Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
INTRODUCTION
MATERIAL AND METHODS
RESULTS
DISCUSSION
Acknowledgements
CONFLICTS OF INTEREST
References
Figures

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(2):193-208. doi:10.7150/ijbs.7.193 This issue Cite
Research Paper
Interactions of MCP1 with Components of the Replication Machinery in Mammalian Cells
Elsa Bronze-da-Rocha1,2,4, ![]() , Chii-Mei Lin4, Tsutomu Shimura3,4, Mirit I. Aladjem4
, Chii-Mei Lin4, Tsutomu Shimura3,4, Mirit I. Aladjem4
1. Departamento de Ciências Biológicas, Laboratório de Bioquímica, Faculdade de Farmácia da Universidade do Porto, Portugal
2. Instituto de Biologia Molecular e Celular da Universidade do Porto, Portugal
3. Department of Pathology, Institute of Development Aging and Cancer, Tohoku University, Seiryo-machi, Sendai, Japan
4. Laboratory of Molecular Pharmacology, CCR/NCI/NIH, Bethesda, USA
Received 2010-11-19; Accepted 2011-2-12; Published 2011-2-17
Abstract
Eukaryotic DNA replication starts with the assembly of a pre-replication complex (pre-RC) at replication origins. We have previously demonstrated that Metaphase Chromosome Protein 1 (MCP1) is involved in the early events of DNA replication. Here we show that MCP1 associates with proteins that are required for the establishment of the pre-replication complex. Reciprocal immunoprecipitation analysis showed that MCP1 interacted with Cdc6, ORC2, ORC4, MCM2, MCM3 and MCM7, with Cdc45 and PCNA. Immunofluorescence studies demonstrated the co-localization of MCP1 with some of those proteins. Moreover, biochemical studies utilizing chromatin-immunoprecipitation (ChIP) revealed that MCP1 preferentially binds replication initiation sites in human cells. Interestingly, although members of the pre-RC are known to interact with some hallmarks of heterochromatin, our co-immunoprecipitation and immunofluorescence analyses showed that MCP1 did not interact and did not co-localize with heterochromatic proteins including HP1β and MetH3K9. These observations suggest that MCP1 is associated with replication factors required for the initiation of DNA replication and binds to the initiation sites in loci that replicate early in S-phase. In addition, immunological assays revealed the association of MCP1 forms with histone H1 variants and mass spectrometry analysis confirmed that MCP1 peptides share common sequences with H1.2 and H1.5 subtypes.
Keywords: MCP1, DNA replication, pre-replication complex, histone H1, interacting proteins
INTRODUCTION
Eukaryotic replication starts with the assembly and the formation of the pre-replication complex (pre-RC) at replication origins during late mitosis or early G1 phase. Initiation of DNA replication requires the binding of the origin recognition complex (ORC) to particular DNA sequences termed replication origins [1-3]. ORC serves as a loading dock for the binding of Cdc6 and Cdt1 that then recruit MCM2-7 proteins to form the pre-replication complex that is activated upon binding of MCM10 [3-8]. Phosphorylation by CDKs and Cdc7-Dbf4 promote the attachment of Cdc45 that is required to load DNA polymerase-alpha as well as for chromosome unwinding [9-11]. RPA recruits primase to the origins and stabilizes unwound DNA [3, 10]. Proliferating Cell Nuclear Antigen (PCNA) is needed to enhance the processivity of DNA polymerases [4, 12-14]. The initiation of DNA replication is also regulated by structural transitions in chromosome organization that require the interaction of histone H1 and its variants with nucleosomes [15, 16] via high-mobility group (HMG) proteins that compete with the binding of H1 to chromatin [17] and through posttranslational modifications of histones [7, 16, 18-20].
Metaphase Chromosome Protein 1 (MCP1), an evolutionarily conserved protein with two forms, 33 kDa and 31 kDa [21], was first identified as the target of an antibody present in serum from a mouse with systemic lupus erythematosus (SLE). SLE is an autoimmune disease, in human and animals, characterized by the production of autoantibodies that react with a wide number of cellular antigens, mostly localized within the nucleus [21-23]. MCP1 exhibits a granular nuclear localization in interphase cells, although it is excluded from the nucleolus, and during mitosis MCP1 is tightly bound to condensed chromosomes [24]. Functional studies showed that MCP1 is required for DNA replication, in particular at G1/S transition and during early S-phase, since microinjection of the specific antibody against MCP1 into the nucleus of human HeLa and rat kangaroo Ptk2 cells prevents S-phase progression [25], suggesting that MCP1 is specifically required for the initiation of replication events.
In order to identify proteins that interact with MCP1 in the early events of DNA replication we determined whether MCP1 binding to replication factors varied at different stages of interphase, whether it co-localized with replication components and whether it preferentially binds replication origins.
MATERIAL AND METHODS
Cell lines
Human HeLa (CCL-93021013 cells from ECACC, a human cervix epithelial carcinoma cell line; chromosome number 2n=46) and MO59K cells (ATCC-CRL2365, a human brain, glial cell, glioblastoma, glioma; modal chromosome number = 75 with a range of 65 to 79) were grown as monolayers in GlutaMax Dulbecco's modified Eagle medium (DMEM) supplemented with antibiotics (100 i.u./ml penicillin, 100 μg/ml streptomycin, and 25 μg/ml amphotericin B), and 10% fetal calf serum (FCS), at 37ºC under 5% CO2. Human K562 (ECACC-89121407 cells), a human Caucasian chronic myelogenous leukaemia cell line (chromosome number 2n=46) was grown in RPMI supplemented with antibiotics and 10% FCS, at 37ºC under 5% CO2. Tissue culture reagents were supplied by Gibco, BRL (Gaithersburg, MD, USA).
Cell synchronization and drug treatments
HeLa and MO59K cells were plated at 1x106/ml and were synchronized G1/S with 1 μg/ml aphidicolin (APH), 2.5 mM hydroxyurea (HU) and 2.5 mM thymidine (Thym) for 18 hours or by double thymidine block (DTB). Releasing experiments were performed after washing the drugs, by incubation in culture media without drugs, for 8 hours. In both, synchronization and releasing experiments, cells were washed, pulse labeled with 50 μM of 5-bromo-2'-deoxiuridine (BrdU; Sigma Chemical Co) for 20 minutes, fixed in 70% ethanol overnight at 4ºC or with 3.7% paraformaldehyde (PFA) in HPEM buffer (30 mM Hepes, 65 mM Pipes, 10 mM EGTA, 2 mM MgCl2, pH=6.9, containing 0.5% Triton X-100) at room temperature (RT) for 10 minutes [25]. Results represent three assays performed in duplicated for each cell line.
Centrifugal elutriation and cell cycle analysis
Human K562 cells at different phases of cell cycle were enriched by centrifugal elutriation (Beckman Coulter, Avanti J-20 centrifuge and JE-5.0 rotor with a 40-ml chamber). Rotor speed was maintained at 2,000 rpm at 4ºC, and the medium flow was controlled by a Cole-Parmer Masterflex pump. Cells were equilibrated in the chamber for 10 minutes with a constant flow of 20 ml/min for efficient separation and the removal of cellular debris. Fractions (50 ml) were collected at increasing flow rates ranging from 24 to 50 ml/min and samples of each fraction analyzed by FACS (FACSCalibur flow cytometer, Becton Dickinson, Mountain View, CA) with excitation of 488 nm to establish distribution of cells through the cell cycle. Cell numbers were normalized prior to preparation of nuclear extracts. HeLa cells labeled with 50 μM BrdU, were trypsinized, washed twice with PBS (phosphate-buffered saline) and fixed with ice-cold ethanol 70% overnight. DNA from 1x106 cells/ml was denaturated in 1.5 N HCl for 30 minutes at room temperature, and BrdU was labeled with mouse anti-BrdU-FITC (0.5 μg/ml) antibody (Becton Dickinson & Co). Cells were resuspended in DNA staining solution [0.5 mg/ml RNase (Quiagen, USA), 50 μg/ml propidium iodide (Sigma Chemical Co) in PBS]. DNA content of the cells was analyzed by FACS. Data were acquired in a listmode data file, gated to 25,000 events in cell cycle, using the CellQuest Pro software, version 4.0.2 (Becton Dickinson) included in the system. Cell cycle was analyzed using the CellQuest Pro software program. Results represent three assays performed in duplicated.
Immunoprecipitation and western blotting
HeLa, MO59K, and K562 nuclei preparations were prepared as described [26], and chromatin digestion was performed treating nuclear extracts with micrococcal nuclease 40μg/ml, for 30 minutes on ice. K562 histone protein preparations were performed according to the method previously described [27]. Total HeLa cell extracts were prepared as described [21]. Immunoprecipitation experiments were performed according to published methods [28]. Briefly, isolated nuclei were resuspended in 1 ml of NET buffer (50 mM Tris-HCl pH=7.5, 150 mM NaCl, 1 mM EDTA, 0.1% Nonidet P-40, 0.25% gelatin, 0.5% sodium deoxycholate) containing 0.1 mM PMSF, 5 mM β-glycerophosphate and 1% of a standard protease inhibitor cocktail (Sigma Chemical Co), and incubated with pre-immune sera and protein A Sepharose beads (Santa Cruz Biotechnology, Inc) 30 minutes at 4ºC. Cleared lysate was incubate (2 hours at 4ºC) separately with the antibody of interest or with immunoglobulin G. Antibody complexes were precipitated (1 hour at 4ºC) with protein A or protein G Sepharose beads (Santa Cruz Biotechnology, Inc). Precipitates were extensively washed with NET buffer and resuspended in SDS-sample buffer. After electrophoresis, western blotting was performed using polyvinylidene difluoride (PVDF) membrane (Immobilon-P-Milipore, Bedford, MA) as described previously [28]. Immune signals were detected using the SuperSignal West Pico Chemiluminescent Substrate (Pierce, USA).
Immunofluorescence
Indirect immunofluorescence was performed according to the methods previously described [24, 25]. Cells were fixed in 3.7% PFA in HPEM buffer at RT for 10 minutes. For PCNA detection, cells were treated with hypotonic lyses solution (10 mM Tris-HCl pH=7.4, 2.5 mM MgCl2, 0.5% Nonidet P-40, 1 mM PMSF) for 8 minutes and fixed with 4% PFA for 10 minutes followed by ice-cold methanol for 15 minutes. DNA visualization was performed using 0.5 μg/ml 4',6-diamidino-2-phenylindole (DAPI) in mounting media (Biomeda Corp., CA). All preparations were observed in an Olympus IX 70 microscope using 63x and 100x objectives. Images were processed with Adobe Photoshop 7.0 (Adobe) software.
Chromatin immunoprecipitation (ChIP)
Exponential growing human K562 cells at different phases of the cell cycle were obtained by centrifugal elutriation (Beckman Coulter, Avanti J-20 centrifuge). Analysis of asynchronous cells was performed in a FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA). Cells were fixed with 1% formaldehyde and quenching of cross-link was performed with glycine. Cells were sonicated six times with a 2-mm tip of a sonicator (Sonics & Material, Inc.) at the maximum setting for 20 seconds, at 1 minute intervals. After centrifugation at 14,000 rpm for 20 minutes, the cleared supernatant was incubated in 1X RIPA buffer [10 mM Tris pH=8.0, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1 mM EDTA, 1 mM PMSF an 1% of a standard protease inhibitor cocktail (Sigma Chemical Co)]. To reduce nonspecific binding to protein A, chromatin was pre-cleared with 10 μl plus protein A agarose (Santa Cruz Biotechnology, Inc) for 1 hour at 4ºC with rotation. The pre-cleared chromatin (0.5 ml) was incubated in the presence and absence of 10 μg of anti-MCP1 antibody (mAb402) and was rotated at 4ºC for 12-14 hours. Protein A beads were added to the ChIP mixture and incubated another 4-6 hours. The beads were washed with 1X RIPA buffer, three times with 1X RIPA plus 0.5 M NaCl, twice with Tris-LiCl buffer (10 mM Tris-HCl pH=8.0, 0.25 mM LiCl, 1% NP-40, 1% deoxycholate and 1mM EDTA), and twice with TE (pH=8). A volume of 0.5 ml elution buffer (10 mM Tris-HCl pH=8.0, 200 mM NaCl, 0.5% SDS and 1 mM EDTA), was then added to protein A beads, and this mixture was incubated at 65ºC for 12-14 hours, followed by treatment with RNase and proteinase K. The DNA was extracted with phenol/chloroform/isoamyl alcohol, precipitated, and resuspended in diethylpyrocarbonate (DEPC) water. DNA concentrations of the samples were determined by Pico green fluorescence (Molecular Probes). Quantitative PCR amplification was used to amplify ChIP-enriched with DNA using specific primers to early origins [human beta-globin origin (hGB), human lamin B2 (hLB2)], primers to late origin [human amylase (hA)] and primers for non-origin [human collagen (hC), human gamma globulin (hGG)]. Pair of forward (F) and reverse (R) primers were the following for: (i) hBG-F-(5'-GGTGAAGGCTCATGGCAAGA-3'), hBG-R-(5'-AAAGGTGCCCTTGAGGTTGTC-3'); (ii) hLB2-F-(5'-TGGGACCCTGCCCTTTTT-3'), hLB2-R-(5'-CGTGACGAAGAGTCAGCT-3'); (iii) hA-76-F-(5'-GATTGCCGAATATATGAACCATCTC-3'), hA-163-R(5'-TATGTCTCCAGGCCACATGTG-3'); (iv) hC-120-F-(5'-TCTGAGAAGCCGTCCTCGTTA-3'); hC-200-R-(5'-CCAACAGTGGAGACACCCTTCTA-3'); (v) hGG-F-(5'-GGCAAGAAGGTGCTGACTTCC-3'), hGG-R-(5'-GCAAAGGTGCCCTTGAGATC-3'). Probes were labeled 5' with FAM and 3' with TAMRA. Some experiments were performed with VIC labeled probes with similar results. Probes were the following for: (1) hBG (5'-CCTTTAGTGATGGCCTGGCTCACCTG-3'); (ii) hLB2 (5'-TTCTAGTGAGCCTCCGAC-3'); (iii) hA (5'-CATCAATTCTGAACCCTGCAACACCAATG-3'); (iv) hC-3' (5'-CGCCGCCTGGCCTGTTCCA3'); (v) hGG (5'-TGGGAGATGCCATAAAGCACCTGGA-3'). The hBG and hLB2 are located 5.2kb and 2.4kb, respectively, from the replication origin, while hA is away 104kb. For non-origin sequences the locations are 5.2kb for hGG and 47kb for hC. Real-time PCR and data analysis was performed as described previously [29]. Results represent three assays performed in duplicated.
Identification of proteins by liquid chromatography coupled with tandem mass spectroscopy (LC-MS/MS)
HeLa or K562 nuclear extracts were immunoprecipitated with anti-MCP1 antibody. After, samples were applied in a NUPage Novex 12% gel and electrophoresis was performed in a cold NUPage MOPS-SDS running buffer. The gel was stained with Coomassie brilliant blue (CBB). Stained protein bands were excised from the gel, and in-gel trypsin digestion was performed as previously described [30], except that (i) duration of destaining was extended to completely remove CBB from the diced gel pieces before trypsin digestion and, (ii) extracted and resuspended peptide mixture was desalted through Zip-Tipm-C18 prior to LC-MS/MS. LC-MS/MS was performed according to the manufacturer's specifications. Briefly, the mixture of tryptic peptides was fractionated by capillary high performance liquid chromatography (HPLC) with flow splitter (Surveyor HPLC, Thermo Electron, San Jose, CA). Peptides were eluted at a flow rate of 250 μl/min. by a gradient of 2-60% of solvent B (0.1% formic acid in 100% acetonitrile) in solvent A (0.1% formic acid in water) and the eluted peptide was directly sprayed into the nanospary source of a LTQ mass spectrometer (Thermo Electron, San Jose, CA). Scans were acquired in both MS and data-dependent MS/MS modes, mass range 400 to 1500 (mass-to-charge ratios) to detect the masses of both parent and daughter ions. For data analysis and processing, the Sequest algorithm was used to interpret MS/MS data [31]. The MS/MS data was searched against Vaccinia virus database [31]. Briefly, acceptable Sequest results must have a ΔCn score of ≥0.1, regardless of charge state of the peptide. Acceptable cross-correlation score (Xcorr) must be ≥ 1.5 for a singly charged peptide and ≥ 2.0 for a peptide with a +2 or +3 charge state. A protein that was identified by four or more unique peptides with Sequest scores that met the acceptance criteria, the protein was considered to be present in the sample analyzed.
Antibodies
Primary antibodies: mouse monoclonal anti-MCP1 antibody (mAb402) diluted 1:3000 for western blot and 1:1000 for immunofluorescence, was prepared and affinity immunopurified as described previously [25]; rat monoclonal antibody anti-BrdU (CldU), diluted 1:50 (OBT300, Accurate Chemical & Scientific Corp.). Polyclonal rabbit antibodies: anti-human ORC2, diluted 1:3000 (Biosciences, Pharmingen); anti-human HP1β (Upstate, Lake Placid, NY), diluted 1:200; anti-PCNA (FL-261), diluted 1:100; anti-human β-tubulin (H-23 h5), diluted 1:500, (Santa Cruz Biotechnology, Inc.); anti-human MCM2, diluted 1:2000; anti-human Histone H3 (tri methyl K9 or MetH3K9), diluted 1:5000 (ab8898, AbCam); anti-human Histone H1, diluted 1:100 (Fl219, Santa Cruz Biotechnology, Inc.); anti-human histone H1.2 (ab17677, AbCam), diluted 1:1000; anti-human histone H1.5, diluted 1:1000 (ab24175, AbCam). Affinity purified goat polyclonal antibodies, diluted 1:100 (Santa Cruz Biotechnology, Inc.): anti-human ORC4 (L-15), anti-Cdc6 (C-19), anti-human Cdc45 (C-20), anti-human MCM7 (V-17), anti-human MCM3 (G-19). Secondary antibodies: donkey horseradish peroxidase labeled antibodies, diluted 1:3000 (Santa Cruz Biotechnology, Inc.): anti-goat, anti-rabbit, and anti-mouse. Affinity purified sheep Cy3 conjugated antibodies (Jackson Laboratories), diluted 1:300: anti-mouse and anti-goat. Alexa Fluor488 donkey antibodies, diluted 1:200 (Molecular Probes): anti-rabbit, anti-mouse and anti-rat. Mouse IgG1 (0.5 μg/ml) anti-BrdU FITC conjugated antibody (Becton Dickinson) for FACS.
RESULTS
Identification of MCP1 interacting proteins
Previous microinjection experiments suggested that MCP1 might have a role in initiating replication events [25]. Based on these data, it was interesting to inquire whether MCP1 exhibited specific associations with the proteins required for the establishment of the pre-RC. To assess whether MCP1 interacts with members of pre-RC, we prepared nuclear proteins from HeLa and K562 cell lines at different phases of interphase. HeLa cells were synchronized and collected at early, mid, and late S/G2 phase and the isolated proteins, after DNA digestion, were immunoprecipitated with anti-MCP1 antibody. In this assay, a mouse immunoglobulin was used as control for the immunoprecipitation of HeLa nuclear extracts obtained at G1/S transition.
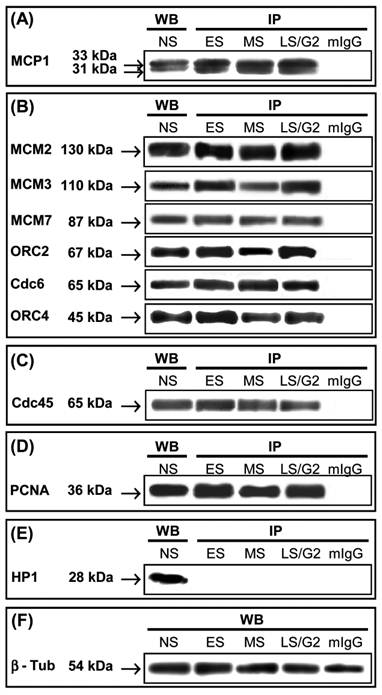
Immunoprecipitation with anti-MCP1 antibody followed by western blot with the same antibody showed the presence of similar amounts of the 33 kDa and 31 kDa of MCP1 forms during the entire S-phase (Fig. 1A). Western blot analyses using antibodies against Cdc6, ORC2, and ORC4 proteins revealed that these proteins co-immunoprecipitated with MCP1 at all stages of S-phases (Fig. 1B). Immunoprecipitation with anti-MCP1 antibody followed by immunoblotting with antibodies against MCM2, MCM3, and MCM7 also showed that MCP1 interacted or was associated with the complex that contained these members of MCM family (Fig. 1B). We also demonstrated that Cdc45 (Fig. 1C) and PCNA (Fig. 1D) co-immunoprecipitated with MCP1. Western blot of nuclear extracts with anti-HP1β antibody detected HP1β protein but in the extracts immunoprecipitated with anti-MCP1 antibody this protein was not present showing that HP1β was not associated with MCP1 (Fig. 1E). This assay confirmed that MCP1 did not associate with proteins marking sequences that replicate during late S-phase.
Immunoprecipitation (IP) of HeLa nuclear extracts at different phases of the cell cycle with anti-MCP1 antibody. Western blot (WB) of HeLa nuclei from non-synchronized cells with antibodies used the detect MCP1 interacting proteins. (ES) - cells that were synchronized at early S-phase by double thymidine block (DTB); (MS) - cells at middle S-phase collected 4 hours after DTB release; (LS/G2) - cells at late-S/G2 phase obtained 7 hours after DTB release. As a control, HeLa nuclear extracts at G1/S transition were immunoprecipitated with a mouse immunoglobulin (mIgG). (A) Immunoblotting with anti-MCP1 antibody detects the 33 kDa and 31 kDa forms of MCP1; (B) Proteins that interact with MCP1 were detected by western blot with antibodies against MCM2, MCM3, MCM7, ORC2, Cdc6 and ORC4; (C) Cdc45 and (D) PCNA are co-immunoprecipitated with MCP1; (E) HP1β is not associated with MCP1. (F) As a loading control a Western blot of HeLa total cell extracts at the same cell cycle phases was done with anti-βtubulin antibody.
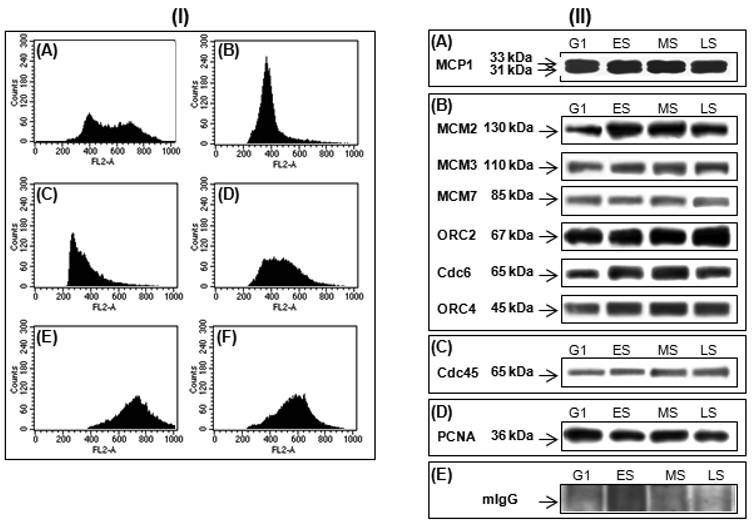
In parallel, the human K562 cells, a non-adherent cell line, were synchronized by centrifugal elutriation (Fig. 2-I). This technique sorted fractions enriched at G1, early-S, middle-S, and late-S phases without any interference due to the stress and checkpoint responses induced by aphidicolin, hydroxyurea, or thymidine block [5]. Using isolated K562 nuclei from G1 and different S-phase fractions, we have shown that MCP1 interacted with Cdc6, ORC2, ORC4, MCM2, MCM3, MCM7 (Fig. 2 II-B), Cdc45 (Fig. 2 II-C) and PCNA (Fig. 2 II-D).
(I) Cell cycle profile of K562 cells before (A) and after elutriation at G1 (B), early-S (C), middle-S, (D), late-S (E) and G2 (F) phases. (II) K562 nuclear extracts synchronized by elutriation at G1, early-S (ES), middle-S (MS), and late-S (LS) were immunoprecipitated with anti-MCP1 antibody and detection of interacting proteins were done by western blot with anti-MCP1 antibody (A), that identify the 33 kDa and 31 kDa forms of MCP1, antibodies against MCM2, MCM3, MCM7, ORC2, Cdc6 and ORC4 (B), Cdc45 (C) and PCNA (D), that recognize the corresponding proteins, and a mouse IgG (mIgG) (E) was used as a control.
To determine whether the two forms of MCP1 are required to interact with pre-RC proteins, we have performed the reciprocal immunoprecipitations with antibodies against some members of the pre-RC components (Cdc6, ORC2, ORC4, MCM2, MCM3, and MCM7), as well as Cdc45 and PCNA. Co-immunoprecipitation data with nuclei preparations obtained at G1/S transition clearly showed that only the 31 kDa form of MCP1 was co-immunoprecipitated with Cdc6, ORC2, ORC4, MCM2, MCM3, MCM7, Cdc45 and PCNA (Fig. 3). Reciprocal immunoprecipitations of nuclei with anti-HP1β and MetH3K9 antibodies followed by western blot with anti-MCP1 antibody revealed that none of the two forms of MCP1 interact with HP1β or MetH3K9 (Fig. 3).
Reverse co-immunoprecipitation (IP) of HeLa nuclear extracts, synchronized at early S-phase by double thymidine block (DBT), with antibodies against Cdc6, ORC2, ORC4, MCM2, MCM3, MCM7, Cdc45, PCNA, HP1β, MetH3K9 and MCP1 followed by immunoblotting with anti-MCP1 antibody. The 31 kDa form of MCP1 is co-immunoprecipitated with all antibodies used except antibodies against HP1β and MetH3K9. Western blot (WB) immunoprecipitation (IP) with anti-MCP1 antibody recognizes the 33 kDa and 31 kDa MCP1 polypeptides in HeLa nuclear extracts synchronized at early S-phase by DBT.

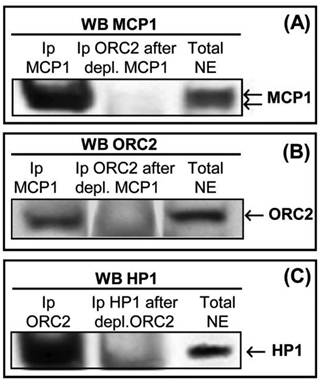
HP1β, which is known to co-immunoprecipitate with some ORC proteins, namely in Drosophila and Xenopus, associates with late replication foci [32-35]. We immunoprecipitated and depleted K562 nuclear extracts to confirm the association between ORC2 and MCP1, and between ORC2 and HP1β. Western blot with anti-MCP1 antibody (Fig. 4A) detected: (i) the presence MCP1 in nuclear extracts (right) and after immunoprecipitation with anti-MCP1 antibody (left); (ii) MCP1 is absente in nuclear extracts that were initially depleted of MCP1 and after immunoprecipitated with ORC2 antibody (middle). Immunoblotting with anti-ORC2 antibody (Fig. 4B) showed: (i) the presence ORC2 in nuclear extracts (right) and after immunoprecipitation with anti-MCP1 antibody (left); (ii) the absence of ORC2 in nuclear extracts that were firstly depleted of MCP1 and after immunoprecipitated with ORC2 antibody (middle). Western blot with anti-HP1β antibody (Fig. 4C) revealed that: (i) HP1β was present in total nuclear extracts (right) and in nuclear extracts immunoprecipitated with ORC2 antibody (left); (ii) HP1β was not present when the extracts were immunoprecipitated with anti-HP1β antibody after depletion of ORC2 (middle), confirming the association between ORC2 and HP1β.
(A) Western blot with anti-MCP1 antibody using K562 nuclear extracts that were previously immunoprecipitated with MCP1 (left), firstly depleted of MCP1 followed immunoprecipitation with ORC2 antibody (middle), and total nuclear extracts not immunoprecipitated (right). (B) Western blot with anti-ORC2 antibody using K562 nuclear extracts that were previously immunoprecipitated with MCP1 (left), initially depleted of MCP1 followed immunoprecipitation with ORC2 antibody (middle), and total nuclear extracts not immunoprecipitated (right). (C) Western blot with anti-HP1β antibody using K562 nuclear extracts that were previously immunoprecipitated with ORC2 (left), depleted of ORC2 followed immunoprecipitation with HP1β antibody (middle), and total nuclear extracts not immunoprecipitated (right).
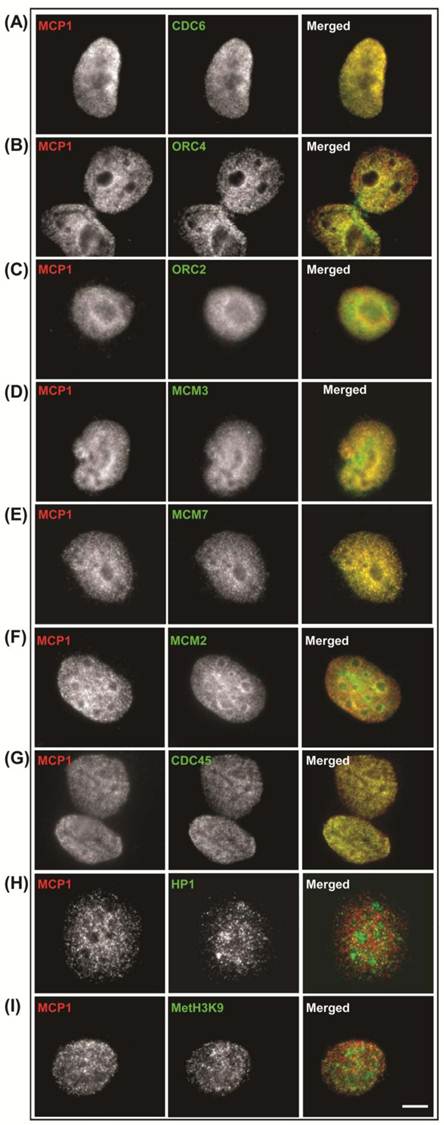
In non-synchronized cultures, we have determined the cellular localization of MCP1 and several components of the pre-RC including Cdc6, ORC2, ORC4, MCM2, MCM3 and MCM7, and Cdc45. An immunofluorescence analysis was also performed with the heterochromatic markers HP1β and MetH3K9, which are known to bind ORC and associate with late replication foci [32-35]. MCP1 location is nuclear; it has a granular distribution throughout the nucleus, is not localized within the nucleolus but has a peripheral prominence around the nucleolus. MCP1 and Cdc6 co-localization showed a speckled nuclear pattern in the nucleus of interphase MO59K cells, but neither protein was present at the nucleolus (Fig. S1-A). Co-localization of MCP1 with ORC4 and ORC2 was essentially nuclear with a peripheral accentuation around the nucleoli (Fig. S1-B and C). For MCP1 and MCM proteins (MCM3, MCM7, and MCM2), the co-localization had a granular nuclear distribution, but was not detected in the nucleolus (Fig. S1-D, E and F). Cdc45 also exhibited speckled nuclear localization and both Cdc45 and MCP1 showed regions of co-localization (Fig. S1-G). MCP1 did not co-localize with the heterochromatic proteins HP1β (Fig. S1-H) and MetH3K9 (Fig. S1-I). These results showed that MCP1 is co-localized with some of the proteins required for the early events of DNA replication but not with proteins that mark DNA sequences replicating in late S-phase.
Co-localization patterns, in non-synchronized cultures, in human MO59K cells of MCP1 (red) and Cdc6 (A), ORC4 (B), ORC2 (C), MCM3 (D), MCM7 (E), MCM2 (F), Cdc45 (G), HP1β (H), and MetH3K9 (I) proteins (green). See text for details. Barr = 10μc.
Characterization of MCP1 by mass spectroscopy
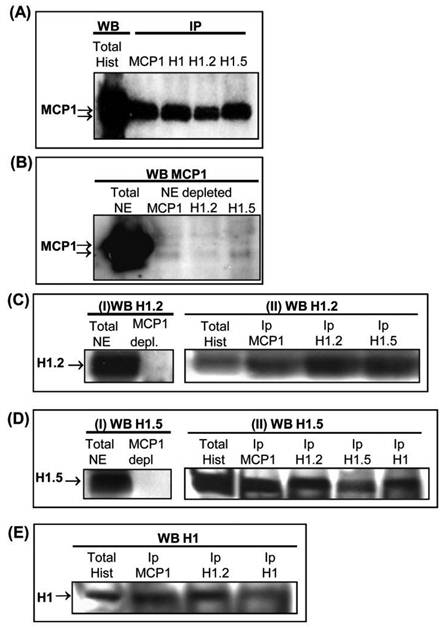
Characterization of 31 kDa and 33 kDa forms of MCP1 by mass spectrometry (LC-MS/MS) revealed that this protein has high levels of homology with histone H1, variants H1.2 and H1.5. To evaluate whether MCP1 could interact with H1 variants, histone preparations and nuclear extracts from K562 cells were immunoprecipitated with anti-MCP1 and anti-histone H1 variant antibodies. Immunoprecipitations of histone preparations were performed with anti-MCP1, anti-histone H1, anti-H1.2 and anti-H1.5 antibodies followed by western blot with anti-MCP1. Results clearly showed the presence of two proteins with apparent molecular weights of 31 kDa and 33 kDa (Fig. 5A) in all histone preparations that were immunoprecipitated with antibodies against H1, H1.2 and H1.5. In addition, the anti-MCP1 antibody did not recognize the 31 kDa and 33 kDa MCP1 forms when nuclear extracts were initially immunodepleted of MCP1, H1.2 and H1.5 variants, using the respective antibodies (Fig. 5B), suggesting the association between these proteins. In fact, reverse depletion experiments using anti-MCP1 antibody showed that MCP1 is associated to these two histone variants since both anti-H1.2 (Fig. 5-I-C) and anti-H1.5 (Fig. 5-I-D) antibodies did not recognize their particular antigens in the nuclear extracts. In the same way, immunoblotting with using H1.2, H1.5 and H1 antibodies in total histone preparations that were immunoprecipitated with MCP1 or anti-histone antibodies revealed the interaction between histones variants and MCP1 (Fig. 5-IIC, II-D and E).
(A) K562 histone preparations were immunoprecipitated (IP) with anti-MCP1, histone H1 (total), histone H1.2 and histone H1.5 antibodies followed by western blot (WB) performed with anti-MCP1 antibody. The right lane, used as a control, shows the reactivity of MCP1 with anti-MCP1 antibody (B) Immunoblotting with anti-MCP1 antibody of K562 total nuclear extracts that were firstly depleted of MCP1, H1.2 and H1.5 using the respective antibodies. (C) (I) Western blot with anti-H1.2 antibody of K562 total nuclear extracts not depleted (left) and depleted (right) of MCP1; and (II) Western blot with anti-H1.2 antibody of total histone preparations not precipitated and previously immunoprecipitated with anti-MCP1, anti-H1.2 and anti-H1.5 antibodies. (D) (I) Immunoblotting with anti-H1.5 antibody of K562 total nuclear extracts not depleted (left) and depleted of MCP1 (right) and (II) Western blot with anti-H1.5 antibody of total histone preparations not precipitated and previously immunoprecipitated with anti-MCP1, anti-H1.2, anti-H1.5 and anti-H1 (total) antibodies. (E) Western blot with anti-H1 antibody of total histone preparations not precipitated and previously immunoprecipitated with anti-MCP1, anti-H1.2 and anti-H1 antibodies.
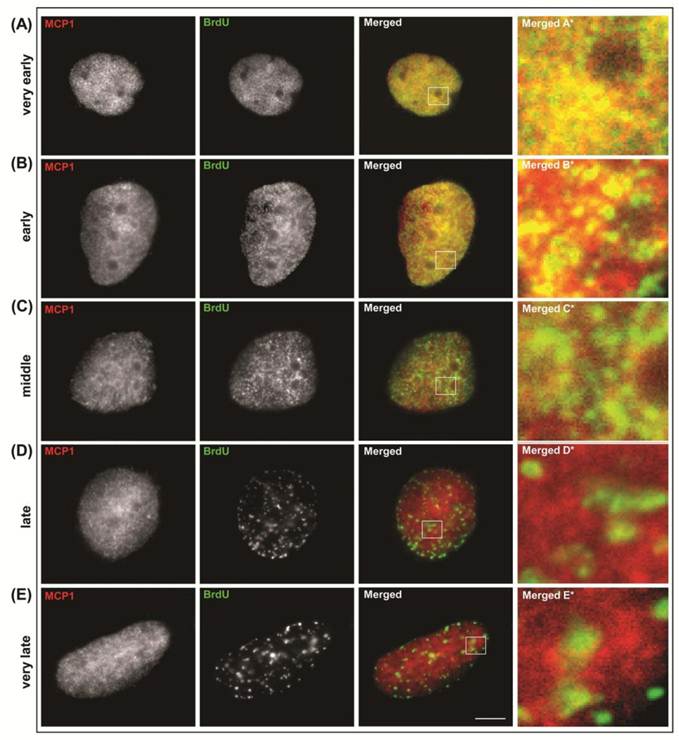
MCP1 co-localizes with early replication foci
Our previous data showed that depletion of MCP1 prevents DNA replication at early S-phase [25]. In the current study we have determined whether MCP1 interacted with replication origins in early and late replicating loci. To address these questions, immunofluorescence was performed with simultaneous labeling of MCP1 and BrdU-substituted DNA (Fig. S2) in human cervix epithelial carcinoma (HeLa cells) and human glioblastoma (MO59K cells) to check whether the patterns obtained with these two cell lines were comparable. BrdU incorporation facilitated the identification of cells in S-phase (those cells actively incorporate BrdU) and the categorization of cells into five different stages throughout S-phase [12]. Classification of cells in S-phase relies on the observation that cells display replication sites as small spots throughout the nucleus at very early S-phase (BrdU pattern I). Later on during early S-phase (BrdU pattern II) replication foci are larger, discrete and stronger near the nuclear boundary. During middle S-phase (BrdU pattern III) replication sites are located at the nuclear periphery and near the perinucleolar region. As cells progress through S-phase, the late replication foci (BrdU pattern IV) are larger, less in number, and at few discrete sites at the periphery of the nucleus at late S-phase. Very late S-phase (BrdU pattern V) is characterized by the presence of smaller number and larger areas of replication sites in the interior of the nucleus and some less significant sites at the periphery of the nucleus [12]. In both cell lines, our results showed that MCP1 co-localized with BrdU replication foci at very early and early S-phases in human cells (Fig. S2-A and B). During middle S-phase, the co-localization patterns of MCP1 and BrdU were restricted to some areas around the nucleolus (Fig. S2-C). However, MCP1 did not co-localize with BrdU in cells at late stages of DNA replication (Fig. S2-D and E).
Immunolocalization of MCP1 (red) and BrdU (green) in human MO59K cells. Classification of cells in S-phase relies on the observation that cells display replication sites as small spots throughout the nucleus (A) at very early S-phase (BrdU pattern I). Later on during early S-phase (BrdU pattern II) replication foci are larger, discrete and stronger near the nuclear boundary (B). During middle S-phase (BrdU pattern III) replication sites are located at the nuclear periphery and near the perinucleolar region (C). As cells progress through S-phase, the late replication foci (BrdU pattern IV) are larger, less in number, and at few discrete sites at the periphery of the nucleus at late S-phase (D). Very late S-phase (BrdU pattern V) is characterized by the presence of smaller number and larger areas of replication sites in the interior of the nucleus and some less significant sites at the periphery of the nucleus (E). Barr = 10μc.
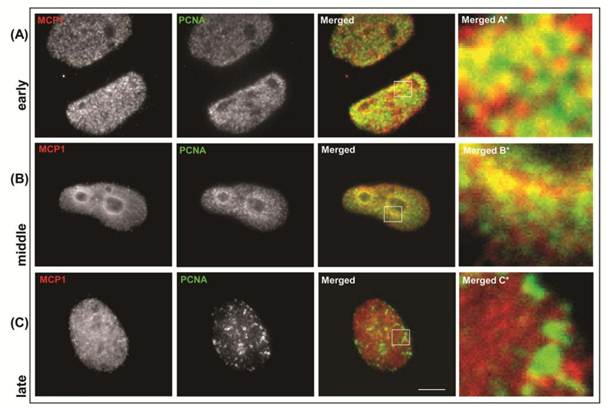
We also analyzed the patterns of co-localization of MCP1 and PCNA, which is a marker of ongoing replication forks [12,14]. PCNA associates with replication foci at the onset of S-phase and co-localizes with the early-replicating chromatin. As cells progress through S-phase, PCNA is redistributed to later-firing compartments of euchromatic replicons and associates transiently with late replication domains [14]. PCNA is present in the nucleus throughout the cell cycle but is associated with chromatin only during S-phase [4, 12]. After labeling non-synchronized cells with antibodies against MCP1 and PCNA we observed that MCP1 and PCNA co-localized at early S-phase (Fig. S3-A), similarly to what was detected with BrdU staining. In agreement, at middle S-phase, MCP1 and PCNA displayed a partial co-localization that was restricted to a few areas around the nucleolus (Fig. S3-B), and at late S-phase, they did not co-localize (Fig. S3-C). These data indicated that MCP1 was present at the early replication foci and suggested that MCP1 might be a component of the pre-RC.
Immunolocalization of MCP1 (red) and PCNA (green) in non-synchronized human MO59K cells. MCP1 associates with PCNA at early S-phase (A) replicating foci. In middle S-phase (B) MCP1 partially co-localizes with PCNA. At late S-phase (C), MCP1 does not co-localize with PCNA. Barr = 10μc.
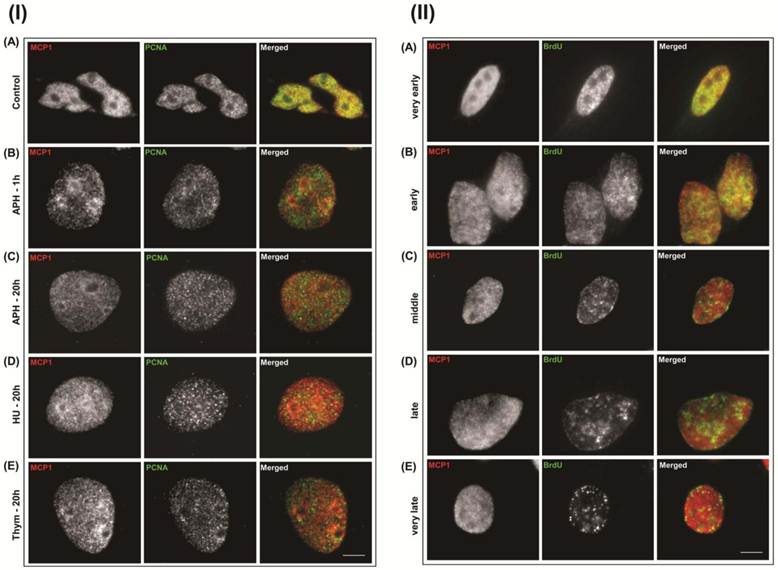
Although PCNA is not a member of the pre-RC it is present at initiating replication forks [4, 12-14]. To determine whether MCP1 associated with initiating or elongating replication forks, we used DNA replication inhibitors that are commonly used to synchronize cells at G1/S transition. Aphidicolin (APH) blocks fork progression but does not prevent the initiation of DNA replication and the formation of short nascent strands [4, 36] as it is an inhibitor of DNA polymerase α and δ [37]. Hydroxyurea (HU) acts primarily as an inhibitor of ribonucleotide reductase, allows the early origin to be fired, but stalls the forks because of the lack of nucleotide precursors [36]. Thymidine (Thym) stops DNA replication since it inhibits precursor production and prevents elongation leading to the accumulation of cells at the very beginning of S-phase [36]. Immunolocalization of MCP1 and PCNA revealed that MCP1 and PCNA did not show any pattern of co-localization after exposure of human cells for 20 hours to 5 μg/ml aphidicolin (Fig. S4 I-C), 2.5 mM hydroxyurea (Fig. S4 I-D), or 2.5 mM thymidine (Fig. S4 I-E). A different pattern emerged when cells were treated with low doses of APH for shorter periods. PCNA and MCP1 did not co-localize when cells were treated for 1 hour with 1.0 μg/ml aphidicolin (Fig. S4 I-B). Nevertheless, under the same conditions MCP1 co-localized with BrdU foci that exhibited a very early (Fig. S4 II-A) and early S-phase pattern (Fig. S4 II-B), and showed a few regions of co-localization at middle S-phase (Fig. S4 II-C). As in untreated cells (Fig. S2-C and D), MCP1 and BrdU did not co-localize according to the characteristic BrdU patterns of late (Fig. S4 II-D) and very late (Fig. S4 II-E) S-stages. These data demonstrated that, unlike PCNA, MCP1 localized to early replication foci even when replication was transiently inhibited.
(I) Immunolocalization, of MCP1 (red) and PCNA (green) in human MO59K control cells shows that both proteins co-localize at early S-phase (A). MCP1 and PCNA do not co-localize after treatment with 1 μg/ml aphidicolin (APH) for 1 hour (B), or after 20 hours of incubation with 5 μg/ml aphidicolin (C), 2.5 mM hydroxyurea (HU) (D), or 2.5 mM thymidine (Thym) (E). (II) Immunolocalization patterns of MCP1 (red) and BrdU (green) in human MO59K cells after treatment with 1 μg/ml aphidicolin for 1 hour. MCP1 associates with BrdU replicating foci at very early (A) and early S-phases (B), at middle S-phase MCP1 partially co-localizes with BrdU (C), and at late (D) and very late S-phases, MCP1 and BrdU do not co-localize (E). Barr = 10μc.
MCP1 binds replication origin sequences
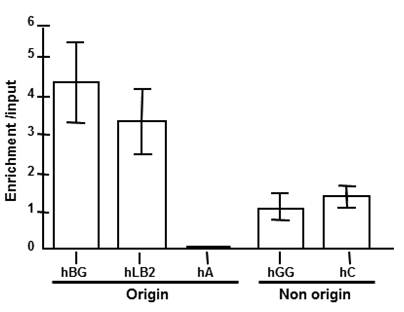
The presence of MCP1 at early replication foci and its absence from late foci suggested that MCP1 might bind DNA sequences that regulate DNA replication. To evaluate this hypothesis, we performed chromatin-immunoprecipitation (ChIP) using DNA sequences that are located at various distances from known replication origins. For this assay we used a human erythroleukaemia cell line K562 because in these cells the human beta-globin gene locus replicates early in S-phase [38] whereas amylase and collagen replicated late [39, 40]. The human lamin B2 locus also replicates early in those erythroid cells. Probes used included two genes from the beta-globin locus (beta-globin is origin proximal and gamma-globin is distal), the origin-proximal region of the human lamin B2 locus and the late origin human amylase locus. Quantitative analysis MCP1-immunoprecipitated chromatin (Fig. 6) revealed enrichment over the input of 3.2-fold for human lamin B2 and 4.3-fold for beta-globin sequences. The gamma-globin and collagen regions show little enrichment (0.9 to 1.4-fold), and late origin sequences for amylase locus were depleted. As primers were selected according to locations known either as initiation or non-initiation regions as positive and negative markers for origin and non-origins sites, these data suggest that MCP1 binds preferentially to the early origins sequences.
Chromatin immunoprecipitation (ChIP) from K562 cells using anti-MCP1 antibody followed by real time PCR analysis of the abundance of specific DNA sequences in MCP1-bound chromatin. Probes included the human beta-globin (hBG), human lamin B2 (hLB2), human amylase (hA), human gamma-globin (hGG) and human collagen (hC). The human lamin B2 and beta-globin loci replicate early in K562 cells, and the human amylase replicate late. The human gamma-globulin and human collagen were used for a non-origin control.
DISCUSSION
Despite the recent advances in understanding the processes of initiation and of DNA replication, the specific mechanisms that determine the assembly of the components of the pre-RC and those that regulate the chromatin structure remain to be elucidated.
The present work aimed to determine whether MCP1 interacts with several proteins required for the initiation of DNA replication. This was evaluated by immunoprecipitation in cells that were synchronized at G1 and at different S-phases, using drug treatment or elutriation. By reverse co-immunoprecipitation assay, we showed that the interactions between MCP1 and several proteins of the pre-RC, Cdc45 and PCNA were done through the 31 kDa form. Importantly, MCP1 did not interact with heterochromatin proteins like HP1β, a key component of condensed DNA that also binds ORC [32] and is strongly implicated in gene silencing and centromeric cohesion [34]. It also did not interact with MetH3K9, which is implicated in HP1β-nucleosome interaction and genomic stability [33]. Moreover, immunofluorescence revealed the presence of patterns of co-localization between MCP1 and proteins that were co-immunoprecipitated, but not with those that did not interact with MCP1.
Data from ChIP analysis suggested a preferential association of MCP1 with origins that replicate early in S-phase or sequences that replicate early when compared with late origin and sequences that replicate in late S-phase. In metazoans, sequences associated to early and late replication foci are not well characterized, but there are several DNA sequences confined to initiation that requires the cooperation or can be affected by other distal sequences, gene transcription and epigenetic modifications [1, 2]. Accordingly, we also detected the presence of MCP1 at early-replicating loci, as shown by the co-localization patterns displayed by MCP1/BrdU and MCP1/PCNA. Early foci can be detected in the decondensed euchromatin region, as hundreds of small replication foci, each with 5-6 replicons. Late loci are larger replication factories, containing clusters of hundreds of replicons that could be visualized in the condensed heterochromatic region in late S-phase [2, 12]. These findings are in conformity with the previous microinjection experiments that demonstrated the requirement of MCP1 for the G1/S transition and early S-phase [25]. In contrast to PCNA, MCP1 was also detected at early replication foci, after short period and low concentration of drugs that inhibit the progression of replication forks. Displacement of PCNA from replications forks, after exposure to low doses of APH had been previously shown [41] and allow the repair of transient breaks [42]. Because MCP1 interacted with ORC proteins, but not with HP1β, and as some ORC members are also partners of HP1β, as previously determined [32, 35, 43] and also supported by data of this work, we cannot rule out that ORC forms different complexes with various proteins. Here, depletion experiments performed in K562 cells, followed immunoprecipitation and immunoblotting, indicated that ORC2 can bind HP1β and ORC2 can bind MCP1. Moreover, immunoprecipitation and reverse co-immunoprecipitation assays, using nuclear extracts from HeLa cells, also confirmed the interaction between MCP1 and ORC2, and the lack of association between MCP1 and HP1β. In that way, when ORC2 was depleted from the extracts we were not able to detected HP1β, but this does not imply that all ORC2 was bound to HP1β. So, there could be two sub-fractions of ORC2, one bound to MCP1 and another bound to HP1β. Probably those complexes form at different times during the cell cycle or are dependent of different types of chromatin architecture or environment. These facts might imply that ORC2 interacts with MCP1 on early origins and with HP1β on late origins. The functional importance of such complex switching and its possible role in determining replication timing can be implied and explored in later studies.
In spite of MCP1 being present mainly at early replicating foci, we showed that these interactions with the pre-RC components are maintained during S-phase and also at G1 to G2 phases. MCP1 levels are stable throughout the G1, S and G2 phases, and the protein-protein interaction seems to be constant throughout interphase. These observations were not in a chromatin bound context, which is variable for Cdc6, MCM Cdc45 or PCNA [5, 11, 44, 45]. In fact, the expression and localization of these proteins change during S-phase, respectively for ORC2-6 [1, 6, 46] and PCNA [4, 12, 45], but the interaction described here occurs between proteins obtained from nuclear extracts after DNA digestion. Thus, it is possible that MCP1 interacts with pre-RC components that are located on early-replicating loci poised to start DNA replication. This hypothesis is consistent with earlier observations that MCP1 sequestering causes the inhibition of S-phase progress [25]. An additional role for MCP1 might be to participate in structural chromatin changes that are required not only for DNA replication but also for other important cellular functions. This suggestion is supported by our observation that MCP1 interacts with histone H1 and its variants. However, unlike histone depletions, in which function of a missing variant may be compensated by other members of the H1 family or additional proteins of the chromatin [15], the lack of MCP1 prevents DNA replication [25]. Recently, the presence of H1.2 and H1.5 was shown at DNA replication and transcription sites [47].
The nuclear localization of MCP1 [24] suggests that MCP1 might be a structural component of chromatin and it might attach some proteins of replication factory or might participate in complexes that contribute to chromatin remodeling that is required for replication, transcription or repair. In the present work we only analyzed the interactions of MCP1 with several components associated to replication machinery and histones, but a proteomic approach would allow the characterization the other MCP1 partners, as described for PCNA [45]. We cannot rule out other functions for MCP1, since it was identified with an antibody from mice with SLE [21], and might be implicated in other relevant cellular events similarly to others proteins recognized by autoimmune sera, like Ku or PCNA [22, 23, 45, 48, 49]. The molecular mechanisms underlying the proposed models will be the subject of our future investigation.
Acknowledgements
We thank Barbara Taylor for expert help in cell sorting and analysis of cell cycle fractions; Sumega Singhania for help in centrifugal elutriation; Dr. Nga Y. Nguyen for the useful advice in mass spectrometry; Dr. Jennifer Seiler for providing the MCM2 antibody; Dr. Paula Almeida Coelho for critical reading and discussion of the manuscript. This work was supported by fellowships from the Luso-American Foundation of Portugal (FLAD), Calouste Gulbenkian Foundation of Portugal and Fulbright Commission of Portugal to Elsa Bronze-da-Rocha, and by the Intramural Research Program of the NIH, Center for Cancer Research, National Cancer Institute.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
References
1. DePamphilis ML. Cell cycle dependent regulation of the origin recognition complex. Cell Cycle. 2005;4:70-79
2. Aladjem MI. Replication in context: dynamic regulation of DNA replication patterns in metazoans. Nat Rev Genet. 2007;8:588-00
3. Chesnokov IN. Multiple functions of the origin recognition complex. Int Rev Cytol. 2007;256:69-109
4. Dimitrova DS, Todorov IT, Melendy T, Gilbert DM. Mcm2, but not RPA, is a component of the mammalian early G1-phase prereplication complex. J Cell Biol. 1999;146:709-22
5. Méndez J, Stillman B. Chromatin association of human origin recognition complex, Cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of pre-replication complexes at late mitosis. Mol Cell Biol. 2000;20:8602-12
6. DePamphilis ML, Blow JJ, Ghosh S. et al. Regulating the licensing of DNA replication origins in metazoa. Curr Opin Cell Biol. 2006;18:231-39
7. Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128:721-33
8. Bochman ML, Schwacha A. The Mcm2-7 complex has in vitro helicase activity. Mol Cell. 2008;31:287-293
9. Zou L, Stillman B. Assembly of a complex containing Cdc45p, replication protein A, and Mcm2p at replication origins controlled by S-phase cyclin-dependent kinases and Cdc7p-Dbf4 kinase. Mol Cell Biol. 2000;20:3086-96
10. Masuda T, Mimura S, Takisawa H. CDK-and Cdc45-dependent priming of the MCM complex on chromatin during S-phase in Xenopus egg extracts: possible activation of MCM helicase by association with Cdc45. Genes Cells. 2003;8:145-61
11. Pacek M, Walter JC. A requirement of MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication. EMBO J. 2004;23:3667-76
12. O'Keefe RT, Henderson SC, Spector DL. Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific-satellite sequences. J Cell Biol. 1992;116:1095-110
13. Hübscher U, Maga G, Sparasi S. Eukariotc DNA polymerases. Annu Rev Biochem. 2002;71:133-63
14. Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665-79
15. Bustin M, Catez F, Lim JH. The dynamics of histone H1 function in chromatin. Mol Cell. 2005;17:617-20
16. Falbo KB, Shen X. Histone modifications during DNA replication. Mol Cell. 2009;28:149-54
17. Catez F, Yang H, Tracey KJ. et al. Network of dynamic interactions between histone H1 and high-mobility-group proteins in chromatin. Mol Cell Biol. 2004;24:4321-28
18. Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693-05
19. Schneider R, Grosschedl R. Dynamics and interplay of nuclear architecture, genome organization, and gene expression. Genes Dev. 2007;21:3027-43
20. Alexandrow MG, Hamlin JL. Chromatin decondensation in S-phase involves recruitment of Cdk2 by Cdc45 and histone H1 phosphorylation. J Cell Biol. 2005;168:875-86
21. Bronze-da-Rocha E, Machado C, Staines NA, Sunkel CE. Systemic lupus erythematosus murine monoclonal DNA-binding antibodies recognize cytoplasmic and nuclear phosphorylated antigens that display cell cycle redistribution in HEp-2 cells. Immunology. 1992;77:582-591
22. Tan EM. Antinuclear antibodies: diagnostic markers for autoimmune diseases and probes for cell biology. Advances Immunol. 1989;44:93-151
23. Balczon R. Autoantibodies as probes in cell and molecular biology. Proc Soc Exp Biol Med. 1993;204:138-54
24. Bronze-da-Rocha E, Catita JA, Sunkel CE. Molecular cloning of Metaphase Chromosome Protein 1 (MCP1), a novel human autoantigen that associates with condensed chromosomes during mitosis. Chromosome Res. 1998;6:85-95
25. Bronze-da-Rocha E, Nóvoa A, Cunha C. et al. The human autoantigen MCP1 is required during early stages of DNA replication. Chromosome Res. 2000;8:669-11
26. Bernat RL, Borisy GG, Rothfield NF, Earnshaw WC. Injection of anticentromere antibodies in interphase disrupts events required for chromosome movement at mitosis. J. Cell Biol. 1990;111:1519-33
27. Schnitzler GR. Isolation of Histones and Nucleosome Cores from Mammalian Cells. Current Protocols in Molecular Biology. New York: John Wiley & Sons. 2003
28. Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning - A Laboratory Manual; 2nd Edition. New York: Cold Spring Harbor Laboratory Press. 1989
29. Lin CM, Fu H, Marinovsky M. et al. Dynamic alterations of replication timing in mammalian cells. Curr Biol. 2003;13:1019-28
30. Vassilev A, Kotaro KJ, Shu H. et al. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/YES-associated protein localized in the cytoplasm. Genes Dev. 2001;15:1229-41
31. Eng JK, McCormack AL, Yates JRI. An approach to correlate mass spectral data of peptides with amino acid sequences in a protein databases. J Am Soc Mass Spectrom. 1994;5:976-89
32. Pak DT, Pflumm M, Chesnokov I. et al. Association of the origin recognition complex with heterochromatin and HP1 in higher eukaryotes. Cell. 1997;91:311-23
33. Eissenberg JC. Molecular biology of the chromo-domain: an ancient chromatin comes of age. Gene. 2001;275:19-29
34. Festenstein R, Pagakis SN, Hiragami K. et al. Modulation of Heterochromatin Protein 1 Dynamics in Primary Mammalian Cells. Science. 2003;299:719-21
35. Shareef MM, Badugu R, Kellum R. HP1/ORC complex and heterochromatin assembly. Genetica. 2003;117:127-34
36. Jackson DA. S-phase progression in synchronized human cells. Exp Cell Res. 1995;220:62-70
37. Ikegami S, Taguchi T, Ohashi M. et al. Aphidicolin prevents mitotic cell division by interfering with the activity of DNA polymerase-alpha. Nature. 1978;275:458-60
38. Epner E, Forrester WC, Groudine M. Asynchronous DNA replication within the human beta-globin gene locus. Proc Natl Acad Sci USA. 1988;85:8081-85
39. Aladjem MI, Rodewald LW, Kolman JL, Wahl GM. Genetic dissection of a mammalian replicator in the human beta-globin locus. Science. 1998;281:1005-9
40. Cimbora DM, Schübeler D, Reik A. et al. Long-distance control of origin choice and replication timing in the human beta-globin locus are independent of the locus control region. Mol Cell Biol. 2000;20:5581-5591
41. Shimura T, Torres MJ, Martin MM, Rao VA, Pommier Y, Katsura M, Miyagawa K, Aladjem MI. Bloom's syndrome helicase and Mus81 are required to induce transient double-strand DNA breaks in response to DNA replication stress. Mol Biol. 2008;375:1152-64
42. Shimura T, Martin MM, Torres MJ. et al. DNA-PK is involved in repairing a transient surge of DNA breaks induced by deceleration of DNA replication. Mol Biol. 2007;367:665-80
43. Hiragami K, Festenstein R. Heterochromatin protein 1: a pervasive controlling influence. Cell Mol Life Sci. 2005;62:2711-26
44. Alexandrow MG, Hamlim JL. Cdc6 chromatin affinity is unaffected by serine-54 phosphorylation, S-phase progression, and over expression of cyclin A. Mol Cell Biol. 2004;24:1614-27
45. Naryzhny SN. Proliferating cell nuclear antigen: a proteomics view. Cell Mol Life Sci. 2008;65:3789-3808
46. Prasanth SG, Prasanth KV, Siddiqui K. et al. Human Orc2 localizes to centrosomes, centromere and heterochromatin during chromosome inherence. EMBO J. 2004;23:2651-63
47. Talasz H, Sarg B, Lindner HH. Site-specifically phosphorylated forms of H1.5 and H1.2 localized at distinct regions of the nucleus are related to different processes during the cell cycle. Chromosoma. 2009;118:693-09
48. Sawalha AH, Harley JB. Antinuclear antibodies in systemic lupus erythematosus. Curr Opin Reumathol. 2004;16:534-40
49. Casiano CA, Mediavill-Varela M, Tan EM. Tumor-associated antigens arrays for the serological of cancer. Mol Cell Proteomics. 2006;5:1745-59
Figures
Author contact
![]() Corresponding author: Elsa Bronze-da-Rocha, Departamento de Ciências Biológicas, Laboratório de Bioquímica, Faculdade de Farmácia da Universidade do Porto, Rua Aníbal Cunha 164, 4050-047 Porto, Portugal. Voice: 00 351 222 078 907. Fax: 00 351 222 003 977 ; E-mail: elsa.rochaup.pt
Corresponding author: Elsa Bronze-da-Rocha, Departamento de Ciências Biológicas, Laboratório de Bioquímica, Faculdade de Farmácia da Universidade do Porto, Rua Aníbal Cunha 164, 4050-047 Porto, Portugal. Voice: 00 351 222 078 907. Fax: 00 351 222 003 977 ; E-mail: elsa.rochaup.pt