Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(6):753-761. doi:10.7150/ijbs.7.753 This issue Cite
Research Paper
Temporal Expression of Mutant LRRK2 in Adult Rats Impairs Dopamine Reuptake
Hongxia Zhou1,* ![]() , Cao Huang1,*, Jianbin Tong1, Weimin C Hong2, Yong-Jian Liu2,
, Cao Huang1,*, Jianbin Tong1, Weimin C Hong2, Yong-Jian Liu2, ![]() , Xu-Gang Xia1,
, Xu-Gang Xia1, ![]()
1. Department of Pathology, Anatomy & Cell Biology, Thomas Jefferson University, 1020 Locust Street, Philadelphia, PA 19107, USA
2. Department of Neurobiology, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213, USA
* These authors contributed equally to this work.
Received 2011-5-23; Accepted 2011-6-2; Published 2011-6-9
Abstract
Parkinson's disease (PD) results from progressive degeneration of dopaminergic neurons. Most PD cases are sporadic, but some have pathogenic mutation in the individual genes. Mutation of the leucine-rich repeat kinase-2 (LRRK2) gene is associated with familial and sporadic PD, as exemplified by G2019S substitution. While constitutive expression of mutant LRRK2 in transgenic mice fails to induce neuron death, transient expression of the disease gene by viral delivery causes a substantial loss of dopaminergic neurons in mice. To further assess LRRK2 pathogenesis, we created inducible transgenic rats expressing human LRRK2 with G2019S substitution. Temporal overexpression of LRRK2G2019S in adult rats impaired dopamine reuptake by dopamine transporter (DAT) and thus enhanced locomotor activity, the phenotypes that were not observed in transgenic rats constitutively expressing the gene throughout life time. Reduced DAT binding activity is an early sign of dopaminergic dysfunction in asymptomatic subjects carrying pathogenic mutation in LRRK2. Our transgenic rats recapitulated the initiation process of dopaminergic dysfunction caused by pathogenic mutation in LRRK2. Inducible transgenic approach uncovered phenotypes that may be obscured by developmental compensation in constitutive transgenic rats. Finding in inducible LRRK2 transgenic rats would guide developing effective strategy in transgenic studies: Inducible expression of transgene may induce greater phenotypes than constitutive gene expression, particularly in rodents with short life time.
Keywords: LRRK2, Parkinson's disease, rats, genetic model, dopamine transporter, dopaminergic neurons
Introduction
Parkinson's disease (PD) is a common neurodegenerative disease caused by progressive degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc). How neurons degenerate in PD is largely unknown. While most PD cases are sporadic, approximately 10% of the cases have genetic mutation in the individual genes, including α-synuclein, parkin, DJ-1, pink1, and leucine-rich repeat kinase 2 (LRRK2) [1-6]. Unlike mutation in the other PD genes, mutation of LRRK2 causes late-onset Parkinsonism indistinguishable from idiopathic PD [7-10], suggesting a role for LRRK2 in sporadic PD. Indeed, a common mutation in LRRK2 (G2019S substitution) occurs in sporadic PD [10], underscoring the importance of LRRK2 for understanding PD pathogenesis.
LRRK2 is a member of the newly defined ROCO protein family, which belongs to the Ras-GTPase superfamily [5, 6]. Limited information from orthologous proteins in Dictyostelium Discoideum suggests a role for ROCO proteins in regulating cytoskeletal structures [11]. Like the other ROCO proteins, LRRK2 contains a conserved GTPase-like domain (Roc) and a mixed lineage kinase-like kinase domain [5, 12]. G2019S mutation located within the kinase domain of LRRK2 augments kinase activity implicated in LRRK2 neurotoxicity [12-15]. Mutation of LRRK2 within its Roc domain (R1440C substitution) also enhances its kinase activity [16], suggesting an intrinsic role of kinase activity in the neurotoxicity. To understand LRRK2 pathogenesis, a critical step is establishing animal models reproducing PD phenotypes observed in patients. Similar to the other genetic mouse models of PD [17-21], mutant mice with endogenous LRRK2 deleted or with mutant LRRK2 introduced fail to develop dopaminergic neuron death [22-29], although some strains display dystrophy of dopaminergic neurites and disturbance to dopaminergic neurotransmission [28, 29]. Like transgenic mice, LRRK2 transgenic Drosophila of different strains also develops diverse phenotypes [30, 31]. Phenotypic expression in LRRK2 transgenic animals largely depends on transgenic strains [22-31]. Even though chromosomal positional effect on transgene expression is ameliorated by using a bulk transgene, such as bacterial artificial chromosome (BAC), BAC transgenic mice expressing mutant LRRK2 develop varying phenotypes without neuronal loss [28, 32]. Such findings in transgenic animals underscore the importance of diverse models for dissecting LRRK2 pathogenesis.
To further assess LRRK2 mutation, we developed transgenic rats expressing mutant human LRRK2 in a controlled manner. Temporal, but not constitutive, expression of human LRRK2 with G2019S substitution enhanced locomotor activity in aged rats. Microdialysis analysis of living rats attributed the enhanced locomotor activity to impaired dopamine reuptake by dopamine transporter (DAT). As a result of compromised DAT activity, amphetamine-evoked dopamine release and amphetamine-elicited locomotor activity were reduced in mutant LRRK2 transgenic rats. Intriguingly, reduced DAT binding is the earliest indication of dopaminergic dysfunction in asymptomatic subjects carrying a pathogenic mutation in LRRK2 [33]. Thus, our transgenic rats recapitulated the initiation process of dopaminergic dysfunction caused by mutation in LRRK2.
Materials and Methods
Transgenic rat production and behavioral analyses
LRRK2 and tTA transgenic rats were produced by pronuclear injection as described previously [34]. TRE-LRRK2G2019S transgenic rats were crossed with CAG-tTA transgenic rats to produce the double transgenic offspring that expressed mutant human LRRK2 in the absence of Doxycycline (Dox). To mimic constitutive gene expression, breeding rats and their offspring were not given Dox through their lifetime. To achieve temporal expression of LRRK2 transgene in adulthood, breeding rats and their offspring were constantly given Dox in drinking water (50µg/ml) until the double transgenic offspring reached 5 months of age. LRRK2 transgenic rats and the control rats were subject to open field activity assay (Med Associates), which measured the total distance and the total stereotypic move the rat made in 20 minutes. Elicited activities were measured at 5 minutes after administration of amphetamine (1mg/kg, I.p.) or 20 minutes after administration of nomifensine (1mg/kg, I.p.). Animal use followed NIH guidelines and the animal use protocol was approved by the Institutional Animal Care and Use Committees at Thomas Jefferson University.
Microdialysis and HPLC analysis
As described previously [35], the extracellular level of striatal dopamine was measured by intra-brain microdialysis combined with HPLC in free-moving rats. Anesthetized rats were implanted with a guide cannula the day before dialysis. On the following day, dialysis probe (BASi) was inserted through the guide cannula into the left striatum (coordinates: 1 mm anterior to bregma; 2.6 mm left from sagittal line; 4 mm ventral to the skull). Continuous perfusion was maintained with modified Ringer's solution (147 mM Na+, 4 mM K+, 1.3 mM Ca2+, 1 mM Mg2+, and 155.6 mM CI-). Basal dopamine release was established within two hours when comparable levels of released dopamine were obtained in three consecutive samplings (20 minutes each). Amphetamine (1.3 mM) and nomifensine (200 µM) were applied to dialysis solution for 40 minutes and evoked dopamine release was measured subsequently. Dopamine concentration in dialysis solution was instantly measured by HPLC. By the end of dialysis, the right striatum of dialyzed rats was dissected and homogenized in 0.1 M perchloric acid and cleared tissue lysates were measured by HPLC for the concentration of dopamine and dopamine metabolites. Tissue pellets were dissolved in 1M NaOH and protein concentration was determined. Tissue contents of striatal dopamine were adjusted with protein concentration. HPLC analysis was done as described previously [36].
Histology and stereological cell counting
Immunostaining and stereological cell counting were done as described previously [36, 37]. Rat's brain was cut into serial sections of 30 µm on a Cryostat. Every fourth section through the SNpc and every fourth section through the locus coeruleus (LC) were immunostained for TH (Pel-Freez; 1: 1000). TH positive neurons in the SNpc and in the LC were estimated by stereological cell counting. Coronal sections of rat's brain through SNpc and striatum were also immunostained with mouse monoclonal antibody to α-synuclein (1: 500; Chemicon), chicken antibody to ubiquitin (1: 1000; Sigma), or rabbit polyclonal antibody to phospho-tau (1: 500; Sigma).
[3H]-dopamine uptake assay
The assay was performed as described previously [38]. The human neuroblastoma SH-SY5Y cells that stably expressed human DAT-EGFP-C1 were seeded into 96-well plates and were then transfected with LRRK2 constructs using Lipofectamine 2000 (Invitrogen, CA). Two days following transfection, cells were washed with PBSCM (PBS with 0.1mM CaCl2, 1 mM MgCl2), and were incubated with 50 nM [3H]-dopamine plus varying concentrations of unlabeled dopamine for 5 min at room temperature. Retained radioactivity in cells was determined by liquid scintillation spectrometry. Data from triplicate samples were analyzed according to the Michaelis-Menten kinetic equation using GraphPad Prism 4.0.
Results
Enhanced locomotor activity in aged rats expressing mutant LRRK2 in adulthood
To further examine the pathogenesis of mutation in LRRK2, we created transgenic rats overexpressing a mutant LRRK2 with G2019S substitution [34]. Transgenic studies of amyotrophic lateral sclerosis suggest that mice and rats develop diverse phenotypes, even if the same disease gene is overexpressed [37, 39]. Rats confer advantages over mice in behavior tests that are crucial to the modeling of neurological diseases such as PD [40, 41]. To increase the diversity of animal models for PD, we chose rats to model the phenotypes of pathogenic mutation in LRRK2. Since developmental compensation for transgene expression may compromise phenotypic expression in transgenic animals, we used a Tet-inducible system to express mutant LRRK2 transgene temporally in adult rats [34]. As described previously [34], one transgenic line carrying two copies of LRRK2 transgene was established and this line expressed mutant human LRRK2 robustly in the midbrain when crossed onto CAG-tTA transgenic background.
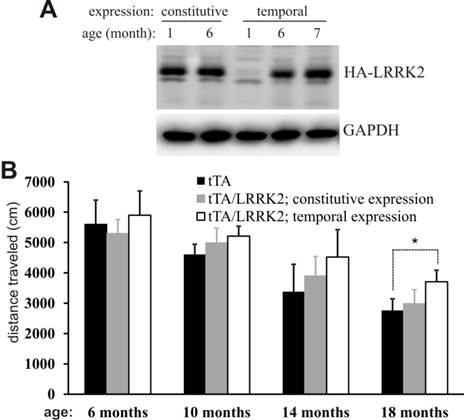
The mutant LRRK2 transgene was driven by the TRE promoter and thus its expression depends on tTA activation and is subject to Dox regulation [34]. In the absence of Dox, mutant LRRK2 transgene was continuously and fully expressed [34], exhibiting a constitutive expression pattern (Fig. 1A). To obtain temporal overexpression of mutant LRRK2 in adult rats, we supplied transgenic rats with Dox in drinking water during embryonic and postnatal development such that the mutant LRRK2 transgene was constantly suppressed from expression in transgenic rats [34]. Dox-treated rats were deprived of Dox at the age of 5 months, and they soon began to express mutant LRRK2 after Dox withdrawal (Fig. 1A). Constitutive overexpression of mutant LRRK2 failed to induce behavioral phenotypes in transgenic rats (Fig. 1B). In contrast to constitutive overexpression, temporal overexpression of mutant LRRK2 caused abnormal locomotor activity in aged, but not in young, transgenic rats (Fig. 1B). The results suggest that temporal gene expression uncovered the phenotypes obscured by developmental compensation.
Unaltered number of dopaminergic neurons and unaltered content of striatal dopamine in LRRK2 transgenic rats
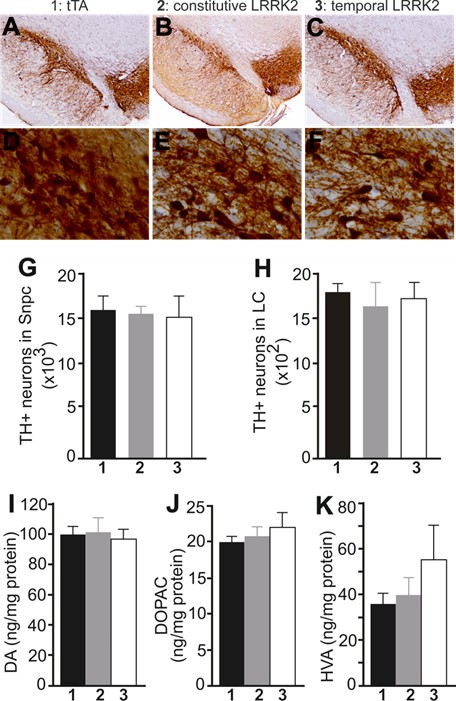
PD is characterized by progressive loss of dopaminergic neurons in the SNpc [42]. We then examined dopaminergic neurons by stereological cell counting. Neither constitutive nor temporal overexpression of mutant LRRK2 altered the number of dopaminergic neurons in transgenic rats at advanced ages (Fig. 2A-1G). Noradrenergic neurons in the locus coeruleus (LC) are often lost at the early stages of PD [43]. We also assessed the TH-positive neurons in the LC and found no loss of the neurons in aged rats constitutively or temporally expressing mutant LRRK2 (Fig. 2H). The axonal terminals of dopaminergic neurons are the primary targets of degeneration in PD [44]. We measured the contents of the neurotransmitter dopamine in the striatum, but did not detect any change to the total striatal dopamine in mutant LRRK2 transgenic rats, although the transgenic rats expressing mutant LRRK2 temporally in adulthood developed abnormal locomotor activity in advanced ages (Figs. 1B and 2I-2K). Proteinopathy is considered a characteristic of PD [42, 45]. In our mutant LRRK2 transgenic rats, we did not detect any inclusion positive for α-synuclein, ubiquitin, or phosphorylated Tau.
Impaired reuptake of dopamine in aged rats overexpressing mutant LRRK2 in adulthood
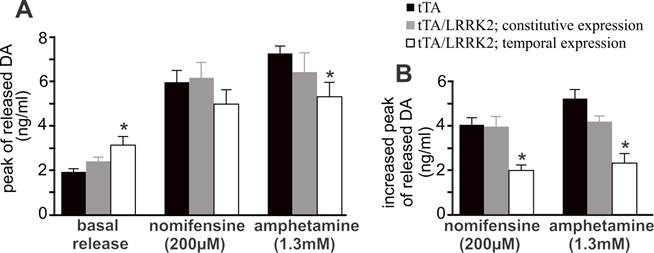
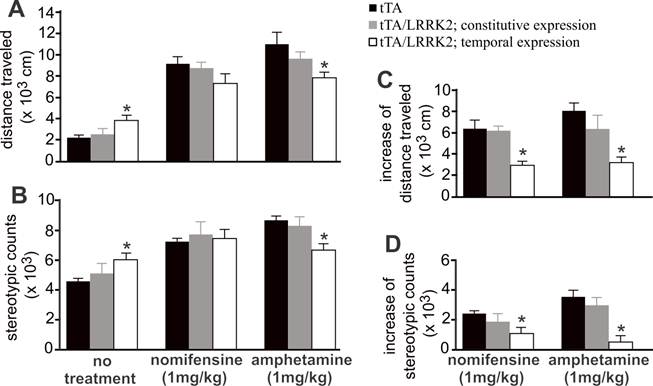
Temporal overexpression of mutant LRRK2 enhanced locomotor activity in aged rats (Figs. 1 and 4), indicating that dopaminergic neurotransmission was altered. In the aged rats, however, the content of striatal dopamine was unaltered (Fig. 2I), suggesting that dopamine synthesis and storage may not be interrupted by temporal overexpression of mutant LRRK2. The extracellular level of dopamine released from the neuronal terminals is virtually related to dopaminergic function. Using intra-brain microdialysis, we assessed dopamine release in the striatum of free-moving transgenic rats and detected a significant increase in dopamine release in temporally, but not in constitutively, mutant LRRK2-expressing rats (Fig. 3). Enhanced locomotor activity in mutant LRRK2 rats is attributable to elevated levels of dopamine in the synaptic cleft. Elevation of extracellular dopamine levels may result from increased release or reduced reuptake of dopamine. Nomifensine, a selective DAT inhibitor, increased dopamine release to comparable levels between the mutant LRRK2 transgenic rats and the control rats (Fig. 3A). This increased release of dopamine upon nomifensine stimulation was significantly reduced by temporal, but not by constitutive, expression of mutant LRRK2 (Fig. 3B).The results suggest that dopamine release was unaltered but that dopamine reuptake was impaired in the mutant LRRK2 rats. Amphetamine is structurally similar to dopamine and has a higher affinity for DAT and vesicular monoamine transporter type 2 (VMAT2) than dopamine. The affinity of amphetamine is higher for DAT than for VMAT2. Amphetamine-evoked release of dopamine was compromised in transgenic rats expressing mutant LRRK2 in adulthood (Fig. 3), confirming that DAT activity was impaired in the mutant rats at advanced ages. Accordingly, amphetamine- and nomifensine-elicited locomotor activity was reduced in aged rats that temporally, but not constitutively, overexpressed mutant LRRK2 (Fig. 4A-4D). Collectively, the results suggest that DAT activity was impaired in aged rats expressing mutant LRRK2 in adulthood.
Temporal overexpression of mutant LRRK2 alters locomotor activity in aged rats. A, Immunoblotting revealed a full recovery of LRRK2 transgene expression in the brainstem after Doxycycline (Dox) withdrawal. Brain tissues were dissected from transgenic rats carrying CAG-tTA and TRE-hLRRK2G2019S-HA transgenes. The double-transgenic rats were untreated with Dox throughout their lives (constitutive expression) or treated with Dox (50 µg/ml in drinking water) from the embryonic stage to 5 months of age (temporal expression). Human LRRK2 was detected with an antibody to HA tag fused to the C-terminal of the mutant human LRRK2. GAPDH immunoreactivity was detected as a control for equal loading. B, Open field activity assay revealed an increase in locomotor activity in aged rats temporally, but not constitutively, overexpressing the mutant LRRK2. Data are means + SEM (n = 10). * p < 0.05.
Overexpression of mutant LRRK2 causes no loss of nigral dopaminergic neurons and striatal dopamine contents in aged rats. A-F, Representative photos of low (A-C) or high (D-F) magnification show the SNpc of transgenic rats at 18 months of age. Transgenic rats carried a CAG-tTA single transgene (1: tTA), or CAG-tTA and TRE-LRRK2G2019S double transgenes (2 and 3: LRRK2). Constitutive expression of mutant LRRK2 (2: constitutive LRRK2) was achieved by withholding Dox throughout rat's lives, and temporal expression of the transgene (3: temporal LRRK2) was achieved by withdrawing Dox from rats at the age of 5 months. G, H, Stereological cell counting revealed no loss of TH-positive neurons in the SNpc and locus coeruleus (LC). Data are means ± SEM (n = 7). I-K, HPLC revealed the contents of striatal dopamine (DA) and DA metabolites (DOPAC and HVA) in transgenic rats. Data are means ± SEM (n = 10). Experimental group number in panels G-K corresponds to that in panels A-F.
Overexpression of mutant LRRK2 impairs DA reuptake in aged rats. A, Intra-brain microdialysis revealed the extracellular levels of DA in the striatum of free-moving rats at 18 months of age. Basal DA release was established in living rats when three consecutive samplings resulted in a sustainable level of DA in dialysates in the absence of stimulation. The peaks of stimulated DA release were detected after nomifensine (200 µM) or amphetamine (1.3 mM) was applied to dialysate. Data are means ± SEM (n = 8). * p < 0.05 compared to CAG-tTA single transgenic rats (tTA). B, Increased peak of stimulated DA release was calculated for each animal by subtracting basal release from stimulated release. * p < 0.05 compared to either tTA single transgenic rats or constitutive LRRK2-expressing rats.
Temporal overexpression of mutant LRRK2 increases spontaneous, but reduces evoked, locomotor activity in aged rats. A, B, Open field activity assay measured locomotor activity in transgenic rats treated or untreated with nomifensine or amphetamine. Transgenic rats (18 months old) were subject to behavior test. C, D, Increased locomotor activity in response to drug stimulation was calculated by subtracting spontaneous activity counts from evoked activity counts. Data are means ± SEM (n = 10). * p < 0.05 compared to CAG-tTA single transgenic rats (tTA).
No direct interaction of mutant LRRK2 with dopamine transporter
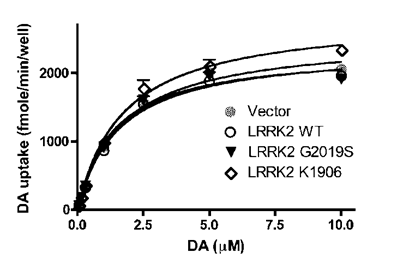
Microdialysis revealed that dopamine reuptake was impaired in aged rats expressing mutant LRRK2 temporally in adulthood (Fig. 3). To further assess the potential interaction of mutant LRRK2 with DAT, we used the neuroblastoma cells SH-SY5Y as an in vitro model of dopaminergic neurons. The SH-SY5Y cells that were stably transfected with DAT-expressing plasmid were transiently transfected with normal or mutant LRRK2-expressing constructs. In the presence of forcedly overexpressed LRRK2 variants, dopamine uptakes were determined and no significant difference between LRRK2 variants tested was observed (Fig. 5). The results suggest that normal and mutant LRRK2 do not interact with DAT directly.
Overexpression of LRRK2 variants has no direct effect on the transport activity of DAT heterogeneously expressed in dopaminergic cells. SH-SY5Y cells stably expressing GFP-DAT were seeded in 96-well plates and transiently transfected with indicated constructs. [3H]-DA uptake was determined 2 days after transfection. Data were analyzed according to the Michaelis-Menten kinetics using GraphPad Prism 4.0 software. The Vmax and Km values derived from the regression curves were not significantly different.
Discussion
Because G2019S mutation in LRRK2 is observed in both the familial and sporadic PD [10], we chose LRRK2G2019S as an example of LRRK2 mutations for transgenic study. Temporal, but not constitutive, overexpression of the mutant LRRK2 induced unexpected phenotypes, including enhanced locomotor activity and impaired dopamine reuptake. Altered locomotor activity in transgenic rats was attributed to elevated levels of extracellular dopamine revealed by intra-brain microdialysis. The extracellular level of dopamine is an outcome of the balance between dopamine release and reuptake. Blocking dopamine reuptake with the DAT-specific inhibitor nomifensine had a reduced effect on extracellular dopamine. DAT-mediated reuptake of dopamine can be reverted by amphetamine. In LRRK2 transgenic rats, amphetamine had a reduced effect on the extracellular level of released dopamine. Pharmacologic analyses suggest that DAT-mediated dopamine reuptake was impaired in mutant LRRK2 transgenic rats. The behavioral change was observed in aged, but not in young, transgenic rats, implying that DAT activity was impaired gradually as mutant LRRK2 was overexpressed. The phenotypes were induced by temporal, but not by constitutive, overexpression of pathogenically mutated LRRK2, although the expression levels of mutant LRRK2 were comparable between temporal and constitutive expression patterns. The findings suggest that damage to DAT activity was an indirect effect of mutant LRRK2. This notion is further supported by in vitro analysis, which did not detect any direct interaction of mutant or normal LRRK2 with DAT. Temporal expression of mutant LRRK2 in adult rats caused gradual damage to DAT-mediated dopamine reuptake.
Neuronal death in PD is considered a dying-back process and the neurites of dopaminergic cells are thought to be the primary targets for degeneration [28, 44]. Most studies of genetic PD models reveal no dopaminergic neuron death, but do reveal damage to dopamine neurotransmission [18, 23, 29, 32, 46]. As impaired D2 autoreceptor function is revealed in DJ-1 knockout and in LRRK2 knockin mice [18, 29], DAT-mediated dopamine reuptake was, for the first time, revealed impaired in a transgenic model for PD. Our results, though unexpected, are consistent with clinical findings in mutant LRRK2 carriers who display reduced DAT binding at asymptomatic stages [33]. Elevation of extracellular dopamine may cause oxidative damage to dopaminergic neurites and is possibly an early event in PD pathogenesis. Compared to constitutive gene expression, temporal overexpression of mutant LRRK2 in adult rats induced a greater phenotype, possibly because it avoided developmental compensation for transgene expression. Our finding may help interpret why transient expression of PD genes by viral delivery often produces great disease phenotypes that could not be reproduced by constitutive expression of the disease genes in transgenic animals [26, 28, 29, 32, 47, 48].
Inducible LRRK2 transgenic rats recapitulated the early pathology observed in asymptomatic subjects who carry pathogenic mutation in LRRK2. However, our transgenic rats did not develop dopaminergic neurodegeneration in their lifetime. The limited copy of mutant LRRK2 transgene (only 2 copies) may yield insufficient disease protein, which was below the threshold to induce dopaminergic neuron death in a rat's lifetime. Phenotypic expression in rats may be improved by increasing expression of the LRRK2 transgene. As PD is considered a multifactorial disease, multiple genetic and environmental factors may interact to induce dopaminergic neuron death [37]. Indeed, varied disease penetrance and varied onset time are observed in the subjects carrying pathogenic mutation in LRRK2 [9], favoring multifactorial pathogenesis in PD. Mutation of a single PD gene, particularly with low yield, may not be sufficient to induce neuron death within the short lifespan of rodents. Based on the multifactorial pathogenesis of PD, a combination of multiple disease factors (including environmental and genetic factors) will improve phenotypic expression in animal models.
Acknowledgements
We thank Ms. Dian Wang and Ms. Xiaotao Wei for technical assistance.
Funding
This work is supported by the Michael J. Fox Foundation (to H.Z. and X.G.X.) and by the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NS064042 and NS072113 to X.G.X and NS058463 to Y.J.L). The content is solely the responsibility of the authors and does not necessarily represent the official view of the NIH institutes.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG. et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276(5321):2045-2047
2. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605-608
3. Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G. et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299(5604):256-259
4. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S. et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304(5674):1158-1160
5. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P. et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601-607
6. Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS. et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44(4):595-600
7. Khan NL, Jain S, Lynch JM, Pavese N, Abou-Sleiman P, Holton JL, Healy DG, Gilks WP, Sweeney MG, Ganguly M, Gibbons V, Gandhi S, Vaughan J, Eunson LH, Katzenschlager R. et al. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson's disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain. 2005;128(Pt 12):2786-2796
8. Wider C, Dickson DW, Wszolek ZK. Leucine-rich repeat kinase 2 gene-associated disease: redefining genotype-phenotype correlation. Neurodegener Dis. 2010;7(1-3):175-179
9. Papapetropoulos S, Singer C, Ross OA, Toft M, Johnson JL, Farrer MJ, Mash DC. Clinical heterogeneity of the LRRK2 G2019S mutation. Arch Neurol. 2006;63(9):1242-1246
10. Nichols WC, Pankratz N, Hernandez D, Paisan-Ruiz C, Jain S, Halter CA, Michaels VE, Reed T, Rudolph A, Shults CW, Singleton A, Foroud T. Genetic screening for a single common LRRK2 mutation in familial Parkinson's disease. Lancet. 2005;365(9457):410-412
11. Bosgraaf L, Van Haastert PJ. Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 2003;1643(1-3):5-10
12. West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102(46):16842-16847
13. Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O'Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15(2):223-232
14. Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9(10):1231-1233
15. Iaccarino C, Crosio C, Vitale C, Sanna G, Carri MT, Barone P. Apoptotic mechanisms in mutant LRRK2-mediated cell death. Hum Mol Genet. 2007;16(11):1319-1326
16. West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16(2):223-232
17. Chen L, Cagniard B, Mathews T, Jones S, Koh HC, Ding Y, Carvey PM, Ling Z, Kang UJ, Zhuang X. Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J Biol Chem. 2005;280(22):21418-21426
18. Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45(4):489-496
19. Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, Wakeham A, You-Ten AJ, Kalia SK, Horne P, Westaway D, Lozano AM, Anisman H, Park DS, Mak TW. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci U S A. 2005;102(14):5215-5220
20. Von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM. Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci U S A. 2004;101(29):10744-10749
21. Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A. 2007;104(27):11441-11446
22. Winner B, Melrose HL, Zhao C, Hinkle KM, Yue M, Kent C, Braithwaite AT, Ogholikhan S, Aigner R, Winkler J, Farrer MJ, Gage FH. Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice. Neurobiol Dis. 2011;41(3):706-716
23. Melrose HL, Dachsel JC, Behrouz B, Lincoln SJ, Yue M, Hinkle KM, Kent CB, Korvatska E, Taylor JP, Witten L, Liang YQ, Beevers JE, Boules M, Dugger BN, Serna VA. et al. Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol Dis. 2010;40(3):503-517
24. Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ3rd, Shen J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A. 2010;107(21):9879-9884
25. Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J Neurosci. 2010;30(5):1788-1797
26. Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC. et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson's-disease-related mutant alpha-synuclein. Neuron. 2009;64(6):807-827
27. Andres-Mateos E, Mejias R, Sasaki M, Li X, Lin BM, Biskup S, Zhang L, Banerjee R, Thomas B, Yang L, Liu G, Beal MF, Huso DL, Dawson TM, Dawson VL. Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine). J Neurosci. 2009;29(50):15846-15850
28. Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, Zhou C, Geghman K, Bogdanov M, Przedborski S, Beal MF, Burke RE, Li C. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat Neurosci. 2009;12(7):826-828
29. Tong Y, Pisani A, Martella G, Karouani M, Yamaguchi H, Pothos EN, Shen J. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci U S A. 2009;106(34):14622-14627
30. Lee SB, Kim W, Lee S, Chung J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun. 2007;358(2):534-539
31. Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T, Ross CA, Montell C, Smith WW. A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A. 2008;105(7):2693-2698
32. Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z. Enhanced Striatal Dopamine Transmission and Motor Performance with LRRK2 Overexpression in Mice Is Eliminated by Familial Parkinson's Disease Mutation G2019S. Journal of Neuroscience. 2010;30(5):1788-1797
33. Nandhagopal R, Mak E, Schulzer M, McKenzie J, McCormick S, Sossi V, Ruth TJ, Strongosky A, Farrer MJ, Wszolek ZK, Stoessl AJ. Progression of dopaminergic dysfunction in a LRRK2 kindred: a multitracer PET study. Neurology. 2008;71(22):1790-1795
34. Zhou H, Huang C, Yang M, Landel CP, Xia PY, Liu YJ, Xia XG. Developing tTA Transgenic Rats for Inducible and Reversible Gene Expression. Int J Biol Sci. 2009;2(5):171-181
35. Xia XG, Schmidt N, Teismann P, Ferger B, Schulz JB. Dopamine mediates striatal malonate toxicity via dopamine transporter-dependent generation of reactive oxygen species and D2 but not D1 receptor activation. J Neurochem. 2001;79(1):63-70
36. Zhou H, Falkenburger BH, Schulz JB, Tieu K, Xu Z, Xia XG. Silencing of the Pink1 gene expression by conditional RNAi does not induce dopaminergic neuron death in mice. Int J Biol Sci. 2007;3(4):242-250
37. Tian T, Huang C, Tong J, Yang M, Zhou H, Xia XG. TDP-43 Potentiates Alpha-synuclein Toxicity to Dopaminergic Neurons in Transgenic Mice. Int J Biol Sci. 2011;7(2):234-243
38. Hong WC, Amara SG. Membrane cholesterol modulates the outward facing conformation of the dopamine transporter and alters cocaine binding. J Biol Chem. 2010;285(42):32616-32626
39. Zhou H, Huang C, Chen H, Wang D, Landel CP, Xia PY, Bowser R, Liu YJ, Xia XG. transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLOS Genetics. 2010;6(3):e1000887
40. Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, Okwuonu G, Hines S, Lewis L, DeRamo C, Delgado O. et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428(6982):493-521
41. Tesson L, Cozzi J, Menoret S, Remy S, Usal C, Fraichard A, Anegon I. Transgenic modifications of the rat genome. Transgenic Res. 2005;14(5):531-546
42. Cookson MR. The biochemistry of Parkinson's disease. Annu Rev Biochem. 2005;74:29-52
43. Forno LS, DeLanney LE, Irwin I, Langston JW. Similarities and differences between MPTP-induced parkinsonsim and Parkinson's disease. Neuropathologic considerations. Adv Neurol. 1993;60:600-608
44. Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. 1973;20(4):415-455
45. Pappolla MA. Lewy bodies of Parkinson's disease. Immune electron microscopic demonstration of neurofilament antigens in constituent filaments. Arch Pathol Lab Med. 1986;110(12):1160-1163
46. Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65(1):66-79
47. Lee BD, Shin JH, VanKampen J, Petrucelli L, West AB, Ko HS, Lee YI, Maguire-Zeiss KA, Bowers WJ, Federoff HJ, Dawson VL, Dawson TM. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson's disease. Nat Med. 2010;16(9):998-1000
48. Krenz A, Falkenburger BH, Gerhardt E, Drinkut A, Schulz JB. Aggregate formation and toxicity by wild-type and R621C synphilin-1 in the nigrostriatal system of mice using adenoviral vectors. J Neurochem. 2009;108(1):139-146
Author contact
![]() Corresponding author: H.Z. (Hongxia.zhouedu), Y.J.L (yjliuedu), or X.G.X (xugang.xiaedu).
Corresponding author: H.Z. (Hongxia.zhouedu), Y.J.L (yjliuedu), or X.G.X (xugang.xiaedu).