Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Prostate cancer and AR
History of AR short form variants
Cloning and identification of...
Function of AR splice variants
Perspectives
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(6):815-822. doi:10.7150/ijbs.7.815 This issue Cite
Review
A New Trick of an Old Molecule: Androgen Receptor Splice Variants Taking the Stage?!
Zhiyong Guo, Yun Qiu ![]()
Department of Pharmacology and Experimental Therapeutics, University of Maryland School of Medicine, Baltimore, Maryland 21201, USA
Received 2011-5-20; Accepted 2011-6-20; Published 2011-7-6
Abstract
Prostate cancer is the second leading cause of cancer-related death in American men. Although most prostate cancers are initially androgen-dependent and respond to androgen ablation therapy, majority of them eventually relapse and progress into incurable castration-resistant (or hormone refractory) prostate cancer. The underlying mechanisms are the focus of intensive investigation for development of more effective treatment. Mounting evidence from both clinical and basic research has demonstrated that the activity of the androgen receptor (AR) is still required for castration-resistant prostate cancer. Multiple mechanisms by which AR is re-activated under androgen-depleted conditions may be involved in the development of castration resistance. The recent identification of AR splicing variants may add another layer of complexity in AR biology. The present review summarizes recent progress in study of AR splicing variants in prostate cancer.
Keywords: androgen receptor, prostate cancer, splicing variants, alternative splicing.
Prostate cancer and AR
Prostate cancer is the second leading cause of cancer-related death in American men [1]. Majority of prostate cancers are initially androgen-dependent and respond to androgen ablation therapy well, with a significant temporary remission. However, over 95% of hormone-responsive tumors eventually relapse and progress into incurable castration-resistant (or hormone refractory) prostate cancer. The underlying mechanisms are the focus of intensive investigation for development of more effective treatment.
The androgen receptor (AR) is a key transcription factor that is activated by androgens and transduces androgen signaling in prostate cells [2, 3]. Mounting evidence from both clinical and basic research has demonstrated that AR activity is still required for castration-resistant prostate cancer [4-7]. The mechanisms by which AR is re-activated under androgen-depleted conditions have yet to be fully elucidated. Mutation and amplification of AR gene, alteration in protein kinases, growth factors, nuclear receptor coactivators, steroid metabolism enzymes and alternative splicing variants have been proposed to modulate AR signaling and may, therefore, contribute to castration resistance [8-15]. In this review, we will focus on the recent progress in study of AR splicing variants in prostate cancer.
History of AR short form variants
The full-length cDNA of the AR gene was first reported in 1988 [16, 17]. The major transcript derived from the AR gene in prostate cells, designated as AR transcript variant 1 (GI: 21322251) in Genbank, encodes a 110-kDa protein with four major functional domains including an N-terminal transactivation domain (NTD), a DNA-binding domain (DBD), Hinge domain (H) and a C-terminal ligand-binding domain (LBD) (Figure 1). Although the LBD is responsible for binding to androgen and some co-factors, it may also serve as a negative regulator of AR transcription activity based on several observations that deletion of LBD generates androgen-independent constitutively active AR mutants [18-20]. However, it was unclear at that time whether such constitutively active AR isoform(s) were naturally expressed in human tissues and if they did exist, what were the functions of these AR short form variants?
Schematic structure of human AR splice variants reported in GenBank. The hatched cassettes stand for the cryptic exons. Solid thick lines represent the transcribed exon sequences. U: unique N- or C-terminal sequence.
For more than a decade, researchers have observed that, in addition to the well-studied 110-kDa AR protein, some lower molecular-weight protein bands are detectable by an antibody for the N-terminal region of AR in some AR-expressing cell lines. However, the explanation for the origins of these AR short form variants was quite controversial. At least four potential mechanisms underlying generation of short form AR proteins were proposed: (1) alternative translation start codons; (2) proteolytic cleavage; (3) premature stop codon resulted from mutation; and (4) alternative transcription start site.
In 1994, Wilson and McPhaul described two forms, 110-kDa and 87-kDa, of AR protein are present in human genital skin fibroblasts [21]. They further showed that the 87-kDa isoform (AR-A) contains an intact C terminus but lacks the normal N terminus found in the 110-kDa isoform (AR-B). They proposed that the AR-A is due to translation initiation of AR protein at the internal Methionine 188 residue of AR-B. They also suggested that AR-A and AR-B may differ in their ability to activate target genes and regulated differently in various cell types, which are reminiscent of the A and B forms of human progesterone receptor [21]. In 2001, Gregory et al. reported that the AR short forms similar to that of the previously described 87-kDa AR-A are derived from in vitro proteolytic cleavage of N- or C-terminal regions of AR during cell extraction and storage [22]. In 2003, Tepper et al. reported an in-frame tandem duplication of exon 3 of AR in CWR22Rv1 cells. This insertional mutation was accompanied by a truncated AR protein of 75-80 kDa. Furthermore, they showed that the short form AR in CWR22Rv1 cells was a C-terminal truncated AR (referred as ARΔLBD) which lacks the LBD. The ARΔLBD exhibits constitutive nuclear localization and DNA binding [23]. In addition, Libertini reported that the calcium-sensitive calpain could remove the AR C-terminal LBD and generate a constitutively active AR protein in in vitro and in vivo analysis [24]. They further showed that this truncated AR is expressed at a higher level in several tumors compared with benign prostate tissues. The truncated AR appears to have three to five times more potent transactivating activity than the full-length AR in reporter assays. In addition, Lapouge reported that a mutation of Q640X identified in the hinge region of AR in metastatic prostate cancer lesions may generate a short form AR protein lacking LBD. This ARQ640X mutant exhibits strong and ligand-independent transcriptional activity [25]. In 2005, Ahrens-Fath and Haendler reported that a novel AR transcript variant, designated as AR transcript variant 2 (GI:58535454) (also referred as AR45) in GenBank, encodes a 45-kDa protein which is composed of a unique seven amino-acid N-terminal sequence, DBD, H and LBD [26]. This transcript may likely be resulted from an alternative transcript start site of the AR gene. AR45 is highly expressed in heart, to a lesser extent, in a variety of tissues including skeletal muscle, uterus, prostate, breast and lung. Overexpression of AR45 may either repress or stimulate AR transcriptional activity depending on cellular context.
Cloning and identification of alternatively spliced AR variants
Since 2008, we and others have cloned and identified multiple alternatively spliced AR variants [11-15]. We initiated our search for potential AR splicing variants based on the observation that a panel of AR shRNAs targeting different exons displayed differential knock-down effects on the 110-kDa and 80-kDa AR proteins, suggesting that AR and its short form variant(s) were derived from more than one transcript. We performed 3'-RACE, and cloned multiple alternatively spliced AR isoforms. These variants are generated from alternative splicing through various mechanisms, including exon skipping, cryptic splicing donor or acceptor usage, cryptic exon inclusion, etc. Because the sequences of two AR transcript variants (the prototype AR and AR45) are already present in GenBank at the time when we deposited our clone sequences, the newly identified splicing variants were designated as AR3 (GI:224181613), AR4 (GI:224181615), AR5 (GI:224181619), AR6 (GI:224181621), and etc [12]. In addition to the isoforms we reported, several other groups also reported a variety of AR splice variants identified in their study. Dehm et al. reported two AR isoforms of AREx1/2/2b and AR1/2/3/2b [11]. Hu et al. described AR-V1, AR-V2, AR-V3, AR-V4, AR-V5, AR-V6, and AR-V7 [13]. Sun cloned ARv567es (GI:270358641) in a castration-resistant xenograft [14]. Watson reported AR-V8 and AR-V9 as well as two mouse Ar vairants mV-1 and mV-4 [15]. These AR isoforms retain NTD and DBD, but lack LBD. Based on currently available sequence data in GenBank, a schematic illustration of AR and its splice variants is summarized in Figure 1. Among the AR isoforms identified so far, AR3 (a.k.a. AR-V7 based on protein sequence homology) appears to be one of the major constitutively active AR isoforms in prostate cancer cell lines and human prostate tissues [12, 13, 27]. Expression of AR3/AR-V7 protein in various human prostate cancer cell lines and tissues has been reported in two independent studies [12, 13]. Meanwhile, existence of a protein product derived from other AR splice variants remains to be validated.
Function of AR splice variants
Given much less abundance of AR splice variants compared to the prototype AR, one of the key issues has been debated in the field ever since their discovery is whether these isoforms are of functional importance or they are just by-products of splicing process of AR gene. Several lines of evidence support important roles of these variants in prostate cancer biology. It has been shown that selective knock-down of AR3 expression (without altering the prototype AR protein level) in CWR-R1 and 22Rv1 cells significantly inhibit their growth under androgen-depleted conditions [12]. The potential role of AR splice variants in driving prostate cancer progression is further supported by several independent clinical correlation studies [12, 13, 27]. The high expression level of AR3/AR-V7 can predict biochemical recurrence as well as cancer-specific survival, suggesting that AR3/AR-V7 may have a strong potential to serve as a prognostic marker independent of AR. These data support the notion that AR3 may play a critical role in prostate cancer biology.
In terms of transcription activity, several AR variants have been shown to induce ligand-independent activation of ARE-driven reporters in the absence of androgens. However, the functions of these AR isoforms should not be simplified as a mere substitution for the prototype AR as they may have significant distinct functions. We and others showed that AR3/AR-V7 and ARv567es isoforms have unique target gene sets distinct from those regulated by the prototype AR [12, 14]. A gene expression profiling study revealed that AR3 shares some overlapping target genes with AR despite its lack of the AF2 domain (LBD) [12]. Therefore, AR3 may play a distinct yet essential role in androgen-independent growth through regulating a unique set of target genes including AKT1, which are not regulated by the prototype AR. These genes are involved in regulation of diverse biological processes including signal transduction, posttranslational modifications, transcription, chromatin remodeling, ion transportation and metabolism, suggesting that AR3 may play a critical role in homeostasis maintenance of its target cells. It is also noteworthy that several genes preferentially regulated by AR3, such as HOXB7, are known to be involved in inhibition of differentiation. AR3 may therefore play a role in inhibiting differentiation or inducing dedifferentiation of prostatic epithelial cells during development of prostate cancer. In addition, another target gene differentially regulated by AR and AR3 is TAF9B, which encodes one of the small subunits of the general transcription complex TFIID and functions as a basal transcriptional co-activator in promoter recognition and transcription complex assembly. TAF9B appears to be positively regulated by AR3 while negatively regulated by AR in both CWR-R1 and 22Rv1 cells, suggesting it may play a role in differential transcription regulation mediated by AR3 and AR. Further experiments are needed to test whether TAF9B plays a role in determination of the promoter specificity associated with AR or AR3. It is very likely that AR and AR3 may function independently, cooperatively or competitively depending upon the context of promoters and cell types.
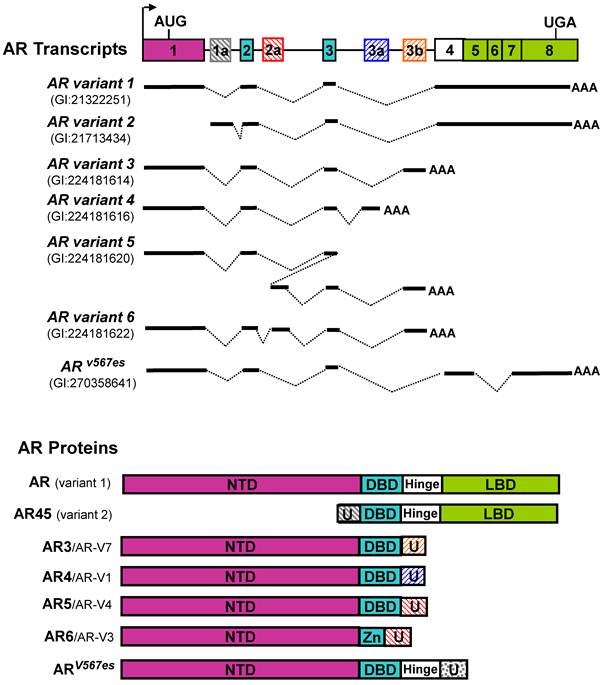
Despite of structural similarity, these AR variants may exert their unique cellular functions, in a cell context dependent manner, due to their presence in different subcellular compartments. For example, in transiently transfected COS-1 or LNCaP cells, AR3 is primarily localized in the nucleus, suggesting a role in regulating gene transcription. AR4 appears to be mainly located in the cytoplasm, suggesting that a cytoplasmic function. Another AR splice variant AR8 is associated with the plasma membrane (Yang and Qiu unpublished data). It is notable that subcellular localization of a given AR isoform could be dynamic and vary in different cell types. For instance, unlike in COS-1 and LNCaP cells, localization of AR3 in transiently transfected DU145 cells is quite heterogeneous and diffused in cytoplasm in about 50% of cells (Figure 2).
Subcellular localization of AR3/AR-V7. COS-1 and DU145 cells were transfected with an AR3 expressing plasmid. At 48h post-transfection, cells were stained with an anti-AR N-terminal antibody (green) and nuclei were visualized with DAPI (blue). In DU145 cells, both nuclear and cytoplasmic staining is observed.
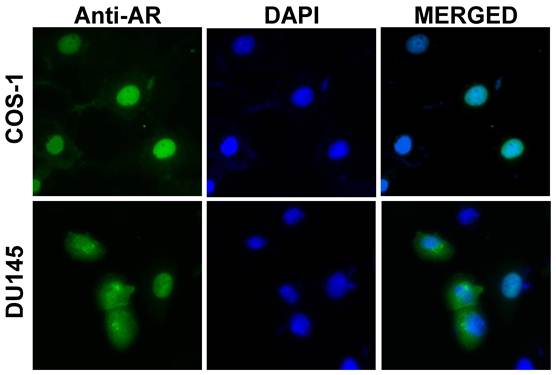
In benign prostate tissue, AR3 protein is mainly present in basal and stromal cells whereas barely detectable in luminal epithelial cells (Figure 3) [12]. However, an increase of AR3 expression is readily detected in luminal cells in the adjacent PIN lesions and majority of malignant luminal cells, suggesting a potential role of AR3 in tumor initiation and development. Although the expression level of AR3 protein is increased, AR3 is mainly found in the cytoplasm in most of hormone naïve tumors while nuclear-positive staining is detected in over 40% of hormone refractory tissues, suggesting that AR3 nuclear translocation may be correlated with androgen independence. It is possible that AR3 transcriptional activity is tightly regulated by its subcellular localization like the prototype AR. It is still unclear what are the mechanisms underlying regulation of AR3 nuclear translocation. The nuclear translocation signal of AR3 could be different from that of AR due to the lack of K630, K632 and K632 residues essential for AR nuclear translocation [28, 29]. The 16-a.a. unique sequence at the C-terminus could reconstitute a new nuclear translocation signal for AR3 (Guo and Qiu unpublished data). We previously showed that Src kinase may regulate AR nuclear translocation independent of androgens, possibly through phosphorylating Y534 in the NTD [30]. Most of Src-induced tyrosine phosphorylation sites are clustered in NTD and also present in AR3 and Src inhibitors could prevent the nuclear translocation of AR3 in CWR-R1 cells (Guo and Qiu unpublished data). Therefore, Src family kinases may be involved in regulating AR3 activity.
Immunohistochemistry analysis on human prostate tissue sections using the AR3 specific antibody (ref [12]). Left, in benign tissues, AR3 is primarily detected in stromal and basal compartments whereas virtually no expression in luminal epithelial cells. Center, in adjacent prostatic intraepithelial neoplasia (PIN), AR3 is detectable in luminal epithelial cells. Right, in hormone-resistant (HR) tumors, nuclear staining of AR3 is enhanced.
It is still under debate whether AR is expressed in basal cells in prostate. Based on our immunohistochemistry analysis using an AR N-terminus antibody, a weak positive staining is detectable in some basal cells. Such relatively weaker signal could easily be neglected when compared to much stronger AR staining in luminal epithelial cells. The presence of AR transcript in purified basal cells is further confirmed by a recent study [31]. The expression of AR3 in basal cells is readily detectable due to the lack of AR3 expression in luminal epithelial cells in benign prostate gland. It should be noted that AR3 staining is quite heterogeneous in most of tumor samples we examined, suggesting that AR3 may only be required for a subpopulation of tumor cells. However, we cannot exclude the possibility that AR3-positive cells may influence their surrounding cells via an indirect manner, such as releasing autocrine/paracrine factors.
Although we are unable to detect direct interaction between endogenous AR3 and AR, ARv567es has been reported to associate with AR under overexpression conditions and modulate AR transcription activity [14]. The existence of various AR isoforms and their potential functions in modulating AR transcription activity may explain why the AR replacement strategy described in our previous study could only partially rescue the AR activity [30]. The AR shRNA that we used in the knockdown experiments could target not only the prototype full-length AR but also the other less abundant AR transcript variants, such as AR2, ARv567es and possibly other unidentified isoforms. It may require restoration of more than just the full-length AR to fully rescue the complicated endogenous AR function, suggesting that optimal AR transcription activity may very likely dependent upon other splice variants in prostate cells.
Several independent studies showed that AR splice variants, AR3/AR-V7 and AR4/AR-V1, are upregulated in hormone refractory prostate tumors compared with hormone naïve counterparts [12, 13, 27]. Increase of the AR3/AR-V7 variant expression in prostate cancer cell lines in response to androgen ablation is also demonstrated [15]. A recent report suggest that increased expression of AR splice variants is associated with amplification of the AR locus in human prostate cancer cell line 22Rv1 and a subset of human prostate tumors [32]. It is noteworthy that, in 22Rv1 cells, despite AR amplification, the level of AR protein is not increased accordingly while the truncated AR splice variants appear to become dominant. AR amplification is reported in about 20% of hormone refractory prostate cancer, however, AR protein levels in hormone-resistant tumors with and without AR amplification are not significantly different [33]. This raises the possibility that “overproduction” of AR variants resulted from AR amplification in castration-resistant tumors actually plays a rate-limiting and essential role in disease progression. The relative expression level of AR and AR isoforms may vary among different prostate cancer cell lines and tissues, possibly regulated by a variety of extracellular stimuli and environmental perturbation. The relative expression level of AR variants to AR is altered under different culture conditions, such as cell density. This implies that intercellular communication via cell adhesion and/or autocrine/paracrine factors may influence the expression of AR isoforms. In addition, the expression level of AR splice variants may also be influenced by the level of AR protein under certain cell culture conditions. To selectively knockdown the prototype AR, we need to consider cell density as well as dose of shRNAs used for knockdown experiments. In our lenti-virus based shRNA knock-down experiments, selective knock-down of AR by the AR shRNA targeting at Exon 6 could be achieved under appropriate experimental conditions [12, 30]. However, we do observe that if very high doses of AR shRNA are used and when cell density is low, the expression of AR3 may be altered (Guo and Qiu, unpublished data). The change of AR3 transcript is likely caused by a posttranscriptional regulation mediated by AR protein as the level of AR pre-mRNA is not affected. Another possible explanation could be that there are more than one AR3 transcripts with different 3'UTR could be present in these cells. Some of them may contain a sequence from Exon 5-8 of the AR gene. However, so far, we have not yet detected such kind of transcripts in CWR-R1 cells. In addition, we cannot exclude the possibility that during the lenti-virus based knockdown process (routinely requires 2-3 days), some kind of secondary effects, such as perturbation of certain components of the splice machinery by viral infection, may indirectly alter AR3 expression. Nevertheless, these observations strongly suggest a functional interaction between AR and AR splice variants.
Taken together, these studies suggest that AR3 may have a distinct biological activity despite partially overlapping with AR. AR3 may primarily play a role in the regulation of androgen-independent biological processes and maintain homeostasis of the prostate gland in concert with AR. Deregulation of AR3 may play a role in the initiation, development and progression of prostate cancer.
Perspectives
Multiple AR transcript variants have been cloned and identified so far. Given that the AR gene is composed of at least eight exons and large introns, it is likely that increasing numbers of the AR splice variants will be identified in future cloning and sequencing effort. Development of sensitive and reliable isoform-specific antibodies for clinical applications could be quite challenging despite of some promising correlation data derived from a small cohort study. Specific detection of each of these variants by PCR-based methods in clinical samples may require more careful design of the amplification primers as every single exon in the AR gene could be a subject of exon skipping during splicing process. The ideal PCR primers should allow the amplified fragment cover all coding exons present in each variant to assure a faithful representation of the corresponding AR isoform. Failure to do so may generate misleading data. Designing the variant specific primer recognizing the unique exon-exon junction would also help to discriminate other similar transcripts generated from usage of cryptic splicing donor/acceptor which may often cause frame shift in the coding region. The expression pattern of AR splice variants could be very dynamic in response to various intracellular and extracellular cues. The dynamic repertoire of AR and AR variants may provide a sophistic regulatory system to orchestrate cellular responses to androgens and other extracellular stimuli. The future challenge would be how to individually characterize each of these AR variants if their expression level is extremely low compared to the prototype AR or their expression may be restricted to certain cell type or stage. Given the relatively low expression of these splice variants, it is important to demonstrate how they exert their unique functions independently, cooperatively or competitively with AR. Due to the nature of heterogeneity in prostate cancer, study on the potential functional interaction as well as independence of AR and AR variants in various prostate cancer cell lines and xenograft models would provide comprehensive understanding of the role of AR variants. Some of these variants may serve as a rate-limiting factor in certain cellular process or highly expressed in certain subtype of cells (such as stem/progenitor and neuroendocrine-like cells) despite apparent low abundance in a pool of mixed cell population. Furthermore, identification of what splicing factors are involved in exon selection in the context of the AR gene could potentially uncover new drug targets as deregulation of splicing of a group of genes are associated with prostate cancer progression. Further investigation on the mechanisms by which AR variants are regulated will allow us to better understand the complicated role of AR in prostate cancer biology and develop new biomarkers and more effective therapeutics to fight prostate cancer.
Acknowledgements
This work was supported by NIH (CA106504) and DOD (W81XWH-08-1-0174) grants to Y.Q. The authors apologize for not being able to cite many important studies related to this subject due to limited space.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277-300
2. McPhaul MJ. Factors that mediate and modulate androgen action. J Investig Dermatol Symp Proc. 2003;8:1-5
3. Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276-308
4. Marcelli M, Cunningham GR. Hormonal signaling in prostatic hyperplasia and neoplasia. J Clin Endocrinol Metab. 1999;84:3463-8
5. Culig Z, Hobisch A, Bartsch G, Klocker H. Androgen receptor--an update of mechanisms of action in prostate cancer. Urol Res. 2000;28:211-9
6. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R. et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33-9
7. Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002;62:1008-13
8. Culig Z, Klocker H, Bartsch G, Hobisch A. Androgen receptors in prostate cancer. Endocr Relat Cancer. 2002;9:155-70
9. Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001-15
10. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34-45
11. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469-77
12. Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H. et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305-13
13. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E. et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16-22
14. Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA. et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715-30
15. Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A. et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107:16759-65
16. Chang CS, Kokontis J, Liao ST. Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science. 1988;240:324-6
17. Lubahn DB, Joseph DR, Sullivan PM, Willard HF, French FS, Wilson EM. Cloning of human androgen receptor complementary DNA and localization to the X chromosome. Science. 1988;240:327-30
18. Simental JA, Sar M, Lane MV, French FS, Wilson EM. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J Biol Chem. 1991;266:510-8
19. Jenster G, van der Korput HA, van Vroonhoven C, van der Kwast TH, Trapman J, Brinkmann AO. Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol Endocrinol. 1991;5:1396-404
20. Rundlett SE, Wu XP, Miesfeld RL. Functional characterizations of the androgen receptor confirm that the molecular basis of androgen action is transcriptional regulation. Mol Endocrinol. 1990;4:708-14
21. Wilson CM, McPhaul MJ. A and B forms of the androgen receptor are present in human genital skin fibroblasts. Proc Natl Acad Sci U S A. 1994;91:1234-8
22. Gregory CW, He B, Wilson EM. The putative androgen receptor-A form results from in vitro proteolysis. J Mol Endocrinol. 2001;27:309-19
23. Tepper CG, Boucher DL, Ryan PE, Ma AH, Xia L, Lee LF. et al. Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res. 2002;62:6606-14
24. Libertini SJ, Tepper CG, Rodriguez V, Asmuth DM, Kung HJ, Mudryj M. Evidence for calpain-mediated androgen receptor cleavage as a mechanism for androgen independence. Cancer Res. 2007;67:9001-5
25. Lapouge G, Erdmann E, Marcias G, Jagla M, Monge A, Kessler P. et al. Unexpected paracrine action of prostate cancer cells harboring a new class of androgen receptor mutation--a new paradigm for cooperation among prostate tumor cells. Int J Cancer. 2007;121:1238-44
26. Ahrens-Fath I, Politz O, Geserick C, Haendler B. Androgen receptor function is modulated by the tissue-specific AR45 variant. FEBS J. 2005;272:74-84
27. Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A. et al. Expression of Androgen Receptor Splice Variants in Prostate Cancer Bone Metastases is Associated with Castration-Resistance and Short Survival. PLoS One. 2011;6:e19059
28. Cutress ML, Whitaker HC, Mills IG, Stewart M, Neal DE. Structural basis for the nuclear import of the human androgen receptor. J Cell Sci. 2008;121:957-68
29. Zhou ZX, Sar M, Simental JA, Lane MV, Wilson EM. A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. Requirement for the DNA-binding domain and modulation by NH2-terminal and carboxyl-terminal sequences. J Biol Chem. 1994;269:13115-23
30. Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O. et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10:309-19
31. Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010;329:568-71
32. Li Y, Alsagabi M, Fan D, Bova GS, Tewfik AH, Dehm SM. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res. 2011;71:2108-17
33. Edwards J, Krishna NS, Grigor KM, Bartlett JM. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br J Cancer. 2003;89:552-6
Author contact
![]() Corresponding author: Yun Qiu, Department of Pharmacology & Experimental Therapeutics, University of Maryland School of Medicine, 655 W Baltimore St, BRB Rm 4-002, Baltimore, MD 21201. Email: yqiuumaryland.edu
Corresponding author: Yun Qiu, Department of Pharmacology & Experimental Therapeutics, University of Maryland School of Medicine, 655 W Baltimore St, BRB Rm 4-002, Baltimore, MD 21201. Email: yqiuumaryland.edu