of IL-8 antibodies resulted in significantly increased caspase-3 activity and decreased cell migration. Data indicate that IL-8 plays a crucial role in the induction of cell migration. Interestingly, Lcn-2-induced cytokines, secretion from RL95-2 cells, could not show the potent cell migration ability with the exception of IL-8. We conclude that Lcn-2 triggered cytokine secretions to prevent RL95-2 cells from undergoing apoptosis and subsequently increased cell migration. We hypothesize that Lcn-2 increased cytokine secretion by RL95-2 cells, which in turn activated a cellular defense system. This study suggests that Lcn-2 may play a role in the human female reproductive system or in endometrial cancer.
Keywords: apoptosis, cytokine secretion, cellular defense, endometrial carcinoma cells
Introduction
Lipocalins comprise a large and ever-expanding group of proteins that exhibit considerable structural and functional variations, both within and between species [1-4]. Many lipocalins, such as PGD2 synthetase, are bi-functional, acting as both an enzyme and a retinoid transporter [5]. To date, however, the functions of many lipocalins remain unclear. Human neutrophil gelatinase-associated lipocalin (hNGAL/24p3) is a 25-kDa glycoprotein; it has the alternative name lipocalin-2 (Lcn-2). Lcn-2 is homologous to a lipocalin (24p3) from mice [6] and was first found in the granules of human neutrophil [7]. Lcn-2 may have similar functions in different species during the estrous cycle. The uterine endometrium is responsive to sex steroid hormones that vary in a regular cyclic manner [8]. During the menstrual cycle, a rapid series of sequences of cellular proliferation, cytokine production and menstrual shedding occurs in the endometrium [9]. It is important to maintain a balance between cell proliferation and cell death during the dynamic remodeling of tissue, and although such mechanisms have been extensively studied, they remain obscure. In the estrus cycle, Lcn-2 is found in the uterus in high amounts during proestrus and estrus, and declines during metaestrus and diestrus [10]. Based on these data, it is suggested that Lcn-2 secretion is closely associated with the cycle of cell proliferation and apoptosis during the remodeling of the endometrium.
Additionally, Lcn-2 is an acute-phase protein [11, 12] expressed in several tissues and organs during inflammation in humans, mice and rats [13]. Expression of Lcn-2 is up-regulated in response to inflammatory stimuli [11]. Lcn-2 is also up-regulated in the uterus during pre-implantation and parturition and is believed to be part of a local inflammatory response associated with birth [10, 12]. Furthermore, Lcn-2 expression is induced by dexamethasone stimulation of the endometrial carcinoma cell line (RL95-2) [14]. Glucocorticoid levels are highest during proestrus when estrogen levels are highest [15]; glucocorticoid is designated as a stress hormone because its levels in circulation rise in response to stress and change in response to specific physiological stimuli [16]. Therefore, based on glucocorticoid fluctuation in the estrus cycle, Lcn-2 may be one of the stress molecules expressed in a stressed uterine microenvironment during the estrus cycle.
Our previous report [14] identified Lcn-2 as an apoptotic factor that initiates elevated intracellular reactive oxygen species (ROS); triggers activation of caspase-8, -9, and -3; and mediates endometrial carcinoma cell apoptosis. Furthermore, Lcn-2 binds to, and is internalized by, RL95-2 cells but not HeLa cells, indicating cell specificity [14]. Lcn-2 plays a dual role in microglia and is also associated with certain types of cancer; it thus may be involved in tumorigenesis [17, 18]. Taken together, these results suggest that Lcn-2 may play a role in inflammation, cell proliferation and cellular apoptosis in different environments. As we know, cell apoptosis and inflammatory response are closely interrelated. This raises the interesting question as to whether Lcn-2 is simply an apoptotic factor, or whether it is perhaps involved in cytokine production. Based on previous studies, Lcn-2 has diverse physiological functions, implying that Lcn-2 plays different roles in different environments. It encouraged us to clarify the possibility of Lcn-2 performing a dual role in regard to cells.
This study aimed to clarify the relationship between Lcn-2 and cytokine expression in RL95-2 cells, and to investigate whether Lcn-2 is involved in cytokine production. In addition to its apoptotic effect, Lcn-2 may be a multifunctional lipocalin in endometrial carcinoma cell physiology. As inhibition of Lcn-2 expression impairs breast tumorigenesis and metastasis [19]; this protein has been known to affect cell migration [20-22]. Based on our present data, Lcn-2-induced changes in the microenvironment may regulate cell migration. The over-expression of this protein in vivo may trigger cell over-growth, which would lead to Lcn-2 being classified as a tumorigenic factor.
Materials and methods
Cell cultures and Lcn-2 treatment
The human endometrial cell line RL95-2 that was used in this study was obtained from American Type Culture Collection. The cells were grown in complete medium containing MEM (Gibco, CA, US) supplemented with 15% (v/v) heat-inactivated fetal bovine serum (FBS) (PAA, Pasching, Austria), 5 μg/ml insulin (Gibco), 100 U/ml penicillin, and 0.1 mg/ml streptomycin (Gibco). Cells (2 × 104 cells/well) were cultured in complete medium with or without 10 μM Lcn-2 in a 48-well plate for the indicated time. After incubation, culture supernatants were collected and analyzed by Western blotting and cytokine array assays (see below). Murine Lcn-2 was purified from mouse uterine fluid. After purification, the purity of Lcn-2 was confirmed via mass spectrometry analysis. Anti-mouse Lcn-2 antiserum was generated in rabbits as described [23].
Cell viability and proliferation assay
RL95-2 cells were seeded into 96-well plates at a concentration of 1.0 × 104 cells/well. Cells were incubated with complete medium in the absence or presence of 10 μM Lcn-2 and collected over the course of 24-72 h. After incubation, cells were washed with serum-free medium and tested for metabolically active cells using the redox dye resazurin using the VisionBlueTMQuick Cell Proliferation Assay Kit (Catalog #K303-500, Biolinkk, New Delhi, India). Upon reduction by viable metabolically active cells, resazurin becomes highly fluorescent (λex = 545 nm; λem = 595 nm). The number of proliferating cells was determined by a cell counter (Invitrogen, CA, US).
Preparation of cell lysates and Western blot analysis
The conditioned medium from RL95-2 cells cultured in the absence or presence of Lcn-2 was subjected to 12% SDS-PAGE and blotted onto a PVDF membrane (PL-BSP0161, PALL, NY, US). Membranes were blocked in blocking buffer, consisting of 5% skim milk, 1× PBS, 0.05% Tween 20 (Sigma, MO, US), and incubated with anti-Lcn-2 rabbit antibody (1:15,000). Membranes were then incubated with secondary anti-rabbit IgG (1:10,000). Protein bands were visualized using an enhanced chemiluminescence kit (Millipore, WBKLS0050, MA, US) and X-ray film.
RNA extraction, reverse transcriptase polymerase chain reaction (RT-PCR) and quantitative real-time PCR (qPCR)
Total RNA was extracted from the control and Lcn-2-treated RL95-2 cells using an RNeasy Mini kit (Qiagen, Hilden, Germany). The total RNA (500ng) was reverse-transcribed in a 20-μl volume using the MMLV reverse transcriptase kit (M6125H, epicenter, WI, US). qPCR and RT-PCR primer pairs used to test for IL-8, IL-6, MCP-1, GRO, Lcn-2 R, LRP-2 and β-actin mRNA are listed in Table 1. To assess mRNA expression, qPCR and data analysis were performed using 7300 System SDS Software (Applied Biosystems, CA, US). Expression of IL-8, IL-6, MCP-1, and GRO mRNA was normalized for each sample using β-actin mRNA as an internal standard. PCR conditions were 40 cycles at 95ºC for 15 s, 60ºC for 30 s, and 72ºC for 1 min. Standardization of the data was performed by subtracting the signal threshold cycles (CT) of the internal standard (β-actin) from the CT of IL-8, IL-6, MCP-1, and GRO. All PCR product identities were sequenced and confirmed. Lcn-2 R and LRP-2 were performed only for RT-PCR.
Primers for polymerase chain reaction
| Primer Sequences | ||
|---|---|---|
| Forward (5'-3') Reverse (5'-3') | ||
| IL-8 | ATgACTTCCAAgCTggCCgT | TCTCAgCCCTCTTCAAAAACTTCTC |
| GRO | AATCCCCggCTCCTgCg | CCCATTCTTgAgTgTggCTATgA |
| IL-6 | ATCTggATTCAATgAggAgACTTgC | gCTCTggCTTgTTCCTCACTACTCT |
| MCP-1 | ACTgAAgCTCgCACTCTCgC | CTTgggTTgTggAgTgAgTgTTC |
| aLcn-2 R | TgTgCTCACCACCAACgC | CCTCAATCTgCCgCTTCACT |
| bLRP-2 | gCCACCCACAgTgATAAgAgAC | ggCAgAgCAAAgCAgAgATgAg |
| β-actin | gCACCAgggCgTgATgg | gCCTCggTCAgCAgCA |
a and b indicate two Lcn-2 receptors, respectively.
Flow cytometry and fluorescence microscopy analysis of DNA damage
Cells were incubated at 37ºC for the indicated time, washed with PBS, and detached by adding 0.25% trypsin-EDTA (Gibco). Cells were resuspended in 400 μl MEM medium containing 2 μg/ml propidium iodide (PI; Sigma, P4170) and 0.5 μg/ml FITC-labeled annexin-V (Sigma) and incubated at room temperature for 10 min in the dark. Flow cytometry data were collected using a Coulter EPICS XL flow cytometer (Beckman-Coulter, CA, US). Fluorescence was initiated by excitation at 488 nm and was measured by emission filters at 525 nm (annexin V-FITC) and 625 nm (PI). To evaluate nuclear morphology, cells were stained with 2 μg/ml of 4', 6-diamidino-2-phenylindole (DAPI; Sigma) for 2 min to visualize DNA and were observed under a fluorescence microscope (AH3-RFCA; Olympus).
Analysis of caspase-3 activity using fluorescent peptide substrate
Caspase-3 activity was determined using PhiPhiLux G1D2, a fluorophore-labeled caspase-specific substrate (OncoImmunin Inc., A304R1G-6, MD, US). The main peptide amino acid sequence was DEVDGI. Caspase-3 activity was measured by fluorescence intensity. Cells were placed in a 48-well plate with different treatments and incubated with 50 μl of substrate solution (10 μM PhiPhiLux G1D2 with 10% FBS) for 1 h at 37ºC. After washing with PBS, the cells were harvested by trypsinization and fluorescence was measured using flow cytometry.
Measurement of cytokine release using antibody arrays
Cytokine release from RL95-2 cells was analyzed using the Human Cytokine Antibody Array I (RayBiotech, H0108001, GA, US). Conditioned medium was collected from RL95-2 cells treated with or without 10 μM Lcn-2 in serum-free medium for the indicated time, centrifuged at 100×g for 5 min at 4ºC to remove floating cells and debris, and stored at -20ºC for further processing. The cytokine array membranes were blocked with blocking buffer ( provided by the Array kit) for 30 min and then incubated for 18 h with 1 ml conditioned medium at 4ºC. Membranes were then washed three times with PBST (PBS containing 0.5% Tween 20) at room temperature with shaking. The membranes were then incubated with 1 ml biotin-conjugated anti-mouse IgG (1:20) for 3.5 h at room temperature and washed as described above, followed by incubation with 1 ml HRP-conjugated streptavidin (1:1,000) for 1.75 h at room temperature. After a thorough wash, membranes were exposed to a peroxidase substrate (detection buffer C and D; RayBiotech) for 1-2 min in the dark prior to imaging with X-ray film. Signal intensities were quantified using Phoretix 2D v2004 software.
Cell migration assay
Cells grown in the logarithmic phase were harvested and resuspended in DMEM/F12 medium supplemented with 10% fetal calf serum (FCS) at a concentration of 2 × 105 cells/ml. After 24-h incubation at 37ºC, 100 μl of the cell suspension was placed into the upper compartment of 12-well plates, with Transwell inserts. Subsequently, medium in the upper compartment was supplemented with 400 μl of conditioned medium (CM) that had been collected at different time intervals from Lcn2-treated RL95-2 cells after 24 to 48 h of incubation in the presence or absence of anti-IL-8. Other antibody neutralizations, which are specific to each cytokine, were also performed individually. The lower compartments were filled with DMEM/F12 containing 10% FCS, and chambers were incubated at 37ºC for 24 h. The filters were gently rinsed with water, and cells on the upper surface of the filters were removed by wiping the surface with a cotton swab. The filters were then fixed with methanol for 10 min and stained with 0.5% crystal violet in 20% methanol for 20 min, followed by two rinses with water. Cells on the lower surface of the membrane were observed and quantified using a light-inverted microscope at 20X magnification. The number of cells that migrated through the insert toward the bottom chamber was counted in ten random fields using phase contrast microscopy. Next, the filters containing the stained cells were removed from the Transwell chambers and individually transferred to a separate well in a 96-well culture plate. The crystal violet dye retained on the filters was extracted with 33% acetic acid (150 μl), and the absorbance of this solution was colorimetrically measured at 570 nm with an ELISA reader.
Statistical analysis
Statistical analysis was conducted using one-way ANOVA with Dunnett's post test using GraphPad InStat version 3.00 for Windows (GraphPad Software).
Results
Lcn-2 effects on RL95-2 cell morphology
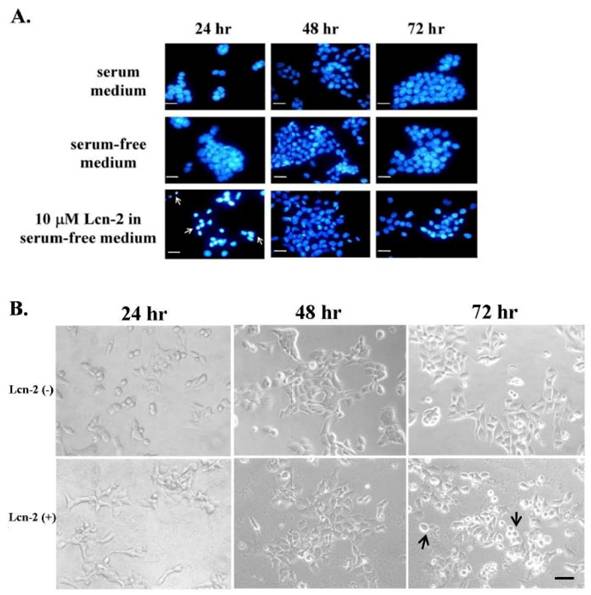
Numerous cytokines are a potent activator of cell survival [24] and are also involved in stress response. To assess possible effects of Lcn-2 on RL95-2 cells, we first observed the cell growth and then tested whether Lcn-2 could play the dual role: apoptosis and cell survival, in these cells. Later, we desired to measure the cytokine secretions. For this purpose, we prolonged the investigation of cell growth under Lcn-2 incubation (> 24h). Using the 10 μM Lcn-2 supplemented culture conditions, we observed DNA fragmentation and condensation as an indicator of apoptosis via microscopic observation. After incubation with or without Lcn-2, cell nuclei were stained with DAPI and observed under a fluorescence microscope (Fig. 1A). According to our previous experiment, a high level of serum in culture media would delay RL95-2 cells' response to Lcn-2 reaction (data not shown). To avoid the serum interference, we verified the cell growth in the media with or without serum. The RL95-2 cells cultured under serum or serum-free media showed normally in nuclei during 72 h culture in this experiment (Fig. 1A, upper and middle panel, respectively). Thus, to avoid possible effects of serum, such as growth factors, serum-free medium was used for subsequent experiments. Interestingly, DNA fragmentation and condensation occurred after 24 h in the presence of Lcn-2 but declined by 48 h (Fig. 1A, bottom panel; arrows). There was an increase in cell numbers in Lcn-2-treated cultures at 48 h as compared with the cell number at Lcn-2-treated for 24 h (Fig. 2A). It indicated that the cell proliferation recovered after 24 h later. The cellular morphology of Lcn-2-treated cells differed markedly from that of untreated cells after 24 h of culture (Fig. 1B). Control cells were round in shape and grouped together (Fig. 1B). At this time, apoptotic cells appeared in Lcn-2 treated cells, compared with the control cells. In addition, the control cells showed tight growth together and Lcn-2-treated cells showed separately in this moment (Fig. 1B). In contrast to 24 h, after 48 h, fewer apoptotic cells could be found and pseudopodia appeared in Lcn-2-treated cells. This coincided with DNA staining data in Fig.1A. It is worth noting that at 72 h incubation more detached cells appeared, indicating that cell migration might have happened or that cells may have died (Fig. 1B, arrows). It is possible that the lower cell number was the result of a shortage of nutrients in the serum-free medium in the 72 h culture. Moreover, cell growth appeared to be restored after persistent incubation of cells in Lcn-2 containing media for >24 h. Together, these results suggest that cell apoptosis seemed to stagnate; moreover, the cells kept growing after persistent exposure of cells in Lcn-2 for more than 24-48 h, but detached after 48-72 h. To avoid cell growth in a nutrient-short condition, we performed our later experiments within 48 h incubation as far as possible.
Lcn-2-induced DNA damage in and cell morphology of RL95-2 cells. RL95-2 cells were cultured on coverslips (1 × 105 cells/coverslip) and incubated with or without 10 μM Lcn-2 for 24-72 h; DNA damage was observed under a fluorescence microscope after DAPI staining. (A) Condensed and fragmented DNA is indicated with arrows. (B) Cell morphology of RL95-2 cells after incubation of cells with or without 10 μM Lcn-2 for 24, 48, and 72 h. Bar = 20 μm.
Cell proliferation and viability of RL95-2 cells after Lcn-2 treatment. RL95-2 cells (2 × 104 cells) were cultured for 36 h in complete medium and then cultured with or without 10 μM Lcn-2 for an additional 12, 24 and 48 h. (A) Cell numbers. (B) Relative percentages of viable cells after Lcn-2 treatment as compared with untreated control cells. The black bar indicates control cells; the blank bar indicates Lcn-2-treated cells. The data are given as the mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001 as compared with the control (n ≥ 4).
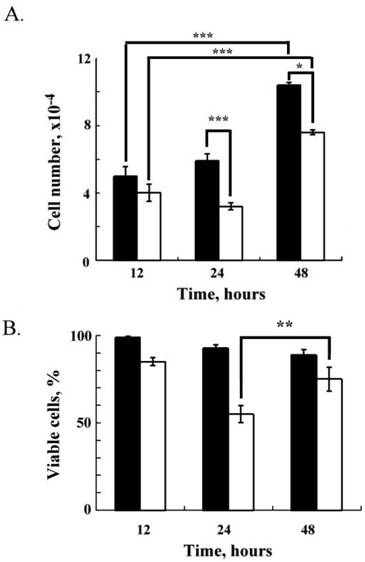
Persistent Lcn-2 effects on RL95-2 cell proliferation and viability
Fig. 2A shows the cell numbers after 12, 24, and 48 h of culture in a medium with or without Lcn-2 (Fig. 2A, blank bar or black bar). In serum-free medium as a control, RL95-2 cells maintained persistent proliferation; the cell numbers were 5 ± 0.6 × 104, 5 ± 0.4 × 104, and 1.04 ± 1.6 × 105 at 12, 24 and 48 h, respectively (Fig. 2A). Thus, these cells proliferated normally in serum-free medium without Lcn-2 for 48 h. In the presence of Lcn-2, cell numbers decreased to 54% (3.6 ± 0.2 × 104) at 24 h (P < 0.001), as compared with the control cells. After a 48 h incubation with Lcn-2, cell numbers did, however, increase to 73% (7.6 ± 1.5 × 104) of the control (P < 0.05), which suggests that cell proliferation, instead of cell apoptosis, occurs in the presence of persistent Lcn-2. Using the redox dye resazurin to analyze cell viability, we determined the percentage of viable cells in these cultures (Fig. 2B). Proportional to the total number of cells, the viable control cells were almost 97±3% at 12 h culture. When RL95-2 cells were incubated with Lcn-2 for 24 h, cell viability decreased to 55 ± 4.9%, as compared with 85 ± 2.2% at 12 h; however, cell viability increased after more than a 24-h incubation, and was maintained at 48 h (75 ± 6.8%). This pattern indicates that under prolonged Lcn-2 incubation, the Lcn-2-resistant cells divided, which seems consistent with the reduction in apoptosis seen after 48 h of Lcn-2 exposure (Fig. 1).
Caspase-3 activity in Lcn-2-treated RL95-2 cells
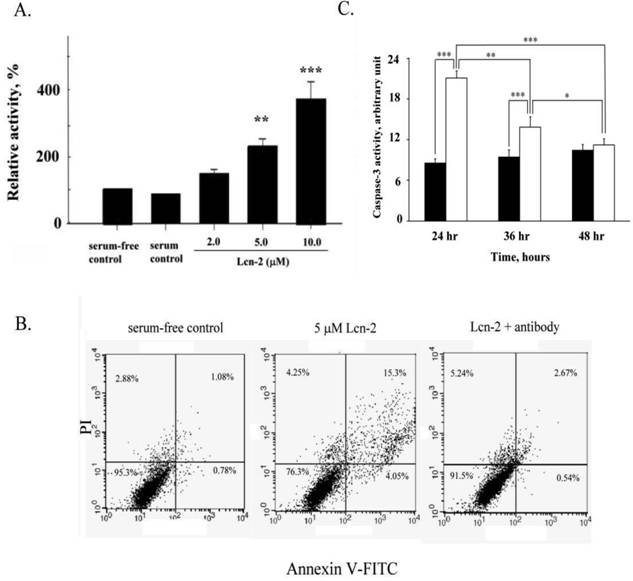
Based on our previous results [14] and the above observations, we confirmed that Lnc-2 induced RL95-2 cell apoptosis under different culture media. Apoptosis was assessed by measuring the activity of the apoptotic enzyme caspase-3 in Lcn-2-treated cells. Caspase-3 activity, relative to that of control cells, increased in a dose-dependent manner after incubation with different concentrations of Lcn-2 for 24 h (Fig. 3A). Even in serum-free medium, caspase-3 activity increased insignificantly, and indicating normal cell growth. We used PI and annexin V-FITC double staining to measure cell death in the presence of 5μM Lcn-2 with or without Lcn-2 antibody. Viable cells increased from 76.3% to 91.5% in the presence of the Lcn-2 antibody (Fig. 3B). Thus the Lcn-2 antibody can neutralize the Lcn-2 reaction, and Lcn-2 may play a specific role in RL95-2 cells. In addition, the caspase-3 activity of Lcn-2-treated cells gradually decreased (Fig. 3C, blank bar), as compared with control viable cells (Fig. 3C, black bar; P<0.001). In 36 h incubation, caspase-3 activity was still at a high level, compared with the control cells (P<0.01). At 48 h, the caspase-3 activity of Lcn-2-treated cells had been markedly reduced and did not show a significant difference as compared with that of the control cells (P > 0.05), less caspase-3 activity was found, compared with 24 h Lcn-2-treated cells (P<0.001; Fig. 3C). The result agrees with the observed increase in cell viability after persistent Lcn-2 exposure. Cell number and cell viability determinations were performed after Lcn-2 exposure for 48 h, and it was ascertained that the reduction in caspase-3 activity did not result from cell lysis (Fig. 2). During 48 h culture under serum-free medium, the caspase-3 activity of the control cells increased slightly, indicating less cell death via apoptosis.
Effects of Lcn-2 on caspase-3 activity in RL95-2 cells. RL95-2 cells were cultured in 48-well plates (2 × 104 cells/well) with different amounts of Lcn-2 for 24 h and then incubated with 50 μl of caspase-3 substrate solution for 1 h at 37ºC. (A) The cleaved substrate has the following excitation and emission peaks: λex = 505 nm and λem = 530 nm. Cells were analyzed by flow cytometry. The data are expressed as the percentage of fluorescence intensity as compared with that of the control cells. (B) For cell apoptosis measurement, RL95-2 cells cultured for 24 h with 5 μM Lcn-2 in the absence or presence of 2.5 μM anti-Lcn-2. Cells were double stained with 2.0 μg/ml annexin V-FITC and 1.0 μg/ml PI for 10 min and analyzed by flow cytometry. (C) Cells were cultured as above with 10 μM Lcn-2 for 24, 36 and 48 h, and then caspase-3 relative activity were measured. The black bar indicates control cells, and the blank bar indicates Lcn-2-treated cells. Data are expressed as the percentage of fluorescence intensity as compared with that of control cells and are presented as the mean ± SEM of multiple experiments (n ≥ 4). ***, P < 0.001.
Confirming the persistence of Lcn-2 in culture
The decrease in caspase-3 activity in RL95-2 cells may have been caused by degradation of Lcn-2 in culture. Thus, we harvested the same volume of conditioned medium from each treatment for each of the incubation times and then measured the amount of Lcn-2 protein by Western blotting. In the serum-free conditioned media (Fig. 4, lanes 1-3), no Lcn-2 was observed in the 24, 36 and 48 h cultures. For the Lcn-2-supplemented conditioned media (Fig. 4, lanes 4-6), Lcn-2 was present in similar amounts across all three time points and showed less protein degradation, as compared with the 20ng purified Lcn-2 (Fig. 4, lane 7). Thus, neither the reduction in cell apoptosis nor the reduction in caspase-3 activity was caused by Lcn-2 degradation.
Quantification of Lcn-2 in RL95-2-conditioned medium. RL95-2 cells were cultured with or without Lcn-2 for the indicated times. Levels of Lcn-2 in conditioned media were determined by 12% SDS-PAGE and Western blotting with anti-Lcn-2 IgG. These data are representative of three independent experiments. (A) Lanes 1-3, conditioned media from RL95-2 cells cultured in serum-free medium for 24, 36 and 48 h, respectively; lanes 4-6, conditioned medium from RL95-2 cells cultured with serum-free medium supplemented with 10 μM Lcn-2 for 24, 36 and 48 h, respectively; lane 7, 20ng of purified Lcn-2; lane 8, cell-free control consisting of an equivalent amount of Lcn-2-supplemented medium incubated for 48 h at 37 ºC without cells. For lane 7, Lcn-2 was loaded on the gel. (B) mRNA levels of two Lcn-2 receptors, LRP-2 and Lcn-2R, were measured by RT-PCR analysis.

Effects of persistent Lcn-2 on cytokine mRNA and protein levels in RL95-2 cells
We next tested whether Lcn-2 stimulated cytokine production in RL95-2 cells, given that Lcn-2 is an acute-phase protein. The culture media were collected from RL95-2 cells culture after incubation with 10 μM Lcn-2. A membrane-bound antibody array technique was then used to characterize the distribution of trace cytokines in the conditioned media. On the array membrane, there were six identical positive control antibodies and four negative controls (Fig. 5A).
Cytokine array analysis of conditioned media from Lcn-2-treated RL95-2 cells. Conditioned medium was collected from RL95-2 cells treated with or without 10 μM Lcn-2 in serum-free medium for the indicated times. Cytokine array membranes were incubated with 1 ml conditioned medium. (A) RayBio® Human Cytokine Antibody Array I Map. (B) The signals of cytokine concentrations in conditioned media; significant signals are labeled with Arabic numerals: 1, MCP-1; 2, IL-6; 3, IL-8; and 4, GRO. Relative intensities of these four cytokines are shown in the lower panels with the intensities of values from control serum-free media set as 1. (C) mRNA levels of cytokines in Lcn-2-treated cells. A total of 4 × 104 cells were incubated with or without 10 μM Lcn-2 in serum-free medium for 24, 36 and 48 h. After incubation, total RNA isolated from cells was reverse transcribed and amplified by RT-PCR using primers for MCP-1, IL-6, IL-8 and GRO. Levels of mRNA were determined by semi-quantitative RT-PCR. The data were calculated by subtracting the signal threshold cycles (CT) of the internal control (β-actin) from the CT of MCP-1, IL-6, IL-8 and GRO, and then the relative expression was obtained by a formula described in the Materials and Methods section. Values are the mean ± SEM of multiple cultures (n ≥ 3). ***, P < 0.001.
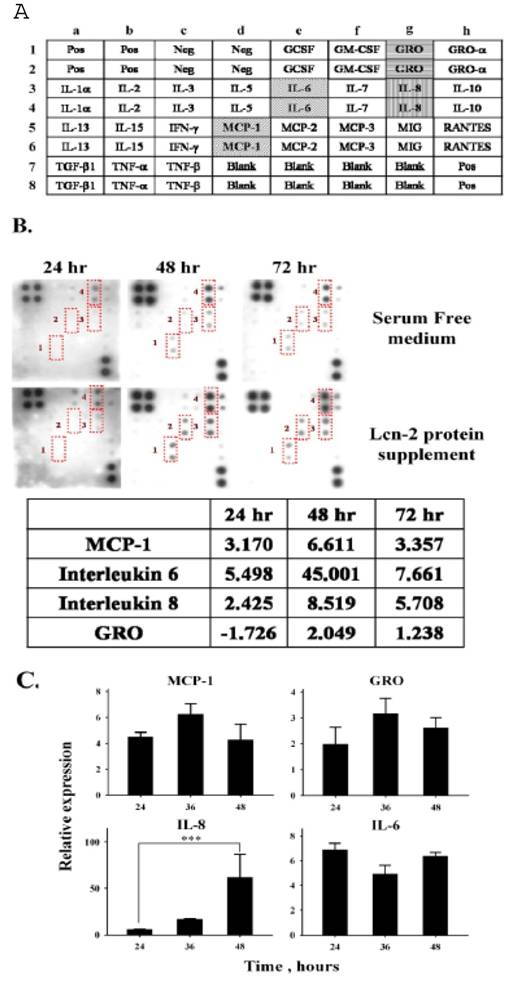
Fig. 5B depicts the results of a typical assay of a 1-ml sample of conditioned medium using the array kit. Under these assay conditions, weak signals were detectable in serum-free medium (Fig. 5B, upper panel); an intense signal was observed only for GRO, with far less-intense signals present for the other cytokines. Four intense signals were detected in conditioned media collected from the 48 and 72 h Lcn-2 cultures: IL-8, IL-6, MCP-1, and GRO (Fig. 5A and B). Changes in cytokines in the conditioned medium should result from Lcn-2-reacted RL95-2 cells. The estimation of the change in these cytokines was based on the relative ratio to the serum-free medium at each time interval, using a non-linear dynamic analysis method with Phoretix 2D software. In the Lcn-2-supplemented medium, the levels of IL-8, IL-6, MCP-1, and GRO were significantly increased 8.5-, 45-, 6-, and 2-fold, respectively, at 48 h (Fig. 5B, bottom panel) as compared with those of the control medium. At 24 h, increases in the expression of these four cytokines may not be sufficient to rescue the cells under apoptotic progression. The cell detachment (Fig. 1B) or nutrient shortage at 72 h might cause less cytokine secretion, and then we were unable to measure the cytokines at high levels. For this reason, we used the 48 h conditional media for the later experiments. Although the data obtained were not quantitative, they were used to obtain a rough estimate of the changes in some detectable cytokines. Even though the fold increase in IL-6 was high, it was secreted at a low basal level in the conditioned medium. The mRNA levels of the four cytokines were detected using semi-quantitative RT-PCR. The expression levels were normalized to β-actin. The relative mRNA levels of Lcn-2-treated cells, divided by the levels in the control cells, equaled the fold increases (Fig. 5C). In Lcn-2-treated cells, the mRNA levels of these four cytokines increased less as compared with those in the control cells, with the exception of IL-8 (P < 0.001). The relative expression mRNA levels of MCP-1, GRO and IL-6 increased to 4.28±1.18, 2.16±0.41 and 6.38±0.31, respectively, compared to the control cells at 48 h. In this respect, mRNA data did coincide with the data from the cytokine array. Possibly, this technique could not accurately measure the small amount of the increase, or these genes were only at a lower basal level. Only the expression and secretion of IL-8 that showed a significant increase at 48 h are easier to detect. IL-8 may act as an autocrine growth factor in endometrial stromal cells [25]. Thus, the secretion of IL-8 may be involved in the reduction in Lcn-2-induced cell apoptosis. To determine the IL-8 effect on Lcn-2-treated RL95-2 cells was our major focus.
Interleukin-8 suppressed Lcn-2-induced apoptosis and improved cell survival
IL-8 expression is significantly increased in late secretory and early-to-middle proliferative phases during the menstrual cycle [26] and may be involved in the growth and proliferation of the endometrium, not only by indirectly stimulating leukocytes to secrete growth factors and cytokines but also by directly stimulating endometrial cell proliferation [25]. Interestingly, Lcn-2 gene expression and protein secretion are present in the proliferative phase of the mouse proestrus stage [10]; its presence may correlate with cytokine secretion during this period. Based on these findings and our data, we hypothesized that Lcn-2-induced IL-8 secretion from RL95-2 cells may correlate with cell apoptosis suppression. To assess the possible effects of IL-8 on Lcn-2-induced RL95-2 cell apoptosis, we analyzed the caspase-3 activity of Lcn-2-treated RL95-2 cells in the presence of IL-8 (Fig. 6A). Because cytokines are active at picomolar levels, IL-8 at picomolar concentrations was used to determine the effect of IL-8 on RL95-2 cells. The data showed no difference in the caspase-3 activity of RL95-2 cells cultured with 20 ng rIL-8 in the absence of Lcn-2 for 36 h. rIL-8 did, however, reduce Lcn-2-induced caspase-3 activity ~20% (from 290 ± 46.1% in control cells to 240 ± 22.5% in rIL-8-treated cells; P < 0.05), as compared with the cells incubated with Lcn-2 alone (Fig. 6A).
Effects of IL-8 on caspase-3 activity in Lcn-2-treated RL95-2 cells. (A) Exogenous rIL-8 (20 ng) was added to Lcn-2-treated RL95-2 cells. (B) IL-8 antibody neutralizing assay. Caspase-3 activities of RL95-2 cells were treated with or without IL-8 antibody for the time period indicated. Cells were then harvested, and enzyme activity was assayed and measured by flow cytometry. 24 h, black bars; 36 h, gray bars; and 48 h, blank bars. The cleaved substrate has the following excitation and emission peaks: λex = 505 nm and λem = 530 nm. Values are the mean ± SEM. *, P < 0.05; ***, P < 0.001 (n ≥ 4).
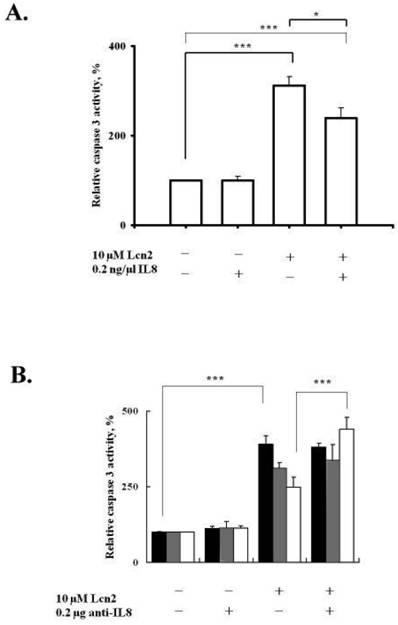
Furthermore, the neutralizing effect of IL-8 antibody on Lcn-2-induced apoptosis was investigated. In the IL8 neutralizing antibody experiment, the caspase-3 activity of cells incubated with Lcn-2 and IL-8 antibody for 48 h was significantly increased by ~35% (from 355 ± 18.6% in the control cells to 462 ± 27.4%; P < 0.001), as compared with Lcn-2 protein treatment only (Fig. 6B). Removing the endogenous IL-8 molecule diminished cell protection from Lcn-2-induced apoptosis. Incubating the cell along with IL-8 antibody did not trigger cellular caspase-3 activity, which is notably different from its effects in control cells. It seems reasonable to suggest that, when Lcn-2 is supplied for 48 h, IL-8 mRNA increases markedly (Fig. 5C). These data suggest that secreted IL-8, which was induced by Lcn-2, suppressed Lcn-2-induced cell apoptosis significantly. Therefore, inhibition of Lcn-2-induced cell apoptosis may be mediated by an IL-8 signal transduction pathway.
Effects of Lcn-2-stimulated RL95-2-conditioned medium on RL95-2 cell migration
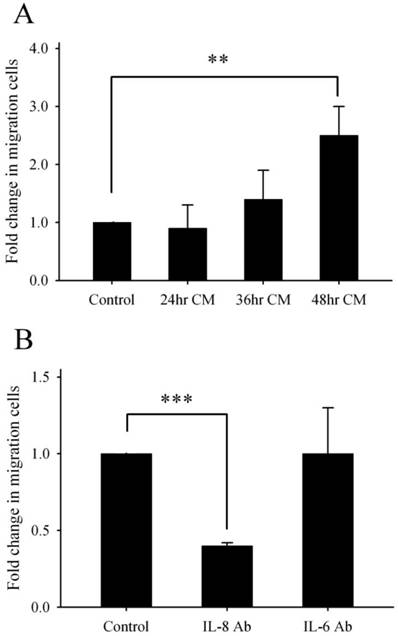
According to the data of Fig. 1B, the detached cells appeared during 48-72 h incubation, the detachment may correlate with cell migration. Lee et al. [27] stated that IL-8 production may regulate cell proliferation, survival and migration. Furthermore, we wanted to evaluate whether Lcn-2 was able to induce IL-8 to promote cell migration. Because previous studies have suggested that IL-8 is involved in cell proliferation and survival, we used a transwell chemotaxis assay to address whether the conditioned media that contained IL-8 affect cell migration of RL95-2 cells. Because of the slow movement of RL95-2 cells, our transwell cell migration assays were performed by following the method of Uchida et al. [28], with migration allowed for 24 h. Migration was determined by the mean number of RL95-2 cells that migrated from the upper to the lower chamber (Fig. 7A). Conditioned media from cultures that had been incubated with Lcn-2 for 48 h increased RL95-2 cell migration significantly (P < 0.01). Because Lcn-2 is induced under serum-free medium [14], we assumed that IL-8 should also be induced under such conditions. Furthermore, we measured the cell migration under serum-free medium with or without IL-8 antibody. As expected, elimination of IL-8 from the medium suppressed cell migration from the upper compartment to the lower compartment (Fig. 7B; P < 0.001). These data indicate that Lcn-2-induced IL-8 could promote the cell migration. The data shown in Fig. 5B is based on four cytokines elevated in Lcn-2 conditional media. Determining whether all of these cytokines correlate with cell migration was also our concern. After neutralization of each cytokine via a specific antibody in 48 h conditional media (48hrCM), we measured the cell migration via transwell assays for 24 h. Actually, the specific antibodies of GRO, MCP-1 and IL-6 in conditional media did not show the correlation with cell migration significantly, even in high doses of these antibodies (data not shown). The IL-6 antibody neutralization assay as representative data was present in Fig. 7B (P>0.05). It indicated that IL-8 production for cell migration is of vital importance.
Effects of Lcn-2-conditioned medium on RL95-2 cell migration. RL95-2 cells (8 × 104) were stimulated with 10 μM Lcn-2 for varying lengths of time, and then culture supernatants were collected from conditioned medium. Cytokine antibodies (anti-IL-8 or anti-IL-6) were added to clarify the effect of these cytokines on 48hrCM-induced cell migration. Cell migration was measured using the transwell assay in the absence or presence of conditioned medium. (A) After incubation for 24 h, RL95-2 cells were stained with 0.5% crystal violet, and cells were counted in ten random fields under a microscope at 20× magnification. (B) OD570 values of cells on the lower surface of the membrane extracted with 33% acetic acid after anti-IL-8 neutralization. These data are representative of three independent experiments. The cell number of RL95-2 incubation under 48hrCM for 24h is as a control experiment. Values are the mean ± SEM. **, P < 0.01; ***, P < 0.001.
Discussion
In RL95-2 cells, apoptosis is induced by Lcn-2 within 24 h of culture [14]. This study evaluated Lcn-2 effects using exogenous Lcn-2 protein, which was purified from mouse uterine fluid, and suggested that, in the presence of Lcn-2, apoptosis may be suppressed and result in improved cell survival. When cells die in vivo, they trigger an inflammatory response leading to the release of a number of molecules that serve as defense and repair mechanisms, and thus the inflammatory response may play a role in limiting further damage to the cells. As an acute-phase protein, Lcn-2 may activate such a cell defense system after the induction of cell apoptosis, via triggering the expression of pro-inflammatory molecules. Numerous reports mention that Lcn-2, as a multifunctional lipocalin in mammals, may play a role as a stress-induced protein that elicits cytokine secretion [29-32]. Exposure of cells to stress results in the generation of ROS and the production of cytokines such as IL-8, which implies that cytokine secretion could be related to intracellular ROS elevation. Furthermore, Lcn-2 elevates intracellular ROS after interacting with RL95-2 cells [14], suggesting that the stimulation of cytokine secretion would be responsive to ROS elevation [33, 34]. The data in this current study show that the production of at least four cytokines increased after 48 h of culture in the presence of Lcn-2. IL-8 mRNA expression showed the most prominent increase among the four cytokines measured and was associated with diminished caspase-3 activity and apoptosis in RL95-2 cells (data not shown).
In mice, IL-8-like molecules are highly expressed in the uterus at the late proestrus and early estrus stages in association with Lcn-2 gene expression (unpublished data). These data suggest that a relationship between Lcn-2 and IL-8 exists during the endometrial cycle. In fact, rIL-8 was associated with reduced Lcn-2-induced apoptosis and caspase-3 activity (nearly 51%, as compared with the absence of IL-8 in the medium). This result may reveal the possibility of an association between Lcn-2 and IL-8 during the menstrual cycle. In the current study, we could not, however, obtain a more significant reduction in caspase-3 activity induced by cell apoptosis to reach a notable level of decrease. This implies that IL-8 may not be a unique factor for cell rescue. In any case, the antibody-neutralizing experiment emphasized that endogenous IL-8 acts as a cell survival factor and that its antibody can block its function. The possibility of other cytokines induced by Lcn-2 that might be involved in the suppression of apoptosis and improved cell survival were also of concern to us. It suggested that IL-8 did play an important role in cell survival, and that IL-8 signal transduction may be one of the mediators to prevent the Lcn-2-induced cell apoptosis. IL-8 directly enhances endothelial cell proliferation, survival and MMP expression [27]. This result suggests that cytokines, such as IL-8, may affect other cell functions, such as cell migration. Here we found that cell migration was increased in RL95-2 cells that were treated with supernatants from cultures of Lcn-2-stimulated RL95-2 cells. Our results are consistent with previous studies that demonstrated that Lcn-2 is expressed during apoptosis [30, 35] and that it has dual functions, including apoptosis and cell survival [29]. In the case of IL-6, MCP-1 and GRO, secretion from Lcn-2-treated RL95-2 cells, each of the cytokines might act as a potent force for cell migration [36, 37]. In the antibody neutralization assay, the results revealed that the elimination of the IL-6, MCP-1 and GRO from the Lcn-2-48hrCM could not affect the cell migration. Due to the expression of IL-6, MCP-1 and GRO at a low level following cell apoptosis, we hypothesized that they may act together in having a synergetic effect on cell survival rather than on cell migration. That is to say, the mutual influence between each cytokine is essential for regulating the physiological function of Lcn-2-treated-cells [38]. Further evidence is necessary to confirm this complicated relationship.
Herein, Lcn-2 was characterized as a bi-functional molecule that plays a role in apoptosis and survival in endometrial carcinoma cells. Perhaps Lcn-2 functions via a different pathway to trigger cell death and/or cell survival at different times or via different Lcn-2 receptors. Alternatively, some unknown molecules may have been elicited in Lcn-2-induced apoptotic cells that then triggered cytokine secretion during apoptosis. Lcn-2 induces the epithelial-to-mesenchymal transition in breast cancer cells and promotes invasion [39], which implies that Lcn-2-induced cell survival may play a role in tumorigenesis. Recently, Lcn-2 expression has been used for the early diagnosis of pancreatic and other cancers [40-42]. Together, these findings reveal the importance of Lcn-2 and its biological significance.
In summary, this report is the first to show that the suppression of Lcn-2-induced apoptosis resulted in increased cytokines secretion and cell migration. We suggest that Lcn-2-induced cytokines secretion may improve cell survival functions. As it is a known cancer marker, Lcn-2 may indirectly correlate with the pathogenesis of uterine diseases, such as uterine tumorigenesis mediated by IL-8 secretion. We inferred that over-expression of Lcn-2 in the uterus may be related to uterine cancer formation via the increased expression of inflammatory molecules. Clearly, more studies are needed to further understand possible roles for Lcn-2 in tumorigenesis in the female reproductive system.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Flower DR. The lipocalin protein family: a role in cell regulation. FEBS Lett. 1994;354:7-11
2. Flower DR. The lipocalin protein family: structure and function. Biochem J. 1996;318( Pt 1):1-14
3. Lopez-Boado YS, Klaus M, Dawson MI, Lopez-Otin C. Retinoic acid-induced expression of apolipoprotein D and concomitant growth arrest in human breast cancer cells are mediated through a retinoic acid receptor RARalpha-dependent signaling pathway. J Biol Chem. 1996;271:32105-32111
4. Garibotti M, Navarrini A, Pisanelli AM, Pelosi P. Three odorant-binding proteins from rabbit nasal mucosa. Chemical senses. 1997;22:383-390
5. Samy ET, Li JC, Grima J, Lee WM, Silvestrini B, Cheng CY. Sertoli cell prostaglandin D2 synthetase is a multifunctional molecule: its expression and regulation. Endocrinology. 2000;141:710-721
6. Akerstrom B, Flower DR, Salier JP. Lipocalins: unity in diversity. Biochem Biophys Acta. 2000;1482:1-8
7. Kjeldsen L, Bainton DF, Sengelov H, Borregaard N. Identification of neutrophil gelatinase-associated lipocalin as a novel matrix protein of specific granules in human neutrophils. Blood. 1994;83:799-807
8. Walmer D.K, Wrona M.A, Hughes C.L, Nelson K.G. Lactoferrin expression in the mouse reproductive tract during the natural estrous cycle: correlation with circulating estradiol and progesterone. Endocrinology. 1992;131:1458-1466
9. Salamonsen LA. Tissue injury and repair in the female human reproductive tract. Reproduction. 2003;125:301-311
10. Huang HL, Chu ST, Chen YH. Ovarian steroids regulate 24p3 expression in mouse uterus during the natural estrous cycle and the preimplantation period. J Endocrinol. 1999;162:11-19
11. Liu Q, Nilsen-Hamilton M. Identification of a new acute phase protein. J Biol Chem. 1995;270:22565-22570
12. Liu Q, Ryon J, Nilsen-Hamilton M. Uterocalin: a mouse acute phase protein expressed in the uterus around birth. Mol Reprod Dev'. 1997;46:507-514
13. Kjeldsen L, Cowland JB, Borregaard N. Human neutrophil gelatinase-associated lipocalin and homologous proteins in rat and mouse. Biochim Biophys Acta. 2000;1482:272-283
14. Lin HH, Li WW, Lee YC, Chu ST. Apoptosis induced by uterine 24p3 protein in endometrial carcinoma cell line. Toxicology. 2007;234:203-215
15. Pollard I, White BM, Bassett JR, Cairncross KD. Plasma glucocorticoid elevation and desynchronization of the estrous cycle following unpredictable stress in the rat. Behavioral biology. 1975;14:103-108
16. Hardy MP, Gao HB, Dong Q, Ge R, Wang Q, Chai WR, Feng X, Sottas C. Stress hormone and male reproductive function. Cell and tissue research. 2005;322:147-153
17. Lee S, Lee J, Kim S, Park JY, Lee WH, Mori K, Kim SH, Kim IK, Suk KA. Dual role of lipocalin 2 in the apoptosis and deramification of activated microglia. J Immunol. 2007;179:3231-341
18. Leng X, Lin H, Ding T, Wang Y, Wu Y, Klumpp S, Sun T, Zhou Y, Monaco P, Belmont J, Aderm A, Akrira S, Strong R, Arlinghaus R. Lipocalin 2 is required for BCR-ABL-induced tumorigenesis. Oncogene. 2008;27:6110-6119
19. Leng X, Ding T, Lin H, Wang Y, Hu L, Hu J, Feig B, Zhang W, Pusztai L, Symmans WF, Wu Y, Arlinghaus RB. Inhibition of lipocalin 2 impairs breast tumorigenesis and metastasis. Can Res. 2009;69:8579-84
20. Shi H, Gu Y, Yang J, Xu L, Mi W, Yu W. Lipocalin 2 promotes lung metastasis of murine breast cancer cells. J Exp Clin Cancer Res. 2008;27:83-92
21. Yang J, Beilenberg DR, Rodig SJ, Doiron R, Clifton MC, Kung AL. et al. Lipocalin 2 promotes breast cancer progression. Proc Natl Acad Sci USA. 2009;106:3913-3918
22. Fougere M, Gaudineau B, Barbier J, Guaddachi F, Feugeas JP, Auboeuf D, Jauliac S. NFAT3 transcription factor inhibits breast cell motility by targeting the lipocalin 2 gene. Oncogene. 2010;29:2292-2301
23. Chu ST, Huang HL, Chen JM, Chen YH. Demonstration of a glycoprotein derived from the 24p3 gene in mouse uterine luminal fluid. Biochem J. 1996;316(Pt 2):545-550
24. Ottaviani E, Malagoli D, Franchini A. Invertebrate humoral factors: cytokines as mediators of cell survival. Prog Mol Subcell Biol. 2004;34:1-25
25. Arici A, Seli E, Zeyneloglu HB. et al. Interleukin-8 induces proliferation of endometrial stromal cells: a potential autocrine growth factor. J Clin Endocrinol Metab. 1998;83:1201-1205
26. Arici A, Seli E, Senturk LM. et al. Interleukin-8 in the human endometrium. J Clin Endocrinol Metab. 1998;83:1783-1787
27. Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinase production and regulated angiogenesis. J Immunol. 2003;170:3369-3376
28. Uchida H, Maruyama T, Ono M, Ohta K, Kajitani T, Masuda H. et al. Histone deacetylase inhibitors stimulate cell migration in human endometrial adenocarcinoma cells through up-regulation of glycodelin. Endocrinol. 2007;148:896-902
29. Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. 2005;123:1293-1305
30. Tong Z, Wu X, Kehrer JP. Increased expression of the lipocalin 24p3 as an apoptotic mechanism for MK886. Biochem J. 2003;372:203-210
31. Caramuta S, De Cecco L, Reid JF. et al. Regulation of lipocalin-2 gene by the cancer chemopreventive retinoid 4-HPR. Int J Cancer. 2006;119:1599-1606
32. Schmidt-Ott KM, Mori K, Li JY, Kalandadze A, Cohen DJ, Devarajan P, Barasch J. Dual Action of Neutrophil Gelatinase-Associated Lipocalin. J Am Soc Nephrol. 2007;18:407-413
33. Ito K, Hanazawa T, Tomita K, Barnes PJ, Adcock IM. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochem Biophys Res Commun. 2004;315:240-245
34. Boncoeur E, Criq VS, Bonvin E. et al. Oxidative stress induces extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase in cystic fibrosis lung epithelial cells: Potential mechanism for excessive IL-8 expression. Int J Biochem Cell Biol. 2008;40:432-446
35. Devireddy LR, Teodoro JG, Richard FA, Green MR. Induction of apoptosis by a secreted lipocalin that is transcriptionally regulated by IL-3 deprivation. Science. 2001;93:829-834
36. Gordon RJ, McGregor AL, Connor B. Chemokines direct neural progenitor cell migration following striatal cell loss. Mol Cell Neurosci. 2009;41:219-232
37. Jovanović M, Vićovac L. Interleukin-6 stimulates cell migration, invasion and integrin expression in HTR-8/SVneo cell line. Placenta. 2009;4:320-328
38. Tseng Y-L, Wub M-H, Yang H-C, Wang C-Y, Lin C-F. Autocrine IL-6 regulates GRO-α production in thymic epithelial cells. Cytokine. 2010;51:195-201
39. Yang J, Moses MA. Lipocalin 2. A multifaceted modulator of human cancer. Cell Cycle. 2009;8:1-6
40. Lim R, Ahmed N, Borregaard N, Riley C, Wafai R, Thompson EW, Quinn MA, Rice GE. Neutrophil gelatinase-associated lipocalin (NGAL) an early-screening biomarker for ovarian cancer: NGAL is associated with epidermal growth factor-induced epithelio-mesenchymal transition. Int J Cancer. 2007;120:2426-2434
41. Bauer M, Eickhoff JC, Gould MN. et al. Neutrophilgelatinase-associated lipocalin (NGAL) is a predictor of poor prognosis in human primary breast cancer. Breast Cancer Res Treatment. 2008;108:389-397
42. Moniaux N, Chakraborty S, Yalniz M, Gonzalez J, Shostrom VK, Standop J, Lele SM, Ouellette M, Pour PM, Sasson AR, Brand RE, Hollingsworth MA, Jain M, Batra SK. Early diagnosis of pancreatic cancer: neutrophil gelatinase-associated lipocalin as a marker of pancreatic intraepithelial neoplasia. Br J Cancer. 2008;98:1540-1547
Author contact
![]() Corresponding author: Institute of Biological Chemistry, Academia Sinica, P.O. BOX 23-106, 10617 Taipei, Taiwan. Tel: 886-2-23665582; Fax: 886-2-336664073; E-mail: stc316sinica.edu.tw
Corresponding author: Institute of Biological Chemistry, Academia Sinica, P.O. BOX 23-106, 10617 Taipei, Taiwan. Tel: 886-2-23665582; Fax: 886-2-336664073; E-mail: stc316sinica.edu.tw