Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(9):1220-1229. doi:10.7150/ijbs.7.1220 This issue Cite
Research Paper
Deletion of the Aryl Hydrocarbon Receptor Enhances the Inflammatory Response to Leishmania major Infection
Guillermo Elizondo1, Miriam Rodríguez-Sosa2, Elizabet Estrada-Muñiz3, Frank J. Gonzalez4, Libia Vega3 ![]()
1. Centro de Investigación y Estudios Avanzados del Instituto Politécnico Nacional. Departamento de Biología Celular. Av. IPN 2508. San Pedro Zacatenco. C. P. 07360, México D. F., México.
2. Facultad de Estudios Superiores-Iztacala, UNAM. Unidad de Biomedicina, C.P. 54090, Tlalnepantla, Estado de México, México.
3. Centro de Investigación y Estudios Avanzados del Instituto Politécnico Nacional. Departamento de Toxicología. Av. IPN 2508. San Pedro Zacatenco. C. P. 07360, México D. F., México.
4. Laboratory of Metabolism, NCI, National Institutes of Health, Bethesda, Maryland 20892, USA.
Received 2011-9-1; Accepted 2011-10-1; Published 2011-10-25
Abstract
The aryl hydrocarbon receptor (AhR) is a ligand-activated receptor that mediates the toxicity of environmental pollutants, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Recently, it has been shown that the AhR plays a role in immune and inflammatory regulation. However, most of these studies are based on the activation of AhR by exogenous ligands. Therefore, in the present study, we addressed the role of this transcription factor, in the absent of exogenous ligand, on the immune response to Leishmania major infection. Our results indicate that inactivation of the AhR results in an alteration of the levels of several cytokines. Lymph node cells from infected Ahr-null animals displayed an increase in IFNγ and IL-12 levels, together with a decrease in IL-4 and IL-10 levels compared to wild-type (wt) mice. Ahr-null mice also presented higher serum levels of the pro-inflammatory cytokine TNF-α prior to parasite inoculation and during infection compared to wt mice. Moreover, a 30% decrease in the population of Treg cells was observed in Ahr-null mice. This decrease was associated with a reduction in Foxp3 mRNA levels. Finally, the alteration in the cytokine profile results in a better resolution of the L. major infection.
Keywords: Aryl hydrocarbon receptor, Inflammatory response, Regulatory T cells, Leishmania major
Introduction
The aryl hydrocarbon receptor (AhR), a member of the basic helix-loop-helix-PAS (bHLH-Per-Arnt-Sim) transcription factor family, is a ligand-activated receptor that mediates the toxicity of environmental pollutants, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) [1, 2]. Upon binding to TCDD, the AhR translocates to the nucleus, dimerizes with AhR nuclear-translocator protein (ARNT), binds dioxin-responsive elements (DREs, also known as xenobiotic-responsive elements or XREs), and upregulates the expression of a battery of genes encoding xenobiotic-metabolizing enzymes, such as cytochrome P450s (CYP1A1, CYP1A2, CYP1B1), NAD(P)H quinone oxidoreductase, and UDP-glucoronosyl-transferase-6 [3]. Although AhR may function as part of an adaptive chemical response, several studies have suggested that this transcription factor could have important functions in liver and cardiac development, cell proliferation, circadian rhythmicity, cholesterol and glucose metabolism [3-6], and more recently, in the ubiquitin-proteasome system [7].
Several studies have demonstrated that AhR is also implicated in immune system regulation. It has been shown that TCDD suppresses both humoral and cellular immunity and induces the expression of several cytokines, such as IL-1, IL-8, and IL-6 [8]. In addition to TCDD, other environmental contaminants that activate AhR, such as benzo[a]pyrene, dimethylbenzanthracene, and 3-methylcholanthrene, induce immunotoxicity [9]. Targeting AhR results in the disruption of the immune system by decreasing lymphocyte accumulation in the spleen and lymph nodes and overriding the thymic atrophy induced by TCDD [4, 10]. Besides that, lipoxin (LX)-A4, an anti-inflammatory molecule, acts as an endogenous AhR ligand [11]. More recently, we have shown that lymphocytes from Ahr-null mice produce more IFNγ and IL-12 than lymphocytes from wild-type (wt) animals [12]. These studies suggest that AhR plays an important role in immune system homeostasis; therefore, the activation status of AhR may modulate the immune system response to an infection and/or immune pathology development. AhR is required for optimal resistance to a Listeria monocytogenes infection in certain animal models [13]. AhR is also involved to resistance to Streptococcus pneumoniae infection when activated by TCDD [14], and it modulates anti-viral immunity [15]. More recently, we showed that AhR plays an important role in the inflammatory response to Toxoplasma gondii [16]. To further determine the role of AhR in immune responses, we infected wt and Ahr-null mice with Leishmania major, an intracellular parasite that causes a well-characterized immune response that is mostly associated with the induction of a Th1-type immune response [17]. After infection, we evaluated several immune endpoints. An exacerbated inflammatory response was observed in the Ahr-null mice, as evidenced by increased swelling and proinflammatory cytokine production when compared to the response of the wt mice to the infection. Our data also indicate that AhR is a regulatory molecule that plays a role in the activation of T cells and in the induction of regulatory T (Treg) cells.
Materials and Methods
Animals. The generation of Ahr-null mice from C57BL/6 mice with a disruption in AhR exon 1 has been previously described [10]. The animals used in this study were Ahr-null and wt male siblings. Mouse breeding and genotyping by PCR were performed as reported elsewhere [12], and wt littermates were used as control mice. All animal studies were performed according to the Guide for the Care and Use of Laboratory Animals, as adopted and promulgated by the U.S. National Institutes of Health and the Mexican Regulation of Animal Care and Maintenance (NOM-062-ZOO-1999, 2001).
Infection with Leishmania major and L. major antigen preparation. L. major promastigotes (LV39 strain) were obtained from the macerates of infected tissue obtained from wt mice and were differentiated by culturing in complete Schneider's medium for 3 days at 37 ºC. For in vivo infection, 3 x 106 stationary phase L. major promastigotes were injected into the footpad of the right back leg of each animal, and the left back leg was inoculated with sterile phosphate balanced saline solution (PBS) as a control. Lesion development was measured using a dial gauge micrometer (Mitutoyo, Kanawa, Japan) at weekly intervals up to 8 weeks after inoculation. The increase in the thickness of the right hind footpad was compared to the uninfected left hind footpad.
L. major antigen was prepared from stationary phase promastigotes grown in vitro. Promastigotes were washed twice in sterile ice-cold PBS by centrifugation at 200 x g for 10 min at 4 °C. Pellets were then frozen in liquid nitrogen and thawed at 37 °C for seven cycles. Protein concentration was determined by the Lowry assay.
Quantification of parasite loads. Mice were sacrificed 8 weeks after infection and parasite burdens were determined by homogenizing footpads of individual mice and carrying out limiting dilution analysis. Briefly, the footpads were excised and homogenized in Schneider's insect medium supplemented with 20% fetal calf serum (FCS), 2 mM L-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin. To estimate the relative parasite load, 200 ml aliquots of the cell suspension (in consecutive 10-fold dilutions) were added to the wells of a 96-well flat-bottomed tissue culture plate in triplicate and were incubated at 28 ºC for 72 h. After incubation, the cultures were examined for the presence of promastigotes using an inverted microscope. The results were expressed as mean parasite titers, using the dilution where no parasites were found and calculating back the amount of parasites present in each footpad.
Serum antibody titers. Blood samples were obtained from the tail vein, and sera were kept at -70 ºC until use. ELISA plates (Nunc Maxisorb, USA) were coated with L. major antigens (50 μg/mL in PBS, pH 9.0), and serial dilutions (1:50, 1:100, 1:500, etc.) of the sera were incubated for 2 h at 37 ºC. The samples were incubated with specific goat anti-IgG (IgG1, and IgG2a) HRP conjugates (Zymed, USA) for 1 h at 37 ºC. A substrate solution containing 2,2'-azino-bis(3-ethylbenzthiazoline)-6-sulfonic acid (ABTS; Sigma-Aldrich, USA) and H2O2 (Merck, Germany) was added, and the absorbance was determined at 450 nm using a microplate reader. The antibody titer of the sera was calculated by identifying the dilution at which no L. major specific antibody was detected (endpoint titer).
Lymph node cells. After sacrifice, the inguinal draining lymph nodes were collected. A single-cell suspension of lymph node cells was prepared for each mouse and immediately used for the respective measurements.
Cytokine and LXA4 measurement. Serum or culture supernatant samples were used to assess cytokine levels by the sandwich ELISA method, as previously described [18]. Total cytokine levels were determined using paired monoclonal antibodies. All antibodies and recombinant proteins were purchased from Peprotech (Mexico). LXA4 levels were measured using a competition EIA kit (Oxford Biomedical Research, EA45, England) following the kit's instructions. Standard curves were plotted as the standard cytokine protein concentration (pg/ml) versus the corresponding mean optical density (O.D.) value of the replicates. The concentrations of the putative cytokine-containing samples were quantified based on the standard curve.
Identification of Treg cells. Flow cytometric analysis of Treg cells was performed using a staining kit (Mouse Treg FlowTM Kit, BioLegend, San Diego, CA) as follows: 1 x 106 cells in wash buffer (DPBS with 1% sodium azide and 1% FCS) were stained with 1 μl of anti-CD4-fluorescein isothiocyanate, anti-CD25-phycoerythrin, and anti-Foxp3-Alexa Fluor 488, according to the manufacturer's instructions. The cells were fixed, and the proportion of cells that were positive for all three markers was evaluated (10,000 events/sample) with a flow cytometer (Becton Dickinson, USA). Non-specific binding was blocked with FcBlock, and the isotype controls were rat anti-mouse IgG-FITC, IgG-PE, or IgG-Alexa Fluor 488. Treg cells were identified by positive staining of activated CD4+ cells (CD25+ expression on the CD4+ population) and further expression of intracellular Foxp3.
PCR analysis of mRNA expression. Lymph node cells were collected, resuspended in Trizol reagent (Sigma-Aldrich), and processed to obtain total RNA. cDNA synthesis was performed with SuperScript One-Step Reverse Transcription-PCR Kit (Invitrogen). In brief, 3 μg of total RNA was mixed with 0.5 µg oligo (dT) 12 to -18 primers, 10 mM of each dNTP and 1x reaction buffer in a final volume of 20 µl. cDNA reactions were incubated at 65 °C for 10 min to denature the RNA template and were quench-cooled for 1 min. Reverse transcriptase (0.5 μl) was added, the samples were incubated at 42 °C for 50 min and 70 °C for 15 min, 0.5 μl of RNaseH was added, and the samples were further incubated for 15 min at 36 °C. cDNA samples were amplified for 30 cycles using Taq polymerase (Invitrogen) and specific primers (Table 1). After amplification, PCR products were separated by gel electrophoresis on 1.5% agarose gels containing ethidium bromide, a nucleic acid gel stain used at 1000X (Amresco), and were visualized with the FLA-5000 chemiluminescence detection system (Fujifilm). The data were normalized to 18rS expression and were analyzed using the Multi-Gauge Image program. All reactions were repeated three times to ensure reproducibility.
Primer sequences and PCR conditions for gene expression analyses.
| Gene (oC) | Oligonucleotide sequence (5´-3´) | Annealing | Cycles | Product size (bp) |
|---|---|---|---|---|
| Foxp3 | F: GGCCCTTCTCCAGGACAGA | 56 | 30 | 112 |
| R: GCTGATCATGGCTGGGTTGT | ||||
| IL-17 | F: TGGCGGCTACAGTGAAGGCA | 56 | 30 | 400 |
| R: ACAATCGAGGCCACGCAGGT | ||||
| 18rS | F: GGACAGGATTGACAGATTGATAG | 60 | 30 | 100 |
| R: CTCGTTCGTTATCGGAATTAAC |
F: Forward, R: Reverse
Statistical analyses. The statistical significance of the data was evaluated using the non-parametric Mann-Whitney U test, Wilcoxon rank test or Student's t-test, as appropriate. In all cases, differences between animal groups were considered to be statistically significant if p < 0.05 (*).
Results
Exposure to polycyclic aromatic hydrocarbons (PAHs) can result in immune system alterations in both the innate and adaptive immune system. These compounds share a common signaling mechanism: the AhR. Therefore, in the present study, we evaluated the role of AhR in the immune response to L. major infection.
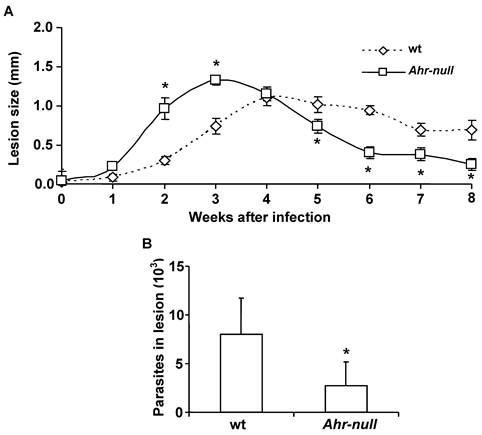
L. major promastigotes were injected into the footpads of wt and Ahr-null mice. One week after the infection, the inflammatory process was initiated in both wt and Ahr-null mice, although inflammatory intensity was higher and started earlier in the Ahr-null mice. Four weeks later, inflammation declined earlier in the Ahr-null mice compared to wt mice (Fig. 1A). The final outcome of L. major infection was determined by evaluating parasite numbers in the lesions 6 weeks after infection. Ahr-null mice presented a decrease in the number of parasites in the footpad lesions when compared to wt mice (Fig. 1B). These differences may be explained by the initial exacerbated inflammatory response observed in Ahr-null mice.
Progression of L. major lesions in wt and Ahr-null mice. Promastigotes were injected into the footpads of the mice, as described in the Materials and Methods. Progression of the lesion growth in the footpads was measured based on the mean lesion size every week (A), and the number of parasites present in the lesion was determined after eight weeks (B). Mean ± SD (n = 15), *p<0.05, Mann-Whitney U test.
Because inflammatory processes are mediated by cytokines, we examined several pro- and anti-inflammatory factors during the infection. Blood samples were obtained every two weeks for two months, and the levels of TNFα, IL-12, IL-10 and LXA4 were measured. Ahr-null mice presented higher levels of the pro-inflammatory cytokine TNF-α when compared to wt mice, both prior to parasite inoculation and during infection (Fig. 2A).
Inflammatory-related factors after infection with L. major. Blood samples were collected from the tail veins of the mice every two weeks, and the levels of TNF-α (A), IL-12 (B), IL-10 (C), and lipoxin A4 (D) were measured in the serum, as described in the Materials and Methods. Mean ± SD (n = 15), *p<0.05, Mann-Whitney U test.
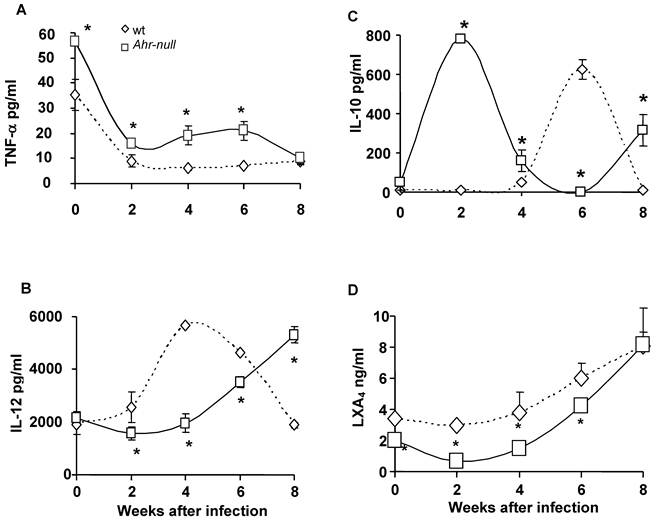
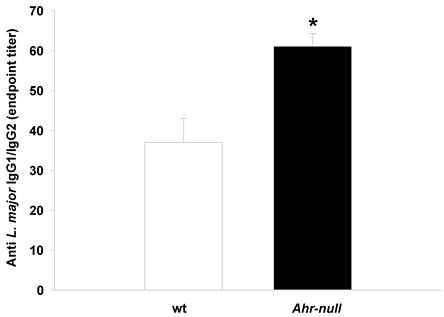
During the first four weeks of infection, the wt mice contained lower levels of IL-10 than the Ahr-null mice (Fig. 2C). In contrast, serum IL-12 levels were higher in the wt animals compared to the Ahr-null mice during the same period (Fig. 2B). Ahr-null mice displayed a decrease in IL-10 levels and an increase in IL-12 levels at four and six weeks post-infection, respectively. The opposite was observed in wt mice, in which an increase in IL-10 levels and a decrease in IL-12 levels were apparent at six weeks post-infection (Figs. 2B and C). The levels of LXA4, an anti-inflammatory molecule, were higher in wt mice before parasite inoculation and during the infection compared to Ahr-null mice. In both animals, a time-dependent increase in LXA4 levels was observed (Fig. 2D). To more completely characterize the immune response to L. major, specific IgGs (IgG1 and IgG2a) were quantified at 3 weeks post-infection. As shown in Fig. 3, Ahr-null mice presented a higher proportion of the endpoint titer of IgG1/IgG2a compared to wt animals.
Antibody response in wt and Ahr-null mice. The production of L. major-specific IgG1/IgG2a is presented as reciprocal endpoint titers. These titers were determined after 3 weeks of infection in serum collected from the tail vein. Mean ± SD (n = 5), *p<0.05, Student's t-test.
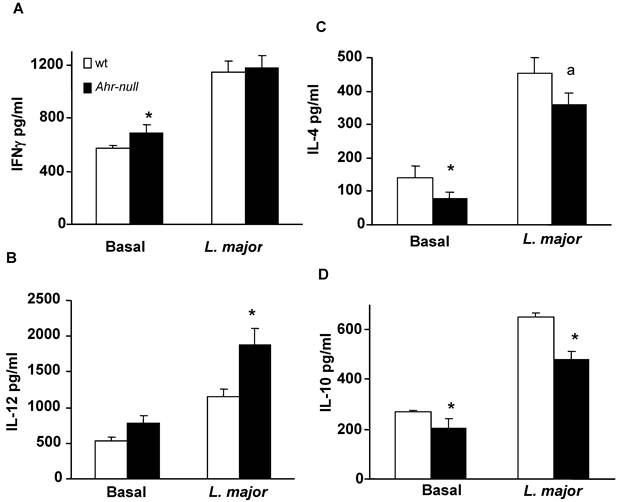
To further characterize the immune response, lymph node cells from infected wt and Ahr-null mice were cultured with L. major antigen, and cytokine levels were determined. IFN-γ levels were higher in Ahr-null lymph node cells compared to wt cells at baseline. IL-12 levels were higher in Ahr-null lymph node cells when compared to wt cells after exposed to L. major antigen (Figs. 4A and 4B). In contrast, the levels of the anti-inflammatory cytokines IL-4 and IL-10 were decreased in Ahr-null cells compared to lymph node cells from wt mice (Figs. 4C, and 4D).
Cytokine profile of lymph node cells from infected animals. Lymph nodes from wt and Ahr-null mice were collected after six weeks, and the cells were cultured with L. major antigen. After five days of culture, levels of IFN-γ (A), IL-12 (B), IL-4 (C) and IL-10 (D) in the supernatants were measured by sandwich ELISA. Mean ± SD (n = 10), *p<0.05, Student's t-test.
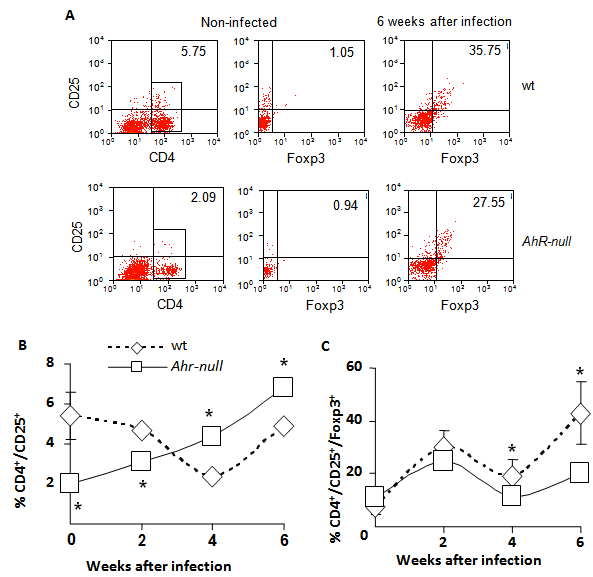
Because inflammatory responses are also modulated by T regulatory (Treg) lymphocytes, we asked whether Ahr-null mice exhibited altered Treg cell counts. Lymphocytes from the draining lymph node were obtained at different time points post-infection, and the CD4+/CD25+/Foxp3+ lymphocyte subpopulation was evaluated by flow cytometry. As shown in Fig. 5C, at four weeks after infection, a slight but significant decrease in Treg cells was observed in Ahr-null compared to wt mice, and the decrease was more evident after six weeks (30% decrease in Ahr-null Treg cells compared to those of wt mice). We also observed an inverse pattern between Ahr-null and wt mice in the induction of activated Th cells. Ahr-null mice presented significantly lower numbers of activated Th cells during the first two weeks of infection. By four and six weeks post-infection, this profile was reversed, and the Ahr-null mice displayed higher numbers of activated Th cells compared to wt mice (Fig. 5B).
Levels of Th and Treg cells from wt and Ahr-null mice. Cells from drained lymph nodes of infected wt and Ahr-null mice were collected and stained for Treg-specific markers, as described in the Materials and Methods. The cells were evaluated for the expression of CD4, CD25, and Foxp3 by flow cytometry. Representative flow cytometric analysis of the CD4+CD25+Foxp3+ cells (A). The proportion of CD25+ Th cells from the total of lymph node cells (B) and the percentage of Treg cells from the double positive population (C) were determined at different time points after infection with L. major. Mean ± SD in triplicate (n = 5), *p<0.05, Student's t-test.
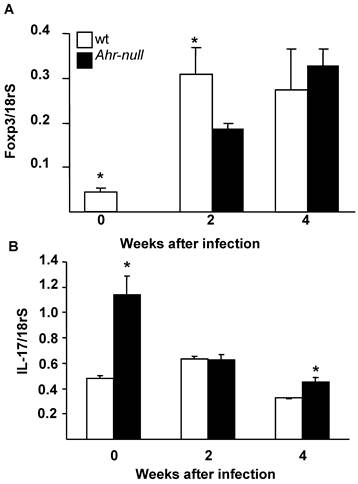
The differentiation of T cells into Treg cells is modulated by the presence of Foxp3 and IL-17; therefore, the levels of Foxp3 and IL-17 mRNA from lymph node cells were determined. Ahr-null cells presented lower levels of Foxp3 mRNA before the infection and two weeks after the infection when compared to lymph node cells from wt mice (Fig. 6A). In contrast, wt cells presented lower levels of IL-17 mRNA before the infection and four weeks post-infection (Fig. 6B).
Levels of Foxp3 and IL-17 mRNA from wt and Ahr-null lymph node cells. Expression of Foxp3 (A) and IL-17 (B) is normalized to the relative expression of 18rS in lymph node cells. Mean ± SD in triplicate (n = 5), *p<0.05, Student's t-test.
Discussion
Several studies have suggested that AhR plays a role in immune and inflammatory regulation. However, most of these studies are based on AhR activation by exogenous ligands, such as TCDD [8, 14, 19]. In the present study, we addressed the endogenous role of this transcription factor, in the absence of exogenous ligand, on the immune response to L. major infection.
As expected, C57BL/6 wt mice presented a Th1 immune response after infection, and high serum levels of IL-12 and low or absent serum levels of IL-10 were observed at the beginning of the infection. In contrast, AhR-null mice developed a mixed immune response with lower levels of IL-12 and higher levels of IL-10 and TNF-α. The proportional distribution of the IgG1 and IgG2 serum isotypes, determined at three weeks post-infection, indicated that the AhR-null mice tended to develop a Th2 immune response after L. major infection. The higher IgG1/IgG2a proportion observed in Ahr-null mice might be a result of lower IL-12 levels because this cytokine enhances IgG2 production, but it reduces IgG1 levels [20].
Initially, the inflammatory process was more pronounced in the AhR-null mice, which may account for the decreased number of parasites in the footpad lesions when compared to wt mice. This may be due to high levels of the pro-inflammatory cytokine TNF-α. The role of TNF-α in leishmaniasis has been studied previously; inactivation of this cytokine leads to aggravation of the infection, whereas TNF-α treatment results in the amelioration of the disease [21, 22].
Consistent with the present results, a recent study showed that smoke-induced lung inflammation is two- to three-fold more severe in Ahr-null mice compared to wt mice. Moreover, the authors reported that untreated Ahr-null mice expressed three- to five-fold higher levels of pro-inflammatory cytokines (including TNF-α) than wt mice [23]. In addition, AhR ligands, such as benzo[a]pyrene, have been shown to markedly diminish TNF-α secretion by monocytes [9]. All together, these data suggest that AhR negatively controls TNF-α expression and/or secretion, indicating that the levels of this cytokine are increased when AhR is not functional. The mechanism by which AhR affects TNF-α expression is not well understood. However, because TNF-α is regulated by NF-κB [24], and AhR and NF-κB can regulate the activity of one another [25], AhR could act as a negative modulator of cytokine expression via its interaction with NF-κB. This idea is supported by the demonstration that lung cells from Ahr-null mice presented higher NF-κB activity compared to wt mice [23]. Moreover, the functional interaction between AhR, RelB and RelA NF-κB subunits has been previously shown [26, 27].
Conversely, it has been shown by several groups that TNF-α is capable of eliciting IL-10 release in an autoregulatory feedback loop [28-30]. Therefore, the high serum levels of IL-10 present in AhR-null mice might be the result of the high TNF-α levels. Consistent with these results, high levels of IL-10 down-regulate the production of IL-12 [31, 32].
During an inflammatory response, compensatory mechanisms are also activated, such as the synthesis of lipoxins. In particular, LXA4, which possesses significant anti-inflammatory activity, has been shown to inhibit TNF-α production and activity [33-35]. In the present study, we evaluated serum LXA4 levels. Our results show a constant increase in LXA4 serum concentrations as the infection progresses in both wt and Ahr-null animals. This response may contribute to the decreased TNF-α levels observed after two weeks of infection. However, the AhR-null mice presented lower levels of LXA4 compared to the wt mice. It has been reported that TCDD, the most potent AhR ligand, increases the expression of prostaglandin endoperoxide H synthase (PGHS)-2 in an AhR-dependent manner [36, 37]. Additionally, the inhibition of (PGHS)-2 results in a reduction of 15-hydroxyeicosatetraenoic acid synthesis by approximately 50%, thereby decreasing 15-epi-LXA4, which provides 88% of the total amount of LXA4 [38]. Therefore, these data, together with our present results, suggest that the AhR might be implicated in the synthesis of LXA4, possibly through the regulation of PGHS-2 gene transcription as previously demonstrated in some human cells [39].
After six weeks of infection, a Th1 response developed in the AhR-null mice, as the serum cytokine profiles indicate. This response correlates with the high IFN-γ and IL-12 levels and the low levels of IL-4 and IL-10 secretion produced by the AhR-null lymph node cells. Moreover, the polarization of the response is more pronounced in the AhR-null cells than in the wt cells.
At four and six weeks post-infection, Ahr-null lymph node cells presented higher levels of CD4+CD25+ cells and lower levels of Treg cells compared to wt cells. During an infection, Treg cells play an important role in controlling immune responses by diminishing potentially damaging effects to self-tissues that can result from an uncontrolled exacerbated immune response. Together with the high TNF-α levels, the decreased number of Treg cells found in Ahr-null mice could contribute to the robust inflammation and the increased elimination of parasite burden. Similar to the present study, it has been reported that the C57BL/6 mice carrying an allele coding for an Ahr molecule with reduced affinity for its ligands (Ahrd mice) presented lower levels of CD4+Foxp3+ T cells [40]. Mutations in Foxp3, a transcription factor, have been shown to impair Treg activity [41], and more recently, it has been shown that AhR regulates Treg and Th17 differentiation in a ligand-specific manner [40]. The same authors demonstrated that this regulation is mediated by AhR through the control of Foxp3 expression. As with this study, a decrease in Foxp3 mRNA levels was observed in Ahr-null lymph node cells when compared to wt cells before infection. Consistent with the negative regulation of Treg and Th17 cells, Ahr-null mice presented higher levels of IL-17 mRNA. After infection, the increase in Foxp3 mRNA levels was accompanied by a decrease in IL-17 mRNA levels. These results also indicate that factors other than AhR are involved in Foxp3 regulation.
In conclusion, our present findings demonstrate that AhR disruption results in altered immune responses to L. major infection; in particular, loss of Ahr results in lowered Treg cell levels, increased TNF-α levels, and a modified timeframe of IL-10 and IL-12 secretion. Our results also show that AhR may play an important role in immune response modulation and suggest the presence of an endogenous ligand. Additional experiments are needed to fully understand and clarify the potential epigenetic mechanisms by which AhR functions, such as an AhR/NF-κB interaction. Finally, our results also identify the AhR as a promising pharmacological target for the treatment of certain autoimmune and immunosuppressive diseases.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Whitlock JPJr. Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol. 1999;39:103-25
2. Okey AB, Riddick DS, Harper PA. The Ah receptor: mediator of the toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and related compounds. Toxicol Lett. 1994;70(1):1-22
3. Gonzalez FJ, Fernandez-Salguero P, The aryl hydrocarbon receptor. studies using the AHR-null mice. Drug Metab Dispos. 1998;26(12):1194-8
4. Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ. Aryl-hydrocarbon receptor-deficient mice are re-sistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol Appl Pharmacol. 1996;140(1):173-9
5. Elizondo G, Fernandez-Salguero P, Sheikh MS, Kim GY, For-nace AJ, Lee KS, Gonzalez FJ. Altered cell cycle control at the G(2)/M phases in aryl hydrocarbon receptor-null embryo fibroblast. Mol Pharmacol. 2000;57(5):1056-63
6. Sato S, Shirakawa H, Tomita S, Ohsaki Y, Haketa K, Tooi O, Santo N, Tohkin M, Furukawa Y, Gonzalez FJ, Komai M. Low-dose dioxins alter gene expression related to cholesterol biosynthesis, lipogenesis, and glucose metabolism through the aryl hydrocarbon receptor-mediated pathway in mouse liver. Toxicol Appl Pharmacol. 2008;229(1):10-9
7. Reyes-Hernandez OD, Mejia-Garcia A, Sanchez-Ocampo EM, Cabanas-Cortes MA, Ramirez P, Chavez-Gonzalez L, Gonzalez FJ, Elizondo G. Ube2l3 gene expression is modulated by activation of the aryl hydrocarbon receptor: implications for p53 ubiquitination. Biochem Pharmacol. 2010;80(6):932-40
8. Kerkvliet NI. AHR-mediated immunomodulation: the role of altered gene transcription. Biochem Pharmacol. 2009;77(4):746-60
9. van Grevenynghe J, Rion S, Le Ferrec E, Le Vee M, Amiot L, Fauchet R, Fardel O. Polycyclic aromatic hydrocarbons in-hibit differentiation of human monocytes into macrophages. J Immunol. 2003;170(5):2374-81
10. Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, Rudikoff S, Ward JM, Gonzalez FJ. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science. 1995;268(5211):722-6
11. Schaldach CM, Riby J, Bjeldanes LF. Lipoxin A4: a new class of ligand for the Ah receptor. Biochemistry. 1999;38(3):7594-600
12. Rodriguez-Sosa M, Elizondo G, Lopez-Duran RM, Rivera I, Gonzalez FJ, Vega L. Over-production of IFN-gamma and IL-12 in AhR-null mice. FEBS Lett. 2005;579(28):6403-10
13. Shi LZ, Faith NG, Nakayama Y, Suresh M, Steinberg H, Czuprynski CJ. The aryl hydrocarbon receptor is required for optimal resistance to Listeria monocytogenes infection in mice. J Immunol. 2007;179(10):6952-62
14. Vorderstrasse BA, Lawrence BP. Protection against lethal challenge with Streptococcus pneumoniae is conferred by aryl hydrocarbon receptor activation but is not associated with an enhanced inflammatory response. Infect Immun. 2006;74(10):5679-86
15. Head J, Lawrence B. The aryl hydrocarbon receptor is a modulator of anti-viral immunity. Biochem Pharmacol. 2009;77(4):642-53
16. Sanchez Y, Rosado Jde D, Vega L, Elizondo G, Estrada-Muniz E, Saavedra R, Juarez I, Rodriguez-Sosa M. The unexpected role for the aryl hydrocarbon receptor on susceptibility to ex-perimental toxoplasmosis. J Biomed Biotechnol. 2010:505694
17. Brown JA, Greenwald RJ, Scott S, Schweitzer AN, Satoskar AR, Chung C, Schopf LR, van der Woude D, Sypek JP, Sharpe AH. T helper differentiation in resistant and susceptible B7-deficient mice infected with Leishmania major. Eur J Im-munol. 2002;32(6):1764-72
18. Vega L, Styblo M, Patterson R, Cullen W, Wang C, Germolec D. Differential effects of trivalent and pentavalent arsenicals on cell proliferation and cytokine secretion in normal human epidermal keratinocytes. Toxicol Appl Pharmacol. 2001;172(3):225-32
19. Thurmond TS, Staples JE, Silverstone AE, Gasiewicz TA. The aryl hydrocarbon receptor has a role in the in vivo maturation of murine bone marrow B lymphocytes and their response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol. 2000;165(3):227-36
20. Gracie JA, Bradley JA. Interleukin-12 induces interfer-on-gamma-dependent switching of IgG alloantibody subclass. Eur J Immunol. 1996;26(6):1217-21
21. de Kossodo S, Grau GE, Louis JA, Muller I. Tumor necrosis factor alpha (TNF-alpha) and TNF-beta and their receptors in experimental cutaneous leishmaniasis. Infect Immun. 1994;62(4):1414-20
22. Liew FY, Li Y, Millott S. Tumour necrosis factor (TNF-alpha) in leishmaniasis. II. TNF-alpha-induced macrophage leishmanicidal activity is mediated by nitric oxide from L-arginine. Immunology. 1990;71(4):556-9
23. Thatcher TH, Maggirwar SB, Baglole CJ, Lakatos HF, Gasiewicz TA, Phipps RP, Sime PJ. Aryl hydrocarbon recep-tor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am J Pathol. 2007;170(3):855-64
24. Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involve-ment of four kappa B-like motifs and of constitutive and in-ducible forms of NF-kappa B. Mol Cell Biol. 1990;10(4):1498-506
25. Tian Y, Ke S, Denison MS, Rabson AB, Gallo MA. Ah re-ceptor and NF-kappaB interactions, a potential mechanism for dioxin toxicity. J Biol Chem. 1999;274(1):510-5
26. Vogel CF, Sciullo E, Li W, Wong P, Lazennec G, Matsumura F. RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol Endocrinol. 2007;21(12):2941-55
27. Vogel CF, Matsumura F. A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-kappaB family. Biochem Pharmacol. 2009;77(4):734-45
28. Wanidworanun C, Strober W. Predominant role of tumor necrosis factor-alpha in human monocyte IL-10 synthesis. J Immunol. 1993;151(12):6853-61
29. van der Poll T, Jansen J, Levi M, ten Cate H, ten Cate JW, van Deventer SJ. Regulation of interleukin 10 release by tumor necrosis factor in humans and chimpanzees. J Exp Med. 1994;180(5):1985-8
30. Daftarian PM, Kumar A, Kryworuchko M, Diaz-Mitoma F. IL-10 production is enhanced in human T cells by IL-12 and IL-6 and in monocytes by tumor necrosis factor-alpha. J Immunol. 1996;157(1):12-20
31. D'Andrea A, Aste-Amezaga M, Valiante NM, Ma X, Kubin M, Trinchieri G. Interleukin 10 (IL-10) inhibits human lym-phocyte interferon gamma-production by suppressing natural killer cell stimulatory factor/IL-12 synthesis in accessory cells. J Exp Med. 1993;178(3):1041-8
32. Tripp CS, Wolf SF, Unanue ER. Interleukin 12 and tumor necrosis factor alpha are costimulators of interferon gamma production by natural killer cells in severe combined immu-nodeficiency mice with listeriosis, and interleukin 10 is a physiologic antagonist. Proc Natl Acad Sci U S A. 1993;90(8):3725-9
33. Hachicha M, Pouliot M, Petasis NA, Serhan CN. Lipoxin (LX)A4 and aspirin-triggered 15-epi-LXA4 inhibit tumor ne-crosis factor 1alpha-initiated neutrophil responses and traf-ficking: regulators of a cytokine-chemokine axis. J Exp Med. 1999;189(12):1923-30
34. Goh J, Baird AW, O'Keane C, Watson RW, Cottell D, Bernasconi G, Petasis NA, Godson C, Brady HR, MacMathuna P. Lipoxin A(4) and aspirin-triggered 15-epi-lipoxin A(4) antagonize TNF-alpha-stimulated neutrophil-enterocyte interactions in vitro and attenuate TNF-alpha-induced chemokine release and colonocyte apoptosis in human intestinal mucosa ex vivo. J Immunol. 2001;167(5):2772-80
35. Souza DG, Fagundes CT, Amaral FA, Cisalpino D, Sousa LP, Vieira AT, Pinho V, Nicoli JR, Vieira LQ, Fierro IM, Teixeira MM. The required role of endogenously produced lipoxin A4 and annexin-1 for the production of IL-10 and inflammatory hyporesponsiveness in mice. J Immunol. 2007;179(12):8533-43
36. Liu Y, Levy GN, Weber WW. Induction of human prosta-glandin endoperoxide H synthase-2 (PHS-2) mRNA by TCDD. Prostaglandins. 1997;53(1):1-10
37. Pineau T, Costet P, Puel O, Pfohl-Leszkowicz A, Lesca P, Al-vinerie M, Galtier P. Knockout animals in toxicology: as-sessment of toxin bioactivation pathways using mice deficient in xenobiotic metabolizing enzymes. Toxicol Lett. 1998;102-103:459-64
38. Claria J, Lee MH, Serhan CN. Aspirin-triggered lipoxins (15-epi-LX) are generated by the human lung adenocarcinoma cell line (A549)-neutrophil interactions and are potent inhibitors of cell proliferation. Mol Med. 1996;2(5):583-96
39. Degner SC, Papoutsis AJ, Selmin O, Romagnolo DF. Targeting of aryl hydrocarbon receptor-mediated activation of cyclooxygenase-2 expression by the indole-3-carbinol metabolite 3,3'-diindolylmethane in breast cancer cells. J Nutr. 2009;139(1):26-32
40. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453(7191):65-71
41. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27(1):68-73
Author contact
![]() Corresponding author: Libia Vega; Centro de Investigación y Estudios Avanzados del Instituto Politécnico Nacional. Departamento de Toxicología. Av. IPN 2508. San Pedro Zacatenco. C. P. 07360. México D. F., México. Phone +52-55-5747-3800 ext. 5472. Fax +52-55-5747-3395. E-mail: lvegamx
Corresponding author: Libia Vega; Centro de Investigación y Estudios Avanzados del Instituto Politécnico Nacional. Departamento de Toxicología. Av. IPN 2508. San Pedro Zacatenco. C. P. 07360. México D. F., México. Phone +52-55-5747-3800 ext. 5472. Fax +52-55-5747-3395. E-mail: lvegamx