International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Results and discussion
Materials and methods
Acknowledgements
Abbreviations
Supplementary Material
References
Introduction
Results and discussion
Materials and methods
Acknowledgements
Abbreviations
Supplementary Material
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(8):1203-1213. doi:10.7150/ijbs.7.1203 This issue Cite
Research Paper
Photoactivation Studies of Zinc Porphyrin-Myoglobin System and Its Application for Light-Chemical Energy Conversion
Chin-Hao Chang1, Yi-Ting Hu1, Chen-Fu Lo2, Liyang Luo3, Hung-Ming Lin1, Cheng-Hsiang Chang1, Ching-Yao Lin2 ![]() , Eric Wei-Guang Diau3
, Eric Wei-Guang Diau3 ![]() , Tung-Kung Wu1
, Tung-Kung Wu1 ![]()
1. Department of Biological Science and Technology, National Chiao Tung University, 30068, Hsin-Chu, Taiwan, Republic of China
2. Department of Applied Chemistry, National Chi Nan University, Puli, Nantou County, Taiwan, Republic of China
3. Department of Applied Chemistry and Institute of Molecular Science, National Chiao Tung University, Hsin-Chu, Taiwan, Republic of China
Received 2011-5-3; Accepted 2011-9-7; Published 2011-10-27
Citation:
Chang CH, Hu YT, Lo CF, Luo L, Lin HM, Chang CH, Lin CY, Diau EWG, Wu TK. Photoactivation Studies of Zinc Porphyrin-Myoglobin System and Its Application for Light-Chemical Energy Conversion. Int J Biol Sci 2011; 7(8):1203-1213. doi:10.7150/ijbs.7.1203. https://www.ijbs.com/v07p1203.htm
Other stylesAbstract
An artificial zinc porphyrin-myoglobin-based photo-chemical energy conversion system, consisting of ZnPP-Mb or ZnPE1-Mb as a photosensitizer, NADP+ as an electron acceptor, and triethanolamine as an electron donor, has been constructed to mimic photosystem I. The photoirradiated product is able to reduce a single-electron acceptor protein cytochrome c, but cannot catalyze the two-electron reduction of acetaldehyde by alcohol dehydrogenase, thus demonstrating a single electron transfer mechanism. Furthermore, the artificial system can bifunctionally promote oxidoredox reactions, depending on the presence or absence of a sacrificial electron donor, thus suggesting its potential application in electrochemical regeneration steps involved in chemical transformation and/or energy conversion.
Keywords: photoelectrochemical energy conversion, zinc protoporphyrin, myoglobin, oxidoredox reaction, biosensitizer
Introduction
In biological photosynthesis, light energy is converted into electrochemical energy, stored as adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide phosphate (NADPH), and subsequently used for carbohydrates and other compound formation. It has been known that porphyrins and proteins with redox activity are key factors of photosynthesis. Artificial analogues of these processes are being investigated as methods for electricity [1-3], chemical energy [4], and hydrogen production [5,6]. For example, porphyrins and related porphyrinoids have been shown to be promising candidates for the fabrication of porphyrin-based dye sensitized solar cells (DSSCs) and organic solar cells, based on the intrinsic properties of the molecules including their vital roles in photosynthesis, intense absorption in the visible region, and diverse structural plasticities for light harvest [7-12]. Recently, Yeh, Diau, and Grätzel et al. reported a porphyrin-based DSSC, YD2, with an overall energy conversion efficiency of 11%, comparable to the state-of-the-art ruthenium-polypyridyl-based ones [13].
In parallel, many achievements on the study of intermolecular photoinduced electron transfer reactions have been constructed by replacing heme with zinc-protoporphyrins (ZnPPs), due to the longer lifetime of the excited triplet state compared with other metalloporphyrins and its function as strong reductants [14-18]. Hu et al. have constructed a myoglobin-based donor-sensitizer-acceptor triad to aid in the understanding of the long-lived charge-separated states and the regulation of the electron transfer pathway [19]. Moreover, reconstitution of apomyoglobin (apoMb) or human serum albumin (HSA) with zinc-protoporphyrin has greatly expanded protein function from oxygen storage or transport to that of a new functionalized enzyme with electron transfer properties and chemical reactivity [20-22]. These results indicated that the protein's environment can control and organize the coordination of prosthetic groups, prevent quenching aggregate, and prolong the lifetime of charge separation [19,23-32].
In light of the important biomimetic application of the photosystems, we present a zinc porphyrin-myoglobin-based photo-chemical energy conversion system which, upon photoirradiation, can catalyze the reduction of NADP+ to NADPH. Two different porphyrin derivatives, ZnPP and 5-(4-carboxy-phenylethynyl)-10,20-biphenyl porphinato zinc (II) (ZnPE1) (Fig. 1), were independently reconstituted into apoMb to form ZnPP-Mb or ZnPE1-Mb complexes and were compared for their light to chemical energy conversion efficiency. The experimental results showed that reconstitution of ZnPP or ZnPE1 into an apoMb protein pocket can prevent the quenching aggregation and extend the lifetime of charge separation. The substitution of ZnPP with ZnPE1 can improve up to a two-fold the efficiency of the light to chemical energy conversion. The ZnPP-Mb or ZnPE1-Mb system carried out a step-to-step one electron transfer mechanism upon photo-induced charge transfer, as demonstrated by the catalysis of a single-electron reduction of ferricytochrome c to ferrocytochrome c by cytochrome c. Furthermore, the artificial system exhibited a bifunctionally promote redox reaction dependent on the presence or absence of a sacrificial electron donor. This study is the first report to describe the reconstituted ZnPP-Mb/ZnPE1-Mb system as a bifunctional photo-induced oxidoredox system to convert solar energy into chemical energy. Therefore, the construction of such a photosystem would in principle serve as a self-regenerative photoelectrochemical cell and could be applied to electrochemical regeneration steps involved in chemical transformation and/or energy conversion as well as for sensor applications.
Fig 1
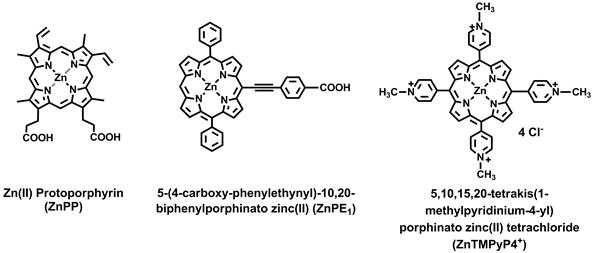
Structure of zinc protoporphyrin (ZnPP), 5-(4-carboxy-phenylethynyl)-10,20-biphenylporphinato zinc (II) (ZnPE1), and 5,10,15,20-tetrakis(1-methylpyridinium-4-yl) porphinatozinc(II) tetrachloride (ZnTMPyP4+).
Results and discussion
Steady-state, picosecond fluorescence, and anisotropy dynamics of ZnPE1-Mb
The dynamics of electronic and orientational relaxations of the ZnPP-Mb complex in aqueous solution have been studied using the time-correlated single-photon counting (TCSPC) technique [31]. Measurement of UV-vis spectra of ZnPE1 in THF and ZnPE1-Mb in KPi solution showed similar red-shift of Soret and Q bands from 439 to 443 nm and 567/615 to 576/628 nm, respectively (Supplementary Material: Figure S1). Time-resolved fluorescence anisotropy decays of the system showed significant change in rotational correlation period from 0.25 ns (ZnPE1 in THF) to ~10 ns (ZnPE1-Mb in KPi), giving the evidence for the successful construction of ZnPE1-Mb complexes (Fig. 2A) in aqueous solution. Picosecond fluorescence transient of ZnPE1-Mb in KPi at 295 K, with excitation at λex = 435 nm and emission at λem = 630 nm, showed a biexponential decay feature with two time coefficients: τ1 = 0.6 ns and τ2 = 2.5 ns (Fig. 2B).
Fig 2
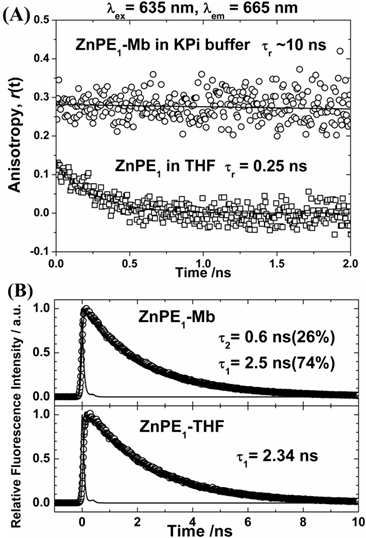
(A) Picosecond fluorescence anisotropy of ZnPE1 in THF (squares) and encapsulated within apomyoglobin in buffer solution (circles) obtained at λem = 655 nm with excitation at λex = 635 nm. (B) Picosecond fluorescence transient of ZnPE1-Mb complex in KPi with λex = 435 nm and λem = 630 nm. For ZnPE1 in THF, the λex = 435 nm and λem = 623 nm.
Because the lifetime of ZnPE1 in THF is 2.34 ns due to the S1-T1 intersystem crossing (ISC) of the zinc porphyrin, the slow decay component of the ZnPE1-Mb transient (τ2 = 2.5 ns) is reasonably assigned to the isolated ZnPE1 molecules inside the heme pocket of apoMb [29,31]. We noticed that porphyrin molecules form aggregates in KPi buffer, and this is the case for both ZnPP and ZnPE1 [31]. Intermolecular energy transfer would become very efficient in porphyrin aggregates [32]. As a result, the fast-decay component of the ZnPE1-Mb transient (τ1 = 0.6 ns) is assigned to the ZnPE1 aggregates outside the active site of the apoMb in buffer solution [3,31].
Assembly of artificial photosynthetic system and measurement of light-chemical energy conversion efficiency
The artificial photosynthetic system consists of four components: the light source (419 nm), photosensitizer (ZnPP-Mb or ZnPE1-Mb), substrate (NADP+) and the sacrificial electron donor (triethanolamine, TEA). We first evaluate the effects of photoirradiation time, dark time, and the sacrificial electron donors on photo-chemical energy conversion efficiency (Supplementary Material: Fig. S2). When the light was omitted from the reaction, no absorbance at 340 nm, assigned to NADP+ reduction, could be detected, indicating the light dependence of the reduction of NADP+. Upon photoirradiation, the absorbance at 340 nm increased along with the decrease and blue shift of the Soret band of ZnPP-Mb from 428 to 413 nm and of ZnPE1-Mb from 443 nm to 426 nm, respectively (Fig. 3). Furthermore, the absorbance at 340 nm increased with time even in the dark. We have previously determined the stability of the ZnPP-Mb complex under femtosecond laser irradiation and found that the absorbance at the maxima of both B and Q bands decreased with increasing duration of excitation and increasing width of the absorption bands [31]. The ZnPP-Mb complexes first absorbed the excitation photons and then relaxed to their lower electronic states via non-radiative transitions, which transfers the excitation energy into the internal energy of the protein. Subsequently, the internal energy of the system might be further transferred into the environment by solvent-induced vibrational relaxation that increases the thermal energy (or temperature) of the local environment. Next, dissociation of the ZnPP-Mb complex might occur under two different circumstances: (1) if dissociation was more rapid than the energy transfer or (2) if dissipation of the thermal energy was too slow. In either situation, the ZnPP-Mb complex decomposed so that ZnPP moved out of the heme pocket of the protein to form aggregates with other free ZnPP species. Therefore, the decrease of Soret band absorbance could be attributed to the conformational change of protein structure and release of ZnPP out of the protein active site pocket, further supporting the essential roles of light in triggering the reaction and affecting the stability of the photosensitizer [31]. After photoirradiation, approximately 90.0% and 44.6% of NADP+ was photochemically reduced by ZnPE1-Mb and ZnPP-Mb, respectively. Further, when ZnPE1-Mb/TEA was first photoirradiated for 6 hrs before adding the NADP+ and kept in the dark for 5 days, reduction of NADP+ occurred with similar efficiency as that of simultaneously adding all components at the beginning.
Fig 3
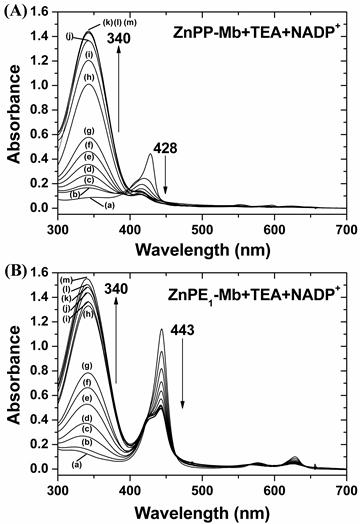
Change of UV-vis absorption spectra for (A) ZnPP-Mb and (B) ZnPE1-Mb co-incubated with TEA and NADP+ at different times of photoirradiation and then kept in the dark for up to 5 days. [ZnPP-Mb] = 25 μM, [ZnPE1-Mb] = 25 μM, [TEA] = 1 M, [NADP+] = 2.5 mM. (a) Before irradiation. (b) - (g) Irradiation time was 1, 2, 3, 4, 5, and 6 h, respectively. (h) - (m) Irradiation time was 6 h and then kept in the dark for 12, 24, 48, 72, 96, 120 h, respectively.
Finally, the highest light-chemical energy conversion efficiency was obtained with optimal concentrations, which contained 1 M TEA, 5 mM NADP+ in 100 mM KPi (pH 9.0), 10 μM ZnPE1-Mb (or 25 μM ZnPP-Mb), photoirradiation for 6 hours, and was then kept in the dark for 5 days. These results indicated that the NADP+ was reduced photochemically following photoirradiation of the solution, consistent with the results previously reported by Nishiyama et al [33,34].
Fig 4
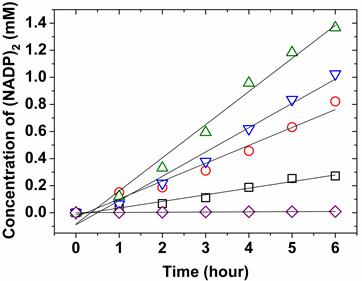
Effect of protein concentration for catalytic kinetics at photoirradiation. 2.5 mM NADP+ with 1 M TEA were independently reacted with 25 μM (○, red) of ZnPP-Mb, 10 μM (□, black) of ZnPP-Mb, 25 μM (▽, blue) of ZnPE1-Mb, or 10 μM (△, green) of ZnPE1-Mb under photoirradiation in 100 mM KPi, pH 9. 2.5 mM NADP+ with 1 M TEA reacted without reconstituted Mb (◇, purple) was used as control.
Next, we compared the effect of protein concentrations on the catalytic velocity of ZnPP-Mb and ZnPE1-Mb, respectively. Figure 4 shows the comparison of catalytic velocity for 10 and 25 μM of ZnPP-Mb or ZnPE1-Mb, respectively, co-incubated with TEA and NADP+ under photoirradiation for 6 hrs. The arithmetically catalytic velocity of 10 and 25 μM ZnPP-Mb is 0.08 and 0.125 mM/hr, whereas that of the 10 and 25 μM ZnPE1-Mb is 0.245 and 0.223 mM/hr, respectively. No apparent reactivity could be detected for 5 mM NADP+ and/or 1 M TEA reacted with apoMb. Interestingly, the catalytic velocity of ZnPP-Mb increased with higher ZnPP-Mb concentration, whereas a decline of catalytic velocity was observed in higher ZnPE1-Mb concentration. Moreover, an approximately two-fold catalytic velocity for ZnPE1-Mb is consistent with the photo-chemical energy conversion efficiency. The higher catalytic velocity of ZnPE1-Mb than that of ZnPP-Mb could be attributed to the higher efficiency of ZnPE1, since measurements of the UV-vis spectra showed that ZnPE1 exhibited similar catalytic velocity and conversion efficiency to that of the ZnPP even with lower solubility of ZnPE1 than ZnPP in KPi buffer. The reconstitution of ZnPE1 into the heme pocket of apoMb prevents aggregation and influences the relaxation of ZnPE1, further improving the energy conversion efficiency [31]. Furthermore, the higher catalytic velocity of ZnPE1-Mb at lower concentrations suggested that the equilibrium between the quench of the excited intermediates and the proceeding of the photochemical conversion was reached at a lower concentration for ZnPE1-Mb than for ZnPP-Mb, since similar phenomenon was also observed when the concentration of ZnPP-Mb was increased from 25 to 40 μM (data not shown).
In parallel, a well-known water soluble porphyrin, 5,10,15,20-tetrakis(1-methylpyridinium-4-yl) porphinatozinc(II) tetrachloride (ZnTMPyP4+), was used as a control model in comparison of catalytic velocity with that of ZnPP and ZnPE1 (Fig. 1). The ZnTMPyP4+ has been used as a photosensitizer and applied in photochemical electron transfer studies [36,37]. When 25 μM of ZnPP, ZnPE1, and ZnTMPyP4+ were independently photoirradiated, a catalytic velocity of 0.08, 0.111, and 0.06 mM/hr were obtained. The results are consistent with the observation that the conversion efficiency of photoinduced NADP+ reduction for ZnPE1 is higher than ZnPP and ZnTMPyP4+. Furthermore, reconstitution of ZnPP or ZnPE1 into apoMb improves the conversion efficiency of photoinduced NADP+ reduction. These results suggest the protein's possible functional role in preventing quenching aggregation and extending the lifetime for charge separation, both necessary for conversion efficiency improvement. Therefore, the conversion efficiency of photoinduced NADP+ reduction in ZnPP or ZnPE1 is better than that of water soluble ZnTMPyP4+.
Electron transfer mechanism of artificial photosynthetic system
The reduction of NADP+ can occur either by two successive one-electron transfer to form 4,4-(NADP)2 and 4,6-(NADP)2, or by a single-step transfer of two-electrons and one hydride to form enzymatically active 1,4-NAD(P)H or inactive 1,6-NAD(P)H [38-41]. Either case can cause an increase of absorbance at 340 nm. To elucidate the ZnPP-Mb or ZnPE1-Mb photoirradiated reaction pathway and product identity, we performed enzymatic analysis using alcohol dehydrogenase, which catalyzes the two-electron conversion of acetaldehyde to ethanol when NADPH is present, and cytochrome c, which acts as a single-electron acceptor to reduce ferricytochrome c into ferrocytochrome c [38-41]. Figure 5A shows the absorbance change of NADPH upon enzymatic reactions. Unexpectedly, when alcohol dehydrogenase was incubated with the photoirradiated product, no apparent decrease of absorbance at 340 nm was observed. In contrast, when the photoirradiated product was added to cytochrome c, a characteristic change of Q band from its oxidized form at 530 nm into reduced form at 523 and 550 nm was observed (Fig. 5B) [42]. These results indicated that the product upon photoirradiation may be a (NADP)2 dimer but not an enzymatically active NADPH monomer.
Fig 5
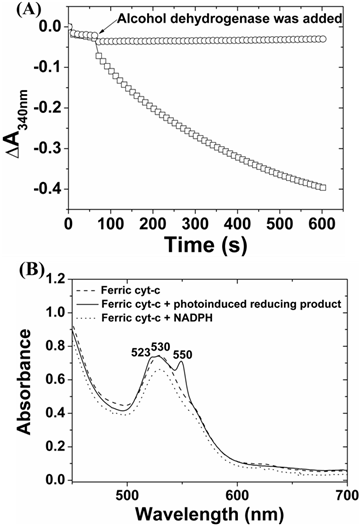
The enzymatic analysis of NADPH (□) or photoinduced reducing product (○) reacted with (A) alcohol dehydrogenase and acetaldehyde and (B) cytochrome c, respectively.
Photo-induced reduction or oxidation of artificial photosynthetic system
The NAD(P)+ and its reduced form NAD(P)H play critical roles in biological systems. In addition, Moore et al. recently constructed a photoelectrochemical biofuel cell which incorporates aspects of both enzymatic biofuel cell and dye-sensitized solar cells [9,43-46]. When the porphyrin-sensitized n-type semiconductor photoanode was photoirradiated to induce charge separation, the porphyrin radical cation could be reduced by NAD(P)H, which transfers two electrons to the photoanode and generates NAD(P)+ as substrate for subsequent hydrogenase enzyme oxidation. Based on the abovementioned information, we also attempted to characterize whether NADH could be oxidized by ZnPP-Mb (or ZnPE1-Mb) under photoirradiation and its potential in constructing a self-regenerative photo-chemical energy conversion system. For the photo-induced oxidation reaction, the reaction buffer was changed from KPi to HEPES to avoid the formation of adduct between the phosphate ion and the pyridine ring of NADH and the subsequent NADH decomposition [47,48]. Figure 6 shows the UV-vis spectra change of ZnPP-Mb + NADH, ZnPP-Mb + NADH + TEA, ZnPE1-Mb + NADH, ZnPE1-Mb + NADH + TEA, NADH and NADH + TEA in 20 mM HEPES (pH 8.5) under continued photoirradiation or dark for 36 hours. As expected, no apparent absorbance change at 340 nm was detected for NADH and NADH + TEA under photoirradiation. Similar results were obtained when all the above-mentioned reactions were performed under dark reactions for 36 hours. Photoirradiation of ZnPP-Mb + NADH and ZnPE1-Mb + NADH resulted in a rapid decrease of absorbance at 340 nm, with a steeper decline for the latter one. These results indicate the light-dependent NADH oxidation reaction in the ZnPP-Mb and ZnPE1-Mb complex. Next, results of photoirradiation of ZnPP-Mb + NADH + TEA showed that absorbance at 340 nm initially decreased but reversely increased after approximately 2 hours. In contrast, when ZnPE1-Mb + NADH + TEA complex was photoirradiated, its absorbance at 340 nm decreased rapidly for the first 2 hours and then leveled slightly thereafter. Moreover, the absorbance decay at 340 nm was slower for ZnPE1-Mb + NADH + TEA than for ZnPP-Mb + NADH + TEA. Finally, the absorbance at 340 nm reached approximately half of the original intensity for both the ZnPP-Mb + NADH + TEA and the ZnPE1-Mb + NADH + TEA after photoirradiation for 36 hours. Furthermore, detailed comparison of the Soret band decay between ZnPP-Mb (or ZnPE1-Mb) with and without TEA showed that the presence of TEA in the reaction solution causes blue-shift and a decrease of the Soret band whereas no shift was observed in the absence of TEA (Supplementary Material: Fig. S3).
Fig 6
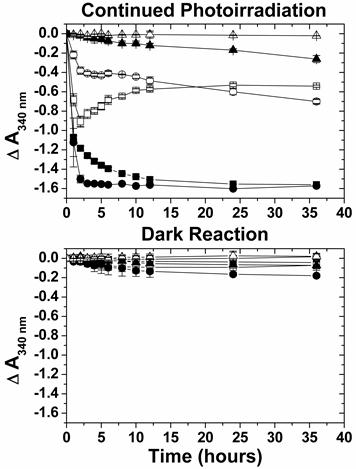
Summary of the absorbance change at 340 nm for ZnPP-Mb + NADH (■), ZnPP-Mb + NADH + TEA (□), ZnPE1-Mb + NADH (●), ZnPE1-Mb + NADH + TEA (○), NADH (▲), and NADH + TEA (△) by comparing with continued photoirradiation for 36 hours and dark reaction. [ZnPP-Mb] = 25 μM, [ZnPE1-Mb] = 25 μM, and [TEA] = 1 M and [NADH] = 2.5 mM. All in 20 mM HEPES (pH 8.5).
The results shown in figure 6 indicate that there are two modes of action, the initial oxidation of NADH to NAD+ and the subsequent reduction of NAD+ to (NAD)2, for the net absorbance change detected at 340 nm. The absorbance profile difference between ZnPP-Mb + NADH and ZnPP-Mb + NADH + TEA as well as ZnPE1-Mb + NADH and ZnPE1-Mb + NADH + TEA suggest that NADH undergoes initial oxidation to form NAD+, which in the presence of TEA is reduced to form (NAD)2 dimers. This in turn causes the accumulation of (NAD)2 dimers and partially contribute to the increase of absorbance at 340 nm. Notably, both NADH and (NAD)2 dimer have similar molar absorption coefficients, 6200 and 6400 M-1.cm-1, respectively [40,49]. Thus, in both systems one unit of absorbance is decreased with the consumption of two NADH to generate one (NAD)2 dimer. Interestingly, the net absorbance profile for the ZnPP-Mb + NADH + TEA system first decreased to less than half of the total absorbance after 2 hours of photoirradiation and then inversely increased to approximately half of the total absorbance after continued photoirradiation for 36 hours. On the other hand, the net absorbance profile of ZnPE1-Mb + NADH + TEA system showed a gentle decline of the 340 nm absorbance to approximately half of its total value with prolonged reaction time. The discrepancy on the absorbance profile between both systems may be attributed to the stronger inhibitory effect of the TEA for ZnPE1-Mb than for ZnPP-Mb on the oxidation rate. Consistent with the hypothesis is the observation that both the oxidation rate and the absorbance profile of ZnPE1-Mb system is TEA concentration dependent which at lower TEA concentration the oxidation rate increased and the absorbance profile converted from that of the ZnPE1-Mb type into that of the ZnPP-Mb type (Supplementary Material: Fig. S4).
Fig 7
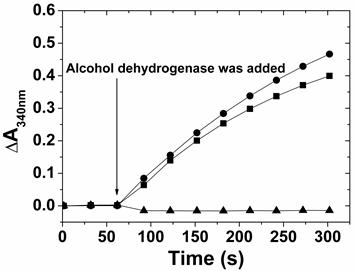
The enzymatic analysis of alcohol dehydrogenase-catalyzed ethanol oxidation, in the presence of NAD+ (●), photoirradiated reactant (▲), and photoirradiated product (■). Ethanol and NAD+ reactant were co-incubated for 60 seconds before adding the enzyme and the absorbance change at 340 nm was recorded for every 30 seconds with a total detection time of 5 mins.
To confirm the identity of the NAD+ produced by the photoirradiation of ZnPP-Mb or ZnPE1-Mb in the absence of TEA, enzymatic oxidation using alcohol dehydrogenase and ethanol as enzyme and substrate, respectively, was performed. Alcohol dehydrogenase can catalyze reversible oxidoredox reactions of ethanol and acetaldehyde, depending on the added NAD+ or NADH in the solution, respectively. When ethanol was oxidized to acetaldehyde, the electron was used to reduce NAD+ into NADH, thus leading to the absorbance increase at 340 nm. Figure 7 shows the comparison of the absorbance change at 340 nm when NAD+, photoirradiated reactant, and photoirradiated product were used as cofactors for the alcohol dehydrogenase-catalyzed ethanol oxidation. As expected, no absorbance was observed for the photoirradiated reactant on alcohol dehydrogenase-catalyzed oxidation reactions, suggesting that NADH cannot be used in ethanol oxidation. On the other hand, the absorbance rate at 340 nm gradually increased when NAD+ or photoirradiated product was added to the alcohol dehydrogenase-catalyzed oxidation reaction. These results demonstrate the feasibility of ZnPP-Mb or ZnPE1-Mb as photoexcited photosensitizers to oxidize NADH and its potential applications in engineering hybrid photoelectrochemical cells.
Hypothetical reaction mechanism of photochemical catalysis of ZnPP-Mb/ZnPE1-Mb-based artificial photosynthetic system for light-chemical energy conversion
The possible reaction mechanism of photo-induced reduction of NADP+ or oxidation of NADH by ZnPP-Mb (or ZnPE1-Mb), in the presence or absence of sacrificial electron donor, respectively, is shown in Scheme 1/Fig. 8. Upon photoirradiation, the absorption of light by the photosensitizer gives rise to an excited state of the ZnPP-Mb* (or ZnPE1-Mb*), which is subsequently reduced by a sacrificial electron donor such as TEA, thus generating a long-lived ZnPP-Mb·- (or ZnPE1-Mb·-) radical anion [50-56]. A marked absorption around 700 nm, which is characteristic of anion radical formation, was determined by photoirradiation of ZnPE1-Mb. The analogous formation of zinc cytochrome c anion radical, upon photoirradiation, has been determined by Shen and Kostić, thus supporting the hypothesis [54]. In parallel, a similar reductive photoredox cycle that uses a tin(IV)- or antimony(V)-porphyrin photosensitizer was also previously reported by Shelnutt and Trudell [51]. Furthermore, Maldotti et al. also reported the utilization of photoexcited iron porphyrin to mimic the catalytic activity of cytochrome P-450 oxygenases both in the reduction of halogenated alkanes and in the oxidation of hydrocarbons [55]. Alternatively, in the absence of the TEA, electron transfer from NADH to ZnPP-Mb* (or ZnPE1-Mb*) may occur to produce the radical ion pair, NADH·+ ZnPP-Mb·-. Consistent with the observation is the reported photo-oxidation of the NADH model compound, 9,10-dihydro-10-methylacridine (AcrH2), in the presence of a sensitizer under visible-light irradiation in MeCN and H2O [50]. For photoinduced reduction of NADP+, following photoinduced charge separation, the resulting anion radical is oxidized by NADP+ in the aqueous solution, ultimately generating (NADP)2 dimer. After reduction of the NADP+ or oxidation of NADH, the photosensitizer radical anion returns to the resting redox state (ZnPP-Mb or ZnPE1-Mb). For photoinduced oxidation, the back transfer of an electron from the ZnPP-Mb·- anion radical to the NADH·+ is prevented by the trapping of the radical ZnPP-Mb·- (or ZnPE1-Mb·-) by oxygen to produce HO·2, and subsequently accompanied by regeneration of ZnPP-Mb (or ZnPE1-Mb). Consistent with the observation is the oxygen dependence of the photo-induced oxidation reaction to generate H2O2 (Supplementary Material: Figure S5) [50].
In conclusion, we have constructed a bifunctional photo-induced oxidoredox system which, upon photoirradiation, can transfer electrons from NAD(P)H or to NAD(P)+ depending on the presence or absence of sacrificial electron donor. The ZnPP-Mb or ZnPE1-Mb protein pocket can prevent the quenching aggregate and extend the lifetime of charge separation. In addition, the efficiency of the NADP+ reduction using ZnPE1-Mb is approximately two-fold greater than that of the system using ZnPP-Mb. In parallel, the ZnPP-Mb or ZnPE1-Mb artificial photosynthetic system carried out a step-to-step one electron transfer mechanism upon photo-induced charge transfer. Finally, the ZnPP-Mb/ZnPE1-Mb artificial photosynthetic system can be realized as a potential bio-photosentizer and applied for the development of renewable energy to replace our reliance on fossil fuels.
Fig 8
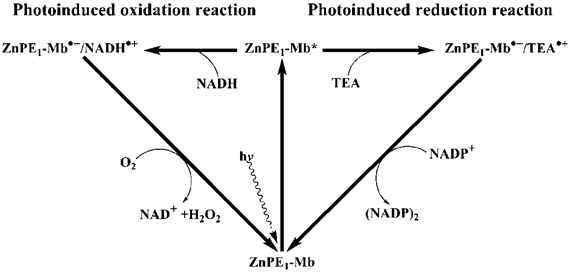
(Scheme 1) Schematic representation of photochemical catalysis of ZnPE1-Mb-based artificial photosynthetic system for light-chemical energy conversion.
Materials and methods
Preparation of reconstituted ZnPP-Mb and ZnPE1-Mb
Apomyoglobin was prepared by the modified butanone method of heme extraction, as previously described [35]. The reconstitution of ZnPP or ZnPE1 into apoMb was performed according to the modified method of Axup et al [57]. Briefly, the apo-protein solution was maintained at ice bath for the duration of the reconstitution process. A 1.5-fold molar excess of ZnPP (or ZnPE1) in DMSO was added dropwise to a 10 mM KPi/DMSO (4:1 v/v) buffer containing apomyolobin at pH 12. After incubation for 15 min, the solution was adjusted to pH 7.0 and allowed to stir for 6 hr at 4 °C. The solution was transferred to a dialysis bag against phosphate buffer (10 mM KPi buffer pH 6.8) overnight. The protein solution was filtered with 0.45 μm cellulose membrane before applied to a Sephadex G-25 column. The column was equilibrated with 100 mM KPi buffer pH 6.8, and eluted with the same buffer at 4 °C. The protein was passed through the column, collected and stored at -20 °C for later experiments.
Photo-induced reduction experiments
The UV-vis absorption spectra were recorded with a standard spectrometer (HP 8453); emission spectra were measured with a fluorescence spectrometer (Hitachi F-700). Photoirradiation reactions were carried out in a photochemical reactor PR-2000 (PANCHUM, Kaohsiung, Taiwan) at room temperature (25 oC) using two RPR 4190 Å lamps that show an emission maximum at 419 ± 15 nm at half bandwidth. The light intensity at the cell is 6 mW/cm2. For photoreduction reaction, photoirradiation was carried out in a final volume of 1 mL of 100 mM phosphate buffer (pH 9.0) containing 25 μM ZnPP-Mb (or 10 μM ZnPE1-Mb), 2.5 mM of NADP+, and 1 M TEA. For photo-induced reduction reaction, the change in the absorbance at 340 nm was detected at 1, 2, 3, 4, 5, 6, 18, 30, 54, 78, 102, and 126 h.
Photo-induced oxidation experiments
For photo-induced oxidation reaction, 25 μM of ZnPP-Mb or ZnPE1-Mb and 2.5 mM NADH were used, but TEA was omitted from the reaction solution. The reaction solution was photoirradiated at 419 nm for 6 hrs and the change in the absorbance at 340 nm, which attributed to (NADP)2 or NAD+ formation, was recorded. The equipment of photo-induced oxidation experiments is the same as that of the photo-induced reduction experiments. For photo-induced oxidation reaction, the absorbance was detected at 1, 2, 3, 4, 5, 6, 8, 10, 12, 24 and 36 h.
Enzymatic product characterization of photo-induced reduction reactions
The general procedure of NAD(P)H enzymatic assay was determined spectrophotometrically using a modified method of Hanson and Jacobson [58]. 10 μL of acetaldehyde was pre-mixed with NADPH or photoinduced reducing product in 1 ml cuvette. 10 μL of 10 U·mL-1 alcohol dehydrogenase was added to the mixture and rapidly mixed. As the enzyme catalyzed acetaldehyde to ethanol, the change of absorbance at 340 nm was monitored for 10 mins. For a single-electron acceptor, cytochrome c, which shows absorption maxima of Q band at 530 nm in the oxidized form and at 523 and 550 nm in the reduced form, was used [59]. 25 μM of cytochrome c was converted into the fully oxidized form by autoxidation for 30 min at pH 3 at 20 °C, then mixed with NADPH or photoinduced reducing product for 10 mins, respectively [60]. The spectra change of cytochrome c reduction reaction was recorded. All changes of UV-vis spectra were recorded by a standard spectrometer (HP 8453).
Enzymatic product characterization of photo-induced oxidation reactions
The enzymatic assay of photo-induced oxidation product was similar with that of photo-induced reduction product but the substrate and coenzyme were replaced with ethanol and NAD+ or photoinduced oxidation product. The change of absorbance at 340 nm was also monitored. All changes of UV-vis spectra were recorded by a standard spectrometer (HP 8453).
Acknowledgements
The authors gratefully acknowledge financial support from National Chiao Tung University, MOE ATU Plan, and National Science Council (NSC-98-2627-M-009-007 and NSC-98-2627-M-009-008).
Abbreviations
ZnPP-Mb: zinc-protoporphyrin myoglobin; ZnPE1-Mb: 5-(4-carboxy-phenylethynyl)-10,20-biphenyl porphinato zinc (II) myoglobin; ATP: adenosine triphosphate; NADPH: nicotinamide adenine dinucleotide phosphate; apoMb: apomyoglobin.
Supplementary Material
Fig.S1-S7.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Zhou JS, Granda ESV, Leontis NB, Rodgers MAJ. Photoinduced electron transfer in self-associated complexes of several uroporphyrins and cytochrome c. J. Am. Chem. Soc. 1990;112:5074-5080
2. McLendon G, Hake R. Interprotein electron transfer. Chem. Rev. 1992;92:481-490
3. Chang CW, Chang CH, Lu HP, Wu TK, Diau EW. Fabrication and photovoltaic characterization of bio-sensitized solar cells using myoglobin-based sensitizers. J. Nanosci. Nanotechnol. 2009;9:1688-1695
4. Long YT, Sutherland TC, Kraatz HB, Lee JS. Photoinduced production of NAD(P)H from an activated fluorescein-DNA monolayer. Chem. Commun. 2004;18:2032-2033
5. Komatsu T, Wang RM, Zunszain PA, Curry S, Tsuchida E. Photosensitized reduction of water to hydrogen using human serum albumin complexed with zinc-protoporphyrin IX. J. Am. Chem. Soc. 2006;128:16297-16301
6. Matsuo T, Asano A, Ando T, Hisaeda Y, Hayashi T. Photocatalytic hydrogen generation using a protein-coated photosensitizer with anionic patches and a monocationic electron mediator. Chem. Commun. 2008;31:3684-3686
7. Hagfeldt A, Grätzel M. Molecular photovoltaics. Acc. Chem. Res. 2000;33:269-277
8. Grätzel M. Dye-sensitized solar cells. J. Photochem. Photobiol. 2003;4:145-153
9. Grätzel M. Conversion of sunlight to electric power by nanocrystalline dye-sensitized solar cells. J. Photochem. Photobiol. 2004;164:3-14
10. Grätzel M. Solar energy conversion by dye-sensitized photovoltaic cells. Inorg. Chem. 2005;44:6841-6851
11. Martı´nez-Dı´az MV, Torres T. Handbook of Porphyrin Science; Volume 10. Singapore: Academic Press. 2010
12. Bottari G, de la Torre G, Guldi DM, Torres T. Covalent and noncovalent phthalocyanine-carbon nanostructure systems: synthesis, photoinduced electron transfer, and application to molecular photovoltaics. Chem. Rev. 2010;110:6768-6816
13. Bessho T, Zakeeruddin SM, Yeh CY, Diau EWG, Grätzel M. Highly efficient mesoscopic dye-sensitized solar cells based on donor-acceptor-substituted porphyrins. Angew. Chem. Int. Ed. 2010;49:6646-6649
14. Rang YJ, Spikes JD. The porphyrin-sensitized photooxidation of horseradish apoperoxidase. Arch. Biochem. Biophys. 1976;172:565-573
15. Bellelli A, Brzezinski P, Arese M, Cutruzzola F, Silvestrini MC, Brunori M. Electron transfer in zinc-reconstituted nitrite reductase from Pseudomonas aeruginosa. Biochem. J. 1996;319:407-410
16. Crnogorac MM, Kostic NM. Redox reactivity and reorganization energy of zinc cytochrome c cation radical. Inorg. Chem. 2000;39:5028-5035
17. Hitomi Y, Hayashi T, Wada K, Mizutani TY, Hisaeda Y, Ogoshi H. Interprotein electron transfer reaction regulated by an artificial interface. Angew. Chem. Int. Ed. 2001;40:1098-1101
18. Liang ZX, Nocek JM, Huang K, Hayes RT, Kurnikov IV, Beratan DN, Hoffman BM. Dynamic docking and electron transfer between Zn-myoglobin and cytochrome b. J. Am. Chem. Soc. 2002;124:6849-6859
19. Hu YZ, Tsukiji S, Shinkai S, Oishi S, Hamachi I. Construction of artificial photosynthetic reaction centers on a protein surface: vectorial, multistep, and proton-coupled electron transfer for long-lived charge separation. J. Am. Chem. Soc. 2000;122:241-253
20. Tsukahara K, Kimura C, Sakurai T. Intracomplex quenching by copper(II) ion of excited singlet and triplet states of zinc myoglobin modified with diethylenetriaminepentaacetic acid. Chem. Lett. 1997;26:601-602
21. Murakami H, Matsumoto R, Okusa Y, Sagara T, Fujitsuka M, Ito O, Nakashima N. Design, synthesis and photophysical properties of C60-modified proteins. J. Mater. Chem. 2002;12:2026-2033
22. Hay S, Wallace BB, Smith TA, Ghiggino KP, Wydrzynski T. Protein engineering of cytochrome b562 for quinone binding and light-induced electron transfer. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17675-17680
23. Harriman A. Luminescence of porphyrins and metalloporphyrins. J. Chem. Soc. Farady I. 1980;76:1978-1985
24. Barboy N, Feitelson J. Quenching of the zinc-protoporphyrin triplet state as a measure of small-molecule diffusion through the structure of myoglobin. Biochemistry. 1987;26:3240-3244
25. Albani J, Albert B. Fluctuation domains in myoglobin. Fluorescence quenching studies. Eur. J. Biochem. 1987;162:175-178
26. Ilari A, Boffi A, Chiancone E. Rigidity of the heme pocket in the cooperative Scapharca hemoglobin homodimer and relation to the direct communication between hemes. Arch. Biochem. Biophys. 1995;316:378-384
27. Aono S, Nemoto S, Ohtaka A, Okura I. Photoreduction of cytochrome c with zinc protoporphyrin reconstituted myoglobin (ZnPP-Mb). J. Mol. Catal. A: Chem. 1995;95:193-196
28. Polm MW, Schaafsma TJ. Triplet state magnetic resonance and fluorescence spectroscopy of metal-substituted hemoglobins. Biophys. J. 1997;72:373-382
29. Yu HZ, Baskin JS, Zewail AH. Ultrafast dynamics of porphyrins in the condensed phase: II; zinc tetraphenylporphyrin. J. Phys. Chem. A. 2002;106:9845-9854
30. Hu YZ, Takashima H, Tsukiji S, Shinkai S, Nagamune T, Oishi S, Hamachi I. Direct comparison of electron transfer properties of two distinct semisynthetic triads with non-protein based triad: unambiguous experimental evidences on protein matrix effects. Chem. Eur. J. 2000;6:1907-1916
31. Luo L, Chang CH, Chen YC, Wu TK, Diau EWG. Ultrafast relaxation of zinc protoporphyrin encapsulated within apomyoglobin in buffer solutions. J. Phys. Chem. B. 2007;111:7656-7664
32. Luo L, Lin CJ, Tsai CY, Lu HP, Li LL, Lo CF, Lin CY, Diau EWG. Effects of aggregation and electron injection on photovoltaic performance of porphyrin-based solar cells with oligo(phenylethynyl) links inside TiO2 and Al2O3 nanotube arrays. Phys. Chem. Chem. Phys. 2010;12:1064-1071
33. Nishiyama K, Uchiyama M, Mie Y, Taniguchi I. Photochemical reduction of NADP+ by zinc protoporphyrin reconstituted myoglobin as a simple model of photosystem I. Chem. Lett. 1999;28:357-358
34. Nishiyama K, Mie Y, Kishita M, Yamada C, Kitagawa R, Taniguchi I. Phototriggered chemical reduction of NADP+ by zinc-reconstituted myoglobin and triethanolamine as a sacrificial donor. Chem. Lett. 2005;34:1032-1033
35. Teale FW. Cleavage of the haem-protein link by acid. Biochim. Biophys. Acta. 1959;35:543
36. Houlding VH, Kalyanasundaram K, Grätzel M, Milgrom LR. Kinetics of triplet decay of water soluble porphyrins. Analysis of ionic strength effects. J Phys Chem. 1983;87:3175-3179
37. Kalyanasundaram K, Neumann-Spallart M. Photophysical and redox properties of water-soluble porphyrins in aqueous media. J. Phys. Chem. 1982;86:5163-5169
38. Burnett RW, Underwood AL. A dimer of diphosphopyridine nucleotide. Biochemistry. 1968;7:3328-3333
39. Jensen MA, Elving PJ. Nicotinamide adenine dinucleotide (NAD+). Formal potential of the NAD+/NADH couple and NAD dimerization rate. Biochim Biophys Acta. 1984;764:310-315
40. Avigliano L, Carelli V, Casini A, Finazzi-Agrò A, Liberatore F. Oxidation of NAD dimer by horseradish peroxidase. Biochem. J. 1985;226:391-395
41. Avigliano L, Carelli V, Casini A, Finazzi-Agrò A, Liberatore F, Rossi A. Oxidation of nicotinamide coenzyme dimers by one-electron-accepting proteins. Biochem. J. 1986;237:919-922
42. Koike H, Katoh S. Heat-stabilities of cytochromes and ferredoxin isolated from a thermophilic blue-green alga. Plant Cell Physiol. 1979;20:1157-1161
43. Garza LDL, Jeong G, Liddell PA, Sotomura T, Moore TA, Moore AL, Gust D. Enzyme-based photoelectrochemical biofuel cell. J. Phys. Chem. B. 2003;107:10252-10260
44. Brune A, Jeong G, Liddell PA, Sotomura T, Moore TA, Moore AL, Gust D. Porphyrin-sensitized nanoparticulate TiO2 as the photoanode of a hybrid photoelectrochemical biofuel cell. Langmuir. 2004;20:8366-8371
45. Hambourger M, Brune A, Dust G, Moore AL, Moore TA. Enzyme-assisted reforming of glucose to hydrogen in a photoelectrochemical cell. Photochem. Photobiol. 2005;81:1015-1020
46. Hambourger M, Gervaldo M, Svedruzic D, King PW, Dust G, Ghirardi M, Moore AL, Moore TA. [FeFe]-hydrogenase-catalyzed H2 production in a photoelectrochemical biofuel cell. J. Am. Chem. Soc. 2008;130:2015-2022
47. Wu JT, Wu LH, Knight JA. Stability of NADPH: effect of various factors on the kinetics of degradation. Clin. Chem. 1986;32:314-319
48. Rover JL, Fernandes JC, de Oliveira Neto G, Kubota LT, Katekawa E, Serrano SH. Study of NADH stability using ultraviolet-visible spectrophotometric analysis and factorial design. Anal. Biochem. 1998;260:50-55
49. Carelli V, Liberatore F, Casini A, Mondelli R, Arnone A, Carelli I, Rotilio G, Mavelli I. Dimers of nicotinamide adenine dinucleotide: New evidence for the structure and the involvement in an enzymatic redox process. Bioorg. Chem. 1980;9:342-351
50. Fukuzumi S, Ishikawa M, Tanaka T. Mechanisms of photo-oxidation of NADH model compounds by oxygen. J. Chem. Soc. Perkin Trans. II. 1989:1037-1045
51. Shelnutt JA, Trudell DE. Photochemical-driven biomimetic oxidation of alkanes and olefins; Volume 437. US: Am Chem Soc. 1990
52. Golovin NM, Seymour P, Jayaraj K, Fu Y, Lever ABP. Perchlorinated phthalocyanines: spectroscopic properties and surface electrochemistry. Inorg. Chem. 1990;29:1719-1727
53. Mack J, Stillman MJ. Photochemical formation of the anion radical of zinc phthalocyanine and analysis of the absorption and magnetic circular dichroism spectral data. Assignment of the optical spectrum of [ZnPc(-3)]. J Am Chem Soc. 1994;116:1292-1304
54. Shen C, Kostić NM. Reductive quenching of the triplet state of zinc cytochrome c by the hexacyanoferrate(II) anion and by conjugate bases of ethylenediaminetetraacetic acid. Inorg. Chem. 1996;35:2780-2784
55. Maldotti A, Andreotti L, Molinari A, Carassiti V. Photochemically driven models of oxygenases based on the use of iron porphyrins. J. Biol. Inorg. Chem. 1999;4:154-161
56. Sinyakov GN, Shul'ga AM, Filatov IV, Dzilinsky K. The electronic structure of transient species in the photochemical reduction of porphyrins. High Energy Chem. 2002;36:255-259
57. Axup AW, Albin M, Mayo SL, Crutchley RJ, Gray HB. Distance dependence of photoinduced long-range electron transfer in Zinc/Ruthenium-modified myoglobins. J. Am. Chem. Soc. 1988;110:435-439
58. Hanson A, Brown A. Three alcohol dehydrogenase genes in wild and cultivated barley: characterization of the products of variant alleles. Biochem. Genet. 1984;22:495-515
59. Margoliash E, Frohwirt N. Spectrum of horse-heart cytochrome c. Biochem. J. 1959;71:570-572
60. Brady RS, Flatmark T. Autoreduction of horse heart ferricytochrome c. Kinetic and equilibrium studies of the over-all reaction. J. Mol. Biol. 1971;57:529-539
Author contact
![]() Corresponding author: T. K. Wu, Department of Biological Science and Technology, National Chiao Tung University, 30068, Hsin-Chu, Taiwan, Republic of China. Tel: +886-3-5729287 Fax: +886-3-5725700 E-mail: tkwmllnctu.edu.tw or C. Y. Lin, Department of Applied Chemistry, National Chi Nan University, Puli, Nantou County, Taiwan, Republic of China. Fax: +886-49-2917956 Tel: +886-49-2910960-4152 E-mail: cylncnu.edu.tw or Eric W. G. Diau, Department of Applied Chemistry and Institute of Molecular Science, National Chiao Tung University, Hsin-Chu, Taiwan, Republic of China. Fax: +886-3-5723764 Tel: +886-3-5712121-31524 E-mail: diaunctu.edu.tw
Corresponding author: T. K. Wu, Department of Biological Science and Technology, National Chiao Tung University, 30068, Hsin-Chu, Taiwan, Republic of China. Tel: +886-3-5729287 Fax: +886-3-5725700 E-mail: tkwmllnctu.edu.tw or C. Y. Lin, Department of Applied Chemistry, National Chi Nan University, Puli, Nantou County, Taiwan, Republic of China. Fax: +886-49-2917956 Tel: +886-49-2910960-4152 E-mail: cylncnu.edu.tw or Eric W. G. Diau, Department of Applied Chemistry and Institute of Molecular Science, National Chiao Tung University, Hsin-Chu, Taiwan, Republic of China. Fax: +886-3-5723764 Tel: +886-3-5712121-31524 E-mail: diaunctu.edu.tw
Citation styles
APA
Chang, C.H., Hu, Y.T., Lo, C.F., Luo, L., Lin, H.M., Chang, C.H., Lin, C.Y., Diau, E.W.G., Wu, T.K. (2011). Photoactivation Studies of Zinc Porphyrin-Myoglobin System and Its Application for Light-Chemical Energy Conversion. International Journal of Biological Sciences, 7(8), 1203-1213. https://doi.org/10.7150/ijbs.7.1203.
ACS
Chang, C.H.; Hu, Y.T.; Lo, C.F.; Luo, L.; Lin, H.M.; Chang, C.H.; Lin, C.Y.; Diau, E.W.G.; Wu, T.K. Photoactivation Studies of Zinc Porphyrin-Myoglobin System and Its Application for Light-Chemical Energy Conversion. Int. J. Biol. Sci. 2011, 7 (8), 1203-1213. DOI: 10.7150/ijbs.7.1203.
NLM
Chang CH, Hu YT, Lo CF, Luo L, Lin HM, Chang CH, Lin CY, Diau EWG, Wu TK. Photoactivation Studies of Zinc Porphyrin-Myoglobin System and Its Application for Light-Chemical Energy Conversion. Int J Biol Sci 2011; 7(8):1203-1213. doi:10.7150/ijbs.7.1203. https://www.ijbs.com/v07p1203.htm
CSE
Chang CH, Hu YT, Lo CF, Luo L, Lin HM, Chang CH, Lin CY, Diau EWG, Wu TK. 2011. Photoactivation Studies of Zinc Porphyrin-Myoglobin System and Its Application for Light-Chemical Energy Conversion. Int J Biol Sci. 7(8):1203-1213.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.