Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Malaria
Leishmaniasis
Trypanosomiasis
Toxoplasmosis
Primary Amebic...
Neosporosis
Coccidiosis
Concluding remarks
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(9):1239-1256. doi:10.7150/ijbs.7.1239 This issue Cite
Review
Macrophage Migration Inhibitory Factor (MIF): A Key Player in Protozoan Infections
Juan de Dios Rosado, Miriam Rodriguez-Sosa ![]()
Unidad de Biomedicina, Facultad de Estudios Superiores Iztacala, Universidad Nacional Autónoma de México (UNAM), 54090 Tlalnepantla, Estado de México, México.
Received 2011-9-1; Accepted 2011-10-1; Published 2011-10-25
Abstract
Macrophage migration inhibitory factor (MIF) is a pleiotropic cytokine produced by the pituitary gland and multiple cell types, including macrophages (Mø), dendritic cells (DC) and T-cells. Upon releases MIF modulates the expression of several inflammatory molecules, such as TNF-α, nitric oxide and cyclooxygenase 2 (COX-2). These important MIF characteristics have prompted investigators to study its role in parasite infections. Several reports have demonstrated that MIF plays either a protective or deleterious role in the immune response to different pathogens. Here, we review the role of MIF in the host defense response to some important protozoan infections.
Keywords: Macrophage migration inhibitory factor, Protozoan infections, Inflammatory response, Control of parasite infections, innate immune response.
Introduction
MIF was originally discovered (in 1966) as a lymphokine, derived from activated T-cells, that inhibited the random migration of macrophages in vitro and was shown to be involved in the mechanism of delayed-type hypersensitivity [1, 2]. Currently, it is known that MIF is a widely expressed and pleiotropic cytokine that functions as a critical upstream mediator of innate immunity and promotes numerous pathophysiological processes [3-11], such as glomerulonephritis [12, 13], arthritis [5, 14], experimental autoinmune encephalomyelitis (EAE) [15], experimental autoinmune myocarditis (EAM) [16], gram-negative [17-23] and gram-positive sepsis [23, 24], colitis [10, 25], asthma [8] diabetes, and pancreatitis [26-28]. MIF is produced and secreted primarily by immune cells, such as lymphocytes, Mø, DC, neutrophils and pituitary cells [29, 30]. MIF secretion is tightly regulated by stress and immune stimuli, including endotoxins, inflammatory cytokines (interferon (IFN)-γ, tumor necrosis factor (TNF)-α) and glucocorticoids [29, 31-33]. Once secreted, MIF exhibits a broad range of immune and inflammatory activities, including the induction of inflammatory cytokines such as TNF-α, IFN-γ, interleukin (IL)-1β, IL-12, IL-6 and, CXCL8 (also known as IL-8), among others [20, 32, 34-40]. MIF favors the expression of the Toll-like receptor 4 (TLR4) gene, which encodes the signal-transducing element of the lipopolysaccharide (LPS) receptor complex [24, 41, 42] through the activation of transcription factors of the ETS family [43]. In addition, MIF counter-regulates the immunosuppressive effects of glucocorticoids [32, 44-46], and it sustains macrophage proinflammatory functions by inhibiting p53 [47]. MIF also promotes the migration and recruitment of immune cells inducing the expression of chemokines (monocyte chemoattractant protein (MCP)-1, and adhesion molecules as intercellular adhesion molecule (I-CAM)-1 and vascular cell adhesion molecule (V-CAM)-1 [48-51].
Parasitic infections are important cause of morbidity and mortality in humans, and the innate and adaptive immune responses triggered by these organisms are critical in determining their outcome. Parasitic-associated factors and host-derived components are also important inflammatory modulators. Recognition of these molecules triggers signaling pathways that influence the host-response to infection and disease progression. In this context, MIF is over-expressed in most parasitic infections; however, the role of MIF in the immune response to parasitic infections has only recently been elucidated, providing valuable information that must to be clarified. The aim of this review is to provide an overview of the current literature regarding the role of MIF in important protozoan infections. We will focus on immune response modulation, the implications of such modulation and the possible mechanisms involved. We will also discuss the differences and similarities in MIF activity infections caused by distinct parasites.
Malaria
Malaria is caused by intracellular parasitic protozoa of the genus Plasmodium (P) and is transmitted by the infected female Anopheles mosquito during blood meals. Malaria is a major cause of severe illness and death in many part of the world. It causes 300 to 500 million new infections per year resulting in approximately 1 to 2 million deaths, and these primarily occur in children under the age of 5 years. Severe anemia and cerebral malaria are the major complications leading to malaria-induced morbidity and mortality [52]. Factors such as host genetic variation, age of first exposure, rate of exposure (endemicity) [53, 54] and the host immune response to P. falciparum [55] play an important role in the generation of symptoms and disease [56, 57].
Remarkable research on the role of MIF in malaria has been recently conducted (Table 1).
MIF in Malaria infection
| Parasite | Model | Disease | in vitro/ in vivo | Treatment | Properties | MIF role | Ref. |
|---|---|---|---|---|---|---|---|
| P. chabaudi | BALB/c mice | SMA | in vitro | rMIF+ Epo | ↓BFU-E ↓CFU-GEMM↑ Parasitemia | Causes erythropoiesis suppression and SMA development | [58] |
| in vivo | ↑MIF ↑SMA | ||||||
| BALB/c mice | SMA | in vitro | rMIF+IFN-γ; TNF-α+Epo | ↓BFU-E ↓CFU-E ↑ERK1/2 ↓P38 | Supressed erythropoietin colony formation and sinergized with TNF-α and IFN-γ modulating MAPK activation. | [60] | |
| MIF-KO BALB/c mice | SMA | in vivo | ↑Parasitemia ↓Mortality ↑ BFU-E ↑ CFU-E | ||||
| P. chabaudi adami556-KA | MIF-KO BALB/c mice | Malaria | In vivo | ↓Parasite burden ↓cumulative parasitemia ↓Peak parasitemia at day 4 post-infeccion | MIF modulates the balance between Th1 and Th2 effector responses, by attenuating the development of Th1 responses, inducing anti-inflammatory cytokines during the infection of P. c. adami. | [59] | |
| In vitro | Anti-MIF | ↓ parasitemia ↓Peak parasitemia at day 4 post-infeccion in WT mice | |||||
| MIF KO | CD4+T cells showed ↑IFNγ, ↓IL-10 at day 4 p.i. ↑ IL-4 (presumable mast cells or eosiniphils) | ||||||
| P. vivax | Adults Adolescents Children | Acute malaria uncomplicated | in vivo | ↑MIF, TNF-α, IFN-γ, IL-12, MCP-1 and IL-10 ↑IgM ↓Hb and ↑MIF | Enhancing the Ab production against the parasite which cause erythrocyte lysis | [62] | |
| P. falciparum | Zambean children | Malaria | in vivo | ↑MIF peripheral blood | Promote polymorphisms, modulate the immune response to hemozoin | [60] | |
| in vitro | Hemozoin | ↓MIF from Mo with 5-CATT (low repeat) ↑MIF from Mo 6/7 CATT | |||||
| Pregnant woman | Placental malaria | in vivo | ↑MIF in IVB ↓MIF in peripheral and cord plasma | Enhancing the local immune response | [63] | ||
| in vivo | ↑MIF in placental plasma ↓IL-12 IL-18 TGF-β1 | Enhancing immune response | [64] | ||||
| in vitro | ↓MIF in PBMC ↑MIF in IVB cells | ||||||
| in vivo | ↑MIF in amniotic ephitelium and IVBMC ↓MIF in villous stroma and extravillous trophoblast | [65] | |||||
| Indian adults | Cerebral malaria | in vivo | ↑Plasma MIF in non-survivors ↓Plasma MIF in patients with mild malaria | Protective role in CM and induce fatal complications | [61] | ||
| Adults Adolescents Children | Acute infec. malaria un-complicated | in vivo | ↑MIF, TNF-α, IFN-γ and MCP-1 in peripheral blood ↑IgM and IgG against parasite correlated to ↓Hb conc. | Enhancing the Ab against parasite which may cause erythrocyte lysis | [62] | ||
| Malawian Children | Cerebral malaria | in vivo | ↓MIF in cerebral vascular walls skeletal muscle and chest | Absence of MIF have a survival advantage in preventing intracranial hemorrhage in systemic infections | [66] | ||
| Gabonese children | Acute malaria | in vivo | ↓MIF in peripheral blood PGE2 and TGF-β1 ↑parasitemia ↑IFN-γ, TNF-α, iNOS and IL-10 | Reduced MIF and TGF-β1 promote an ineffective immune response that enhanced pathogenesis | [67] | ||
| in vitro | ↓MIF mRNA in PBMC | ||||||
| Kenyan children | SMA | in vivo | ↓MIF in plasma ↓MIF mRNA in PBL ↑% PCM related to ↑SMA and ↓MIF in PBMC | A protective response against severe disease | [68] | ||
| in vitro | pfHz or sHz | ↓MIF in PBMC | |||||
| Caucasian Adults | Acute malaria | in vivo | ↓Plasma MIF ↓Peripheral lymphocytes ↑Parasitemia Hb unchanged | Reallocation of lymphocytes to tissues during acute malaria explains the decline in circulating lymphocytes that may causes MIF↓ in plasma | [69] | ||
| Zambian children | malaria | in vivo | ↑6,7 and 8 CATT repeats at MIF -794 allele are correlated to ↑Parasitemia ↑5 CATT repeat at MIF-794 allele is correlated to ↓Parasitemia | Protective effect of MIF -794, 5 CATT allele | [81] | ||
| Kenyan children | Acute malaria | in vivo | ↑risk of developing HDP of C/C compared to G/G genotype | MIF -173 G/C C/C genotypes are associated with an increased risk of HDP, ↓plasma MIF but not increased SMA susceptibility | [82] | ||
| in vitro | pfHz | ↑MIF in PBMC with MIF -173 G/G genotype compared to G/C genotype | |||||
| Kenyan children | SMA | in vivo | ↑longer (7, 8) CATT repeats at MIF -794 alleles associated to ↑SMA and ↓MIF compared to 5, 6 CATT repeats 7C and 8C haplotypes related to ↑SMA ↓MIF compared to 6 G haplotype | MIF promoter at the -173 and -794 polymorphisms can both confer protection or susceptibility to SMA. Carriers of the disease associated alleles have decreased peripheral blood MIF | [83] |
Erythroid (BFU-E), multipotential (CFU-GEMM), and granulocyte-macrophage (CFU-GM) progenitor-derived colony formation. erytropoyetin (Epo); Monocytes (Mo); Intervillous blood (IVB); Peripheral bone marrow cell (PBMC); Peripheral blood lymphocytes (PBL); Pigment containing monocytes (PCM); Mononuclear intrvillous cells (IVBMC); Hingh density parasitemia (HDP); P. falciparum hemozoin (pfHz); Sintetic Hemozoin (sHz); Severe Malaria Anemia (SMA); Cerebral malaria (CM).
In one study, Martiney et al. reported that the ingestion of P. chabaudi-infected erythrocytes or malarial pigment (hemozoin) by macrophages induces the release of MIF, resulting in high circulating levels of MIF in the sera and bone marrow of P. chabaudi-infected BALB/c mice. Furthermore, recombinant MIF administration suppressed the erythroid and other myeloid progenitor development in the presence of erythropoietin an erythropoiesis inhibitory role for MIF [58]. These findings were supported later in vitro studies in which the combined subinhibitory concentrations of MIF, TNF-α and IFN-γ acted synergistically to inhibit erythroid differentiation and hemoglobin production by antagonizing the pattern of mitogen-activated protein (MAP) kinase phosphorylation that normally occurs during erythroid progenitor differentiation. An in vivo study, using P. chabaudi-infected MIF knockout (KO) mice, showed that the absence of MIF is related to an increase in erythropoiesis and decreased mortally. Correlated with a recent study by Malu DT et al, where MIF KO mice showed increased resistance to P. chabaudi adami infection associated with enhanced IFN-γ and reduce IL-4 and IL-10 production by CD4+ T cells, suggesting a regulatory role for MIF on T cell activation, which favors a th2 type susceptible response in WT mice [59]. In addition, increased MIF circulating levels found in Zambian children supports the hypothesis that Plasmodium infection is a potent stimulus for systemic MIF expression in humans, which may be involved in the pathophysiology of malaria-induced anemia [60].
Recent research regarding the role of MIF in the pathogenesis and outcome of cerebral malaria showed that patients with mild malaria and survivors of cerebral malaria had low peripheral blood MIF levels compared to cerebral malaria non-survivors, suggesting that elevated MIF levels may be a risk factor for mortality in cerebral malaria patients [61]. Moreover, patients with P. falciparum and P. vivax infections showed significant increases in MIF, TNF-α, IFN-γ, IL-12, IgM and MCP-1 in the peripheral blood. Also high IgM antibody levels against stage parasite forms were associated with low hemoglobin (Hb) and increased MIF levels, indicating that MIF participates in the inflammatory immune response to malaria. This response, in concert with other inflammatory cytokines and the production of specific antibodies against the parasite, may lead to pathologic responses [62]. However, MIF does not always act systemically. Previous studies by Chaisavaneeyakorn et al reported significantly elevated MIF levels in the inter-villous blood (IVB) plasma in pregnant women with placental malaria compared to both peripheral plasma and cord plasma [63]. Similar results were obtained by Chaiyaroj et a.l, who observed significantly higher MIF production by inter-villous blood mononuclear cells (IVBMC) compared to peripheral blood mononuclear cells (PBMC), as well as high MIF levels in placental plasma compared to paired peripheral plasma [64]. These data were confirmed by immunohistological studies in term placentas, demonstrating that, although MIF is expressed in various cellular compartments, enhanced MIF expression occurs specifically in mononuclear cells and amniotic epithelial cells in placental malaria [65]. These data indicate that increased MIF levels in infected placentas may play an important role in the local immune response, possibly through macrophage activation for parasite clearance. However, not all reports show local or systemic MIF overexpression. Immunohistological studies have reported that, although MIF was found in the nuclei of skeletal muscle cells, it was not found in the cerebral vascular walls of cerebral malaria patients. The lack of MIF overexpression in the cerebral vasculature may prevent excessive cerebral perfusion pressure and the risk of intracranial hemorrhage during systemic inflammation. Interestingly, the role of MIF in nuclei has not yet been determined [66]. Moreover, research in Gabonese children with acute malaria has shown significantly low MIF mRNA levels in PBMCs and low MIF protein levels in peripheral blood, which was associated with low levels of TGF-β1 in presence of TNF-α, IFN-γ, and inducible nitric oxide synthase (iNOS). These results suggest that the reduction of transforming growth factor beta (TGF)-β1 and MIF in children with acute malaria may promote an ineffective immune response culminating in enhanced pathogenesis [67]. These findings are consistent with a later study in 357 Kenyan children, which showed that malaria-induced anemia was associated with decreased circulating MIF levels. The authors also showed low MIF expression in peripheral blood lymphocytes (PBL), peripheral blood mononuclear cells (PBMCs), and in a high percentage of pigment containing monocytes (PCM), concluding that the effective production of MIF during childhood anemia may be protective against severe disease [68] (Fig 1). In addition a recent study in 10 healthy volunteers infected with P. falciparum, showed that a decrease in systemic MIF levels during acute infection is associated with a decline in circulating lymphocytes. Because T cells are the primary source of circulating MIF, these results may be attributed to the reallocation of lymphocytes from the peripheral plasma to infected tissues leading to a decrease in circulating MIF levels [69].
Overview about the role of MIF during Plasmodium infection. Theory 1. Mice infected with plasmodium (1) develop high MIF serum levels (2), MIF inhibits eritropoyesis (3) and promotes pathological complications increasing the risk of developing several malaria anemia (SMA) (4). Plasmodium infected human could develop low (3A) or high levels (3B) of MIF, depending of the (MIF´s polymorphisms expressed). Low levels of MIF correlates with high density parasitemia (HDP) in blood but low risk of developing SMA (5A). However, high expression of MIF induces SMA (5B). Plasmodium infected pregnant woman develop high levels of MIF in placental plasma, amniotic epithelium, mononuclear intervillous blood (IVB), and mononuclear tervillous cells (MIVC). All together indicate that MIF is associated to inhibition of eritropoyesis and promoting SMA, however it is not crucial in the control of the infection.
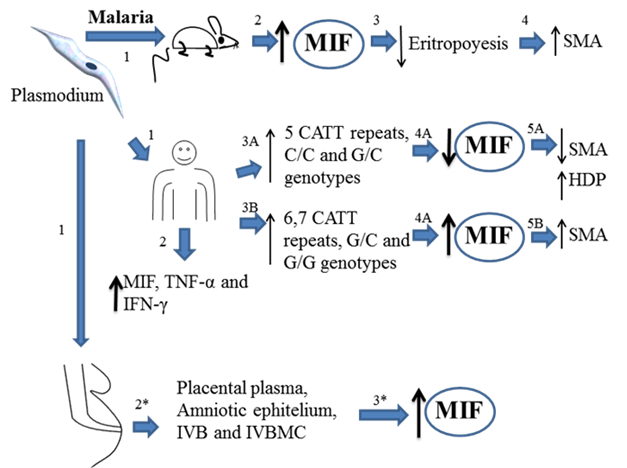
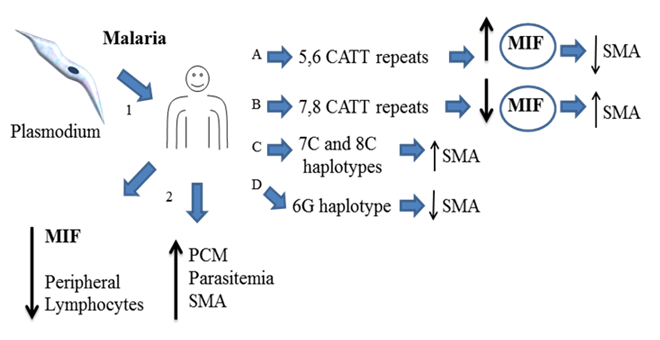
Although much research has been conducted in humans, the role of MIF in malaria remains unclear partly due to genetic variability, as malaria has exerted selective pressure on the human genome [70]. Interestingly, some studies have attributed the variations in MIF circulating levels and susceptibility to severe malaria-induced anemia to the tetranucleotide short tandem repeat polymorphism (STRP) MIF-794 CATT and the single nucleotide polymorphism (SNP) MIF-173 G/C allele [8, 71-83]. The -173 C allele is commonly associated with elevated MIF expression and increased susceptibility to inflammatory diseases [71-75, 77, 78, 80], while longer CATT repeats (>5) at MIF-794 can cause increased susceptibility [80] and irregular MIF production [74, 75, 79] in inflammatory diseases. Related to this, previous studies in Zambian children have shown that monocytes with longer CATT repeats (>5) were associated with high MIF production [60]. Subsequent studies in Zambian and Kenyan children demonstrated that carriers of longer (>5) CATT repeats at the MIF -794 allele and the CC genotype at the MIF -173 allele, were associated with increased susceptibility to high-density parasitemia [81, 82]. Recently, Awandare et al., in a study in Kenyan children, reported that the 7C and 8C MIF-794/-173 haplotypes and an increased number of CATT repeats (>5) in the MIF -794 allele were associated with increased susceptibility to severe malaria anemia (SMA) and a progressive decrease in circulating MIF concentrations. This group also discovered that the 6G haplotype and short CATT repeats were associated with decreased susceptibility to SMA and increased MIF production, suggesting that carriers of the disease-associated alleles may have decreased MIF levels in the blood [83]. Moreover, even though these findings indicate a high prevalence of the short 5/6, 7, and 8 CATT repeats at the -794 and -173 C alleles in Kenyan and Zambian children compared to Caucasians, which points to a selective pressure in these endemic regions by P. falciparum, further research will be necessary to clarify the inconsistencies in previous studies regarding MIF circulating levels and to show the clear involvement of MIF in either malaria resistance or pathology (Fig 2).
Overview about the role of MIF during Plasmodium infection. Theory 2. Human infected with plasmodium (1) down regulates the expression of MIF in peripheral lymphocytes (2), depending of the expressed MIF´s polymorphisms it may favors several malaria anemia (SMA) (B and C). In line with these observations high levels of MIF prevent SMA (A,D). All together indicate that MIF has a protective role against plasmodium infection and low levels of MIF increase the risk of developing SMA.
Leishmaniasis
Leishmaniasis is caused by protozoan parasites belonging to the genus Leishmania and approximately 350 million men, women and children in 88 countries are in danger of contracting the disease. As many as 12 million people may be currently infected, and an estimated 1-2 million new cases occur every year [84]. Generally, infection by these parasites can be divided into three main forms: cutaneous, mucocutaneous and visceral leishmanioses. The disease is prevalent in many tropical and subtropical regions of the world, where it is transmitted via the bite of an infected sandfly [85].
Previous reports have shown that MIF plays a critical role mediating host resistance to Leishmaniasis. Juttner et al. found in vitro that both recombinant murine or human MIF, at 1.5 and 2.5 μg/ml concentrations could activate macrophages to kill Leishmania major [86]. The use of anti-TNF-α antibody or TNF receptor p55-KO mice as well as L-NIL or iNOS-KO mice showed that leishmanicidal activity was mediated by TNF-α autocrine signaling and the production of nitric oxide metabolites by macrophages after MIF stimulation. Moreover, both susceptible BALB/c and resistant C57BL/6 mouse strains expressed high levels of MIF mRNA in lymph nodes draining the site of L. major infection. Related to this, in a study by Satoskar et al., experimental L. major infection in C57BL/6 MIF KO and wild-type (WT) mice showed that MIF KO mice were more susceptible to disease due to significantly larger lesions and greater parasite burdens than WT mice. However, similar proliferative responses and production of IL-4 and IFN-γ cytokines by lymph nodes were observed when stimulated in vitro with L. major antigen (LmAg). In addition, IFN-γ stimulated Mø from MIF KO mice had a defective leishmanicidal activity, which was associated with low superoxide and nitric oxide levels, and high IL-6 levels. This finding suggests that susceptibility of MIF KO mice to L. major infection is due to an impaired macrophage leishmanicidal activity rather than deregulation of Th1 and Th2 responses [87]. Furthermore, Xu et al. demonstrated that oral administration of attenuated Salmonella-transfected plasmids encoding the murine MIF gene and the TNF-α gene enhanced the resistance of BALB/c mice infected with L. major, and this resistance was associated with high iNOS expression compared to other plasmids encoding the MIF, TNF-α or IFN-γ genes alone. This indicates that MIF may play an important role in the resistance to leishmaniasis, likely by synergizing with other inflammatory cytokines, such as TNF-α via the induction of iNOS expression [88]. Interestingly, Kozaci et al. reveled that human patients with acute cutaneous leishmaniasis caused by L. tropica showed high MIF levels compared to healthy subjects [89]. Moreover, a recent study by Santos-Oliveira J.R. et al. in visceral leishmaniasis (VL) patients, the authors demonstrated that high LPS levels from a possible microbial translocation due to damage by visceral leishmaniasis (VL) was correlated with increased pro-inflammatory cytokines (MIF, IFN-γ, TNF-α, IL-2, IL-6, IL-12, IL-17) and low numbers of CD4+ and CD8+ T cells, suggesting a strong lymphocyte activation by the proinflammatory cytokine storm that lead to lymphocyte death by apoptosis, therefore MIF may contribute in the inmuno-pathogenesis by LPS stimulation during VL [90]. In addition, a clinical study by Bimal et al. reported that CD2+ T-cells isolated from patients with active visceral leishmaniasis (VL) produced high levels of MIF after anti-CD25 stimulation in vitro. Moreover, after anti-leishmanicidal treatment, patients with VL showed higher numbers of CD2+ T-cells, indicating a possible role for CD2+ T cells and MIF in the immune response to VL [91]. Related to this, Kar, Metz, and McMahon-Pratt discovered that P-4 antigen-vaccinated BALB/c mice presented without lesions or detectable parasites after Leishmania pifanoi infection. This protection was shown to be CD4+ T cell-dependent and lead to the production of MIF, TNF-α and IFN-γ, but not IL-4. Moreover, the addition of antibodies against either MIF, TNF-α or IFN-γ separately or simultaneously, to macrophages from P-4-immunized mice in vitro, decreased leishmanicidal activity, suggesting that MIF potentially contributes to the control of L. pifanoi infection in P-4-vaccinated mice [92]. Together, these findings demonstrate a crucial role for MIF in the cellular immune response against leishmaniasis. MIF appears to be capable of killing Leishmania parasites by acting directly or indirectly with other cytokines, such as TNF-α and reactive products like nitric oxide. However, additional human studies are needed to confirm this data and, more importantly, to elucidate the molecular mechanisms of how MIF induces its leishmanicidal activity.
MIF in Leishmaniosis
| Parasite | Model | Disease | in vitro/ in vivo | Treatment | Properties | MIF role | Ref. |
|---|---|---|---|---|---|---|---|
| L. major | BALB/c mice C57BL/6 mice | CL | in vitro | rhMIF rmMIF, anti-TNF-α, | ↓Infected Mø ↑ TNF-α by PECS ↓amastigotes in Mø | Induces a leishmanicidal activity in MO mediated by TNF-α and NO | [86] |
| TNFα-KO p55-KO iNOs KO | TNF-α KO TNF-αR55 KO (+ rMIF) L-NIL or iNOS KO+ MIF | ↓MIF | Mediate L. major killing in Mø | ||||
| MIF + IFN-γ | ↑NO by Mø | Mediated leishmanicidal function | |||||
| TGF-β, IL-13 and IL-10 (+ rMIF) | ↓ MIF | ||||||
| Leishmania Ag or Con A | ↑MIF in lymph node cells | ||||||
| in vivo | ↑MIF mRNA in lymph nodes | [62][62] | |||||
| C57BL/6 MIF-KO mice | CL | in vivo | ↑ Large lesions ↑ parasites | Induce in Møs leishmanicidal activity by up-regulating iNOS and NO | [87] | ||
| in vitro | IFN-γ | ↓leishmanicidal activity ↓NO and ↑IL-6 by Mø | |||||
| BALB/c mice | CL | in vivo | Oral S. typhimurium transfected with plasmid encoding IFN-γ or MIF or TNF-α | ↓lesion size and parasites ↑iNOS in spleen cells | Therapeutic effect Synergizes with TNF-α limiting the replication of Leishmania via NO synthesis. | [88] | |
| L. tropica | Turkish patients | CL | in vivo | ↑MIF in serum | Favors cellular immune response | [89] | |
| Leishmania infantum chagasi | Human | Viseral Leishmaniasis (VL) | In vivo | ↑LPS levels in plasma correlated to ↑MIF, IL-6, ↑IL-8 ,↑IFABP levels, ↑sCD14, ↑% of TCD3 HLA-DR and ↑% of CD4+ CD25+ compared to Healthy Subjects (HS) ↓CD4+ and CD8+ T cells but ↑cellular activation. ↑IFN-γ, TNF-α, IL-2, IL-6, IL-12, IL-17 and MIF) ↑IL-4, IL-5, IL-13 and IL-10 ↑Chemokines (IL-8, MCP-1, MIP-1β) compared to HS. | MIF production may contribute to the inflammatory response in VL patients, and the increased susceptibility to the inmunophatogenesis caused by LPS stimulation. | [90] | |
| Anti-Leishmania Therapy | ↓LPS in plasma, ↑CD4+ and CD8+ T cells, ↓sCD14, ↓MIF, ↓IFABP levels and ↓Pro-inflammatory cytokines compared to active VL patients. | ||||||
| L. donovani | Indian patients | VL | in vivo | ↓TCD2+, active VL ↑TCD2+, cured VL | Important role for TCD2+ and MIF in immunity to VL | [91] | |
| in vitro | Anti-cd2 | ↑ MIF, by T cells from active VL | |||||
| L. pifanoi | BALB/C mice | CL | in vivo | P-4 antigen vaccination | ↓lesions ↑CD8+ and CD4+cells ↑MIF, IFN-γ and TNF-α mRNA in lymph node cells | Contributes in the control of infection in P-4 antigen vaccinated mice | [92] |
| P-4 vaccination and CD4+cells depletion | ↓parasite ↓IFN-γ and IL-2 in T cells | ||||||
| in vitro | Ab anti-MIF IFN-γ and TNF-α | ↓Leishmanicidal activity in Møs from P-4 immunized mice |
recombinant human MIF (rhMIF); recombinan murine MIF (rmMIF); specific inhibitor of the inducible nitric oxide synthase (L-NIL); = cutaneous leishmaniosis (CL); Viceral leishmaniosis (VL); (TCD2)
Trypanosomiasis
Trypanosomiasis is caused by protozoan parasites of the genus Trypanosoma. The two major diseases caused by these organisms are sleeping sickness in Africa and Chagas disease in America. Chagas disease is a zoonosis (or an anthropozoonosis) caused by the flagellated protozoan parasite Trypanosoma cruzi. It is transmitted to man by the infected feces of a blood-sucking triatomine bug through the insect sting. An estimated 18 million people are infected worldwide, and most cases occur in Latin America, where Chagas disease is endemic. More than 100 million people are at risk of contracting the disease [93]. Sleeping sickness is a parasitic disease transmitted by the bite of the 'Glossina' insect, commonly known as the tsetse fly. This disease threatens more than 55 million people in 36 countries with an estimated 300,000 new cases per year in Africa alone [94].
There are few, but notable, papers regarding the role of MIF in trypanosomiasis. Our group has found that infected (Trypanosoma cruzi or the Queretaro strain) MIF KO mice displayed high levels of blood and tissue parasitemia, which was associated with severe heart and skeletal muscle immunopathology. Inflamed hearts from T. cruzi-infected MIF KO mice showed high amastigotes nests and expressed high IFN-γ transcripts, but fewer IL-12, p35, IL- 12, p40, IL-23 and iNOS transcripts. Further, these mice succumbed to T. cruzi infection faster than WT mice. This enhanced susceptibility in MIF KO mice was associated with an important reduction of pro-inflammatory cytokines in the sera during early infection. TNF-α, IL-12, IL-18, IFN-γ and IL-1β levels correlated with the impaired spleen cell production of pro-inflammatory IL-12 and IFN-γ, but anti-inflammatory IL-4 and IL-10 cytokine levels were not elevated, demonstrating that the increased susceptibility was not due to high production of anti-inflammatory cytokines. Therefore, MIF may be required for the optimal activation of host innate responses by up-regulating inflammatory cytokines that are crucial for resistance to T. cruzi infection [95]. In relation to this, a recent study by Nishimura K et al reported that MIF, GM-CSF, IL-12 and TNF mRNA are highly expressed in spleen macrophages during Trypanosoma brucei brucei (ILS) infection, in contrast to Trypanosoma gambiense welcome strain (WS) infection, the authors explained that this defference may be due to a possible macrophage activation during ILS infection and a macrophage immunosupression during WS infection, probably by differences in charateristics between WS and ILS, these findings indicate that MIF is involved in the inflammatory immune response against Trypanosomiasis and this involvement may depend on the parasite strain [96]. Additionally, Paiva et al. reported a lethal synergism between T. cruzi infection and LPS-induced shock, where MIF and TNF-α play an important role. Briefly, BALB/c mice infected with the T. cruzi Y strain after an LPS injection showed enhanced susceptibility to endotoxemia. This was in contrast to TNF KO mice treated with anti-MIF, indicating that increased susceptibility to endotoxemia is due to T. cruzi induced MIF and TNF-α, suggesting again that MIF may act synergistically with the TNF pathway to induce pro-inflammatory responses during early infection [97]. Interestingly, hearts and skeletal muscle from infected mice (RA T. cruzi strain) showed early MIF mRNA induction, which was accompanied by enhanced transcripts for cytokines, chemokines and chemokine receptors, such as TNF-α, IFN-γ, CCL5, CXCL9, CCR5, CXCR3 and iNOS. These results lead to the consideration of MIF as part of the primary response against this parasite, and that it acts by collaborating with other cytokines and chemokines in the inflammation processes of infected organs and tissues [98]. In addition, a recent study reported that, in T. brucei infected C57BL/6 mice, MIF, together with important chemokines (CXCL9, 10, CCL2, 3, 4, and 5 mRNA), are significantly induced during the first, and important, peak of parasitemia and the highest expansion of CD11b+Ly6C+ monocytic cells in the liver. However, studies in MIF KO infected mice showed that the egression of CD11b + Ly6C + monocytes from the bone marrow to the blood is MIF-independent, but CCR2-dependent, indicating that MIF does not participate in monocyte recruitment. However, it may be possible that the early induction of MIF in the liver better regulates inflammatory cytokine production. Together, these studies designate an important role for MIF in the induction of a resistant innate immune response to T. cruzi infection. Clinical studies in humans will be necessary to confirm these data [99].
MIF in Trypanosomiosis
| Parasite | Model | Disease | in vitro/ in vivo | Treatment | Properties | MIF role | Ref. |
|---|---|---|---|---|---|---|---|
| T. cruzi Ninoa | WT MIF-KO BALB/c mice | Acute trypanosomiasis | in vivo | ↑ parasitemia ↑Mortality | Activation of innate immunity, inducing inflammatory cytokines | [95] | |
| T. cruzi Queretaro | WT MIF-KO BALB/c mice | Acute trypanosomiasis | in vivo | ↑parasitemia ↑Mortality ↓IL-12, IFN-γ, TNF-α, IL-18, IL-1β, iNOS mRNA ↑pathology in heart and skeletal muscle ↓IgG2a Ab | Activation of innate immunity, inducing inflammatory cytokines | [95] | |
| in vitro | T. cruzi Ag | ↓IL-12 and IFN-γ by spleen cells | |||||
| T. brucei brucei (ILS) | Wistar Rat | Trypanosomiasis | In vivo | Unchanged parasitemia and No. of spleen Møs compared to WS-infected rats. ↑IL-12, ↑TNF, ↑GM-CSF and ↑MIF mRNA but ↑IL-10 expression by spleen Møs from ILS-infected rats (at 4 days p.i.) compared to uninfected ↑ Phagocitic activity, ↑IL-12, ↑TNF↑GM-CSF and ↑MIF mRNA but ↓IL-10 expression by spleen Møs from infected rats (at 4 days p.i.) compared to WS infected rats. | MIF is highly expressed and may participate in the immune response to ILS infection, in contrast to WS infection due to a possible Møs immunosupression during WS infection. | [96] | |
| In vitro | ILS infection | ↑ Phagocitic activity, ↑GM-CSF, ↑M-CSF and ↑MIF but ↓IL-10 mRNA expression by ILS infected spleen Møs commpared to uninfected Møs. Similar results were obtained using HS-P cells | |||||
| ILS infection | ↑ Phagocitic activity but similar GM-CSF, M-CSF and MIF mRNA expression by ILS infected spleen Møs commpared to WS infected Møs. Similar results were obtained using HS-P cells | ||||||
| Cobalt Chloride | ↑GM-CSF and ↑MIF but ↓M-CSF by ILS infected spleen Møs commpared to WS infected Møs treated with cobalt chloride. Similar results were obtained using HS-P cells. | ||||||
| T. brucei gambiese welcome strain (WS) | Wistar Rat | Trypanosomiasis | In vivo | ↑M-CSF mRNA expression by spleen Møs from WS-infected rats (at 4 days p.i.) compared to uninfected ↑IFN-γ, ↑IL-10, ↑M-CSF but ↓ Phagocytic activity ↓MIF ↓GM-CSF ↓IL-12 and ↓TNF mRNA expression by spleen Møs from infected rats (at 4 days p.i.) compared to ILS infected rats ↑IL-12, TNF,IFN-γ and IL-10 mRNA expression by spleen Møs from infected rats (at 4 days p.i.) compared to uninfected | [96] | ||
| In vitro | WS infection | ↑ M-CSF, ↑GM-CSF and ↑MIF mRNA expression by WS infected spleen Møs compared to uninfected Møs. Similar results were obtained using HS-P cells ↓Phagocitic activity but similar GM-CSF, M-CSF and MIF mRNA expression by WS infected spleen Møs commpared to ILS infected Møs. Similar results were obtained using HS-P cells | |||||
| Cobalt chloride | ↓ GM-CSF and ↓MIF but unchanged phagocytic activity and M-CSF expression by WS infected spleen Møs commpared to ILS infected Møs treated with cobalt chloride. Similar results were obtained using HS-P cells. | ||||||
| T. cruzi Y strain | BALB/c mice | Acute trypanosomiosis | in vivo | LPS | ↑LethalShock ↑Mortality | MIF or TNF-R1 induced by T. cruzi are sufficient to cause death of infected mice upon LPS induced shock | [97] |
| T. cruzi RA strain | BALB/c mice | Acute trypanosomiosis | in vivo | ↑MIF, TNF-α, IFN-γ CCL5, CXCL9, CCR5, CXCLR3 ↑ iNOS in hearts ↑MIF,TNF-α, CCL5, CXCL9, IFN-γ, iNOS, CCR5 and CXCLR3 in skeletal muscle | ↑MIF in the primary response against the parasite and it is present in association with a state of muscle inflammation and rhabdomyolysis shortly after infection | [98] | |
| T. brucei | C57BL/6 mice | Acute trypanosomiosis | ↑MIF, CCL2,3,4,5 and CXCL9 | MIF don't participate in the recruitment of CD11+LY6+ cells | |||
IgG2a antibody (IgG2a Ab); CC chemokines ligand (CCL); chemokine receptor (CCR); CX chemokines ligand (CXCL).
Toxoplasmosis
Toxoplasmosis is an infectious disease caused by Toxoplasma gondii, an obligate intracellular parasite member of the phylum Aplicomplexa. Immunocompromised patients are commonly affected in the brain, as well as other organs, such as the lungs, heart and eyes. Moreover, congenital toxoplasmosis is acquired if a women contracts a primary infection while pregnant [100]. The incidence of congenital toxoplasmosis varies between countries. For example, the incidence is as high as 10 per 1,000 individuals in France [101] but only five per 10,000 in the UK [102] and one per 10,000 in the USA [103]. Interestingly, approximately one-third of the world's population is seropositive for this parasite [104].
We found that MIF KO BALB/c mice succumbed faster to challenge with virulent (RH) and avirulent (ME49) strains of T. gondii compared to WT mice. This susceptibility was associated with a high percentage of infected peritoneal macrophages, greater numbers of cysts in brain, severe liver damage and defective early production of proinflammatory cytokines (IL-12, IL-1β, TNF-α, IL-18, IFN-γ and nitric oxide). Moreover, both WT and MIF KO mice showed comparable levels of anti-inflammatory cytokines (IL-4 and IL-10) in the sera. In contrast, we observed that the lack of MIF alters the function of CD11c+ DCs, which produce low IL-12 and TNF-α levels. Similarly, low transcript levels for IL-12, IL-1β and TNF-α were found in bone marrow dendritic cells (BMDCs) that were stimulated in vitro with STAg. Interestingly, brain tissue from patients who died of cerebral toxoplasmosis showed scarce MIF production compared to patients who died of an encephalitic cryptococcal infection. Thus, MIF appears to play a critical role in promoting resistance to toxoplasmosis by regulating the early production of innate pro-inflammatory cytokines. Additionally, lower expression of TNF-αR, IFN-γR and TLR-4 was found in PECs from infected MIF KO mice, suggesting that the absence of MIF renders cells unable to respond rapidly to T. gondii. This is supported by the defective recognition of both proinflammatory cytokines and the parasite in MIF KO mice. These results are consistent with a later study in orally infected MIF KO mice [105], demonstrating that the increased mortality in T. gondii infected (ME49 strain) MIF KO mice is due to uncontrolled parasite replication and dissemination in brain and liver. This phenomenon was associated with low expression of MHC-II and co-stimulatory molecules (CD40, CD80 and CD86) in mononuclear lymphoid nude (MLN) and splenic DCs during the early infection. In addition, low IL-12 production by MIF KO DCs was measured compared to DCs from similarly infected or STAg intravenous-injected WT mice. Moreover, MIF KO BMDCs stimulated in vitro with STAg or LPS showed lower expression of the parasite recognition receptor TLR11, proinflammatory cytokines (IL-12, TNF-α, IL-1β) and nitric oxide. Interestingly, these expression deficiencies were restored upon the addition of exogenous rMIF, indicating that interaction with MIF during oral T. gondii infection is required for DC maturation and the induction of a proinflammatory protective immune response. Additionally, a recent study has demonstrated that MIF production by chorionic villous explants from human placentas is upregulated when stimulated with STAg, particularly in the villous trophoblast. Upon release, MIF induces the dose-dependent expression of ICAM-1, primarily in the syncytiotrophoblast, leading to the adhesion of monocytes (THP-1) on the surface of villous explants. This demonstrates that MIF plays an essential role as an autocrine/paracrine mediator in T. gondii placental infection [51]. Related to this, de Oliveira Gomes, A., et al, reported that MIF is upregulated by first-trimester placental explants and is important to control T. gondii infection due to a decreased parasite load at this gestational stage, in contrast to third trimester placental explants which showed increased susceptibility to infection, suggesting that variations in MIF regulation during gestation have an important role in differential susceptibility to T. gondii infection [106]. These findings evidence the pleiotropic role for MIF, including the maturation of important antigen presenting cells, like DCs, the expression of parasite recognition receptors, the expression of adhesion molecules, like ICAM-1, and inducing pro-inflammatory cytokines. Together, these activities are crucial for the development of a protective innate immune response to T. gondii infection.
MIF in Toxoplasmosis
| Parasite | Model | Disease | in vitro/ in vivo | Treatment | Properties | MIF role | Ref. |
|---|---|---|---|---|---|---|---|
| T. gondii RH ME49 strains | BALB/c MIF-KO | Acute Toxoplas-mosis | in vivo | ↑Mortality compared to MIF +/+ ↓IL-12, IL-1β, TNF-α, IL-18 and IFN-γ in sera ↓CCR5 in PECs ↑Mortality ↑Parasited Mø ↑Cystis in brain ↑ALT and AST (Liver damage) TNF-αR PECS ↓ IFN-γR in splenocytes ↓IL-12 and TNF-α on CD11c+ LOD DCs. | Promotes resistance inducing early production of pro-inflammatory cytokines, and TLR11expression | [121] | |
| in vitro | STAg or LPS | IL-12p35, IL-12p40, IL-12p19, IL-1β and TNF-α in BMDCs | |||||
| T. gondii RH strain | C57BL/6 | in vivo | ↓MIF in sera | ||||
| T. gondii | Human | in vivo | ↓MIF in brains from patients who died by encephalitic | ||||
| T. gondii ME49 strain | Murine BALB/c MIF-KO | Acute Toxoplasm-osis | in vivo | ↑Mortality ↑Brain cysts ↑Parasitemia and ↓Inflammation infiltrates in liver during early infection. ↓MHCII, CD80, CD86 and CD40 expression in MLN DCs and splenic DCs. ↓ MLN DCs producing IL-12 | Modulates the innate immunity to T. gondii oral infection by regulating maturation and cytokine production of DCs | [105] | |
| in vivo | STAg via intravenous | ↓IL-12 in sera ↓IL-12 by splenocytes. ↓ IL-12 by splenic DCs | |||||
| in vitro | STAg or LPS | ↓MHCII, CD86, CD80, CD40 and TLR11 expression in BMDCs | |||||
| STAg or LPS | ↓Allostimulatory ability of DCs (low CD4+ proliferation) | ||||||
| MIF+ STAg or LPS | ↑IL-12, TNF-α, IL-1β, ON and TLR11expression in BMDCs compared to STAg or LPS treatments | ||||||
| T. gondii RH strain | Human | Experimental placental toxoplasm-osis | Ex vivo | STAg | ↑MIF by CVE ↑Intracellular MIF in CVE ↑MIF in the Syncytiotrophoblast of CVE | Induces the ICAM-1 and adhesion of monocytes cells which may be an important mechanism for controlling T. gondii infection in the placental | |
| SPN or rMIF | ↑ICAM-1 in the syncytiotrophoblast of CVE. | ||||||
| SPN + anti-MIF antibodies | ↓ ICAM-1 in the syncytiotrophoblast of CVE. | ||||||
| SPN or SPN+ anti-MIF or rMIF | ↑Monocyte (THP-1) adhesion in the trophoblast of explants cultures | ||||||
| T. gondii RH | Human | Toxoplasm-osis | In vitro | ↑CD74R in syncytiotrophoblast and mesenchymal cells from uninfected first-trimester placental explants (FTPE) compared to Third-trimester placental explants (TTPE) | MIF is up-regulated in first-trimester explants in contrast to third-trimester placental explants and is important to control T. gondii infection. | [106] | |
| ↑MIF expression in cytotrophoblast layer by uninfected FTPE compared to TTPE | |||||||
| IFN-γ or IL-12 | ↑MIF release by uninfected FTPE compared to untreated | ||||||
| IL-10 or TGF-β | Un changed MIF discharge by uninfected FTPE | ||||||
| ↑MIF release by infected FTPE compared to uninfected or infected TTPE | |||||||
| ↑↑MIF release by infected FTPE | |||||||
| IL-10 and TGF-β | ↑MIF release by infected FTPE | ||||||
| ↑MIF expression in the Syncytiotrophoblast layer and mesenchyme from infected FTPE | |||||||
| rMIF | ↓Parasite load in FTPE | ||||||
| IL-12 | ↓Parasite load in FTPE | ||||||
| IFN-γ | ↓Parasite load in FTPE | ||||||
| IFN-γ | ↑MIF release by infected TTPE | ||||||
| ↑MIF expression in cytotrophoblast layer from infected TTPE |
Chorionic Villous explants (CVE); recombinant MIF (rMIF); Soluble Toxoplasma Antigen (STAg); Supernatants from explants cultures exposed to STAg (SNP).
Primary Amebic Meningoencephalitis
The genus Naegleria consists of a group of free-living ameboflagellates (FLA) found in diverse habitats throughout the world, such as freshwater lakes, ponds, domestic water supplies, swimming pools and thermal pools [107-109]. Although over 30 species of Naegleria have been isolated from environmental sources, only Naegleria fowleri has been isolated from humans. N. fowleri causes primary amebic meningoencephalitis (PAM). PAM is a rapidly fatal disease affecting the central nervous system (CNS) that occurs most often in immune-competent individuals, such as children and young adults with a history of swimming and diving in freshwater [110].
Little is known about the role of MIF during amoebic parasite infection; however, a pioneer study by Cursons et al. reported that lymphocytes stimulated with N. fowleri, N. jadini and N. gruberi antigens released MIF, which inhibited the migration of peritoneal exudate cells. Moreover, guinea pigs sensitized to the three species of Naegleria developed typical delayed type hypersensitivity (DTH) reactions when inoculated intradermally with amoeba antigens [111]. In addition Haq, A. et al, showed that Peritoneal cells from hamsters sensitized with Entamoeba histolytica antigens (EHA) produced high MIF levels when stimulated in vitro with amoeba antigens, moreover the production of MIF was found to be greatly increased in animals sensitizing by EAH plus glucan [112]. MIF may be participating in the resistance to PAM and amebiasis; however, this is merely the beginning of a series of studies that aim to reveal the role of MIF, and may be helpful in the development of therapeutic treatments for this disease.
MIF in Amebiosis
| Parasite | Model | Disease | in vitro in vivo | Treatment | Properties | MIF role | Ref. |
|---|---|---|---|---|---|---|---|
| Naegleria flowleri Naegleria jadini | Guinea pigs | PMA | in vivo | Naegleria Ag + Freund adjuvant | ↑Infiltration of mononuclear cells in lesions | Favors the resistance to infection(MIF is involved in the immune response against PMA) | [111] |
| Naegleria gruberi | in vitro | ↑ MIF supernatants of Lymphocytes stimulated with Naegleria Ag | |||||
| Entamoeba histolytica | Hamsters | Amoebiasis | In vitro | Entamoeba histolytica antigens | ↑ MIF in supernatants of Peritoneal cells from E. histolytica-sensitized hamsters | Favors the resistance to infection (MIF is involved in the immune response against amebiasis) | [112] |
| Entamoeba histolytica antigens + glucan | ↑↑ MIF in supernatants of Peritoneal cells from E. histolytica-sensitized hamsters |
Primary Amebic Meningoencephalitis (PMA)
Neosporosis
MIF has achieved great relevance for groups aiming to study the immune response against intracellular parasite infection. Recently, it has been studied in neosporosis [113], an infectious disease caused by Neospora caninum, an obligate intracellular apicomplexa parasite, closely related to T.gondii [114]. Neosporosis is recognized as a major cause of spontaneous abortion in cattle worldwide [114], and it has been proposed that humans are probable hosts for this parasite [115, 116].
Carvalho et al. found that after an in vitro Neospora caninum infection, both human uterine cervical HeLa and choriocarcinoma BeWo cells expressed high MIF levels. In this study, although BeWo cells expressed lower MIF levels than HeLa cells, BeWo cells presented a lower index of infection, decreased cell lysis, reduced parasite replication, more viability and higher TGF-β levels than HeLa cells. Aditionally, pretreatment with IFN-γ reduced infected in BeWo cells, parasite replication in HeLa cells, TGF-β levels in BeWo cells and increased MIF levels in both cell types. Using IDO-specific inhibitors (1-MT), IFN-γ-mediated inhibition of parasite growth in HeLa cells was demostrated to be IDO enzyme dependent. Moreover, pretreatment with N. caninum lysate antigen (NLA) increased MIF levels in HeLa cells, but decreased TGF-β production in BeWo cells [113]. Given the increased MIF production by both cell types in response to IFN-γ and that N. caninum infection and NLA administration correlates with a low infection index and parasite replication, these results suggest that MIF may play also a role in the inmune response against N. caninum infection in the placenta.
Coccidiosis
Avian coccidiosis is an intestinal infection caused by apicomplexan protozoa belonging to at least seven different species of Eimeria [117]. In nature, eimerian infections occur when hosts ingest sporulated oocysts, resulting in a mild or severe disease, characterized by anorexia, digestive disturbances, dehydration, blood loss, and increased susceptibility to other disease agents [118]. Coccidiosis is an important disease economically due to morbidity, mortality and reduced production efficiency of poultry and other livestock. It is estimated to be responsible for monetary losses greater than $3 billion annually [119].
Previous studies regarding the pleiotropic role of MIF have revealed its potential use as a therapeutic agent for a variety of human parasite infections; however, its role in domestic animals like poultry has only recently been investigated. Interestingly Hong et al. found increased transcripts for MIF and other chemokines (MIP-1β, K203, IL-8 and limphotactin), pro-inflammatory cytokines (IFN-γ, IL-1β, IL-6, IL-12, IL-15 and IL-17) and anti-inflammatory cytokines (IL-3, IL-4, IL-10, IL-13 and GM-CSF) in intraepithelial lymphocytes (IELs) from the intestinal jejunum of chickens during experimental Eimeria maxima infection. In addition, the authors reported high expression of the surface markers K1, CD3, CD4, CD8, TCR1 and TCR2 from different IELs subpopulations [120], which were similar to those found in experimental Eimeria acervulina and Eimeria tenella infections from an additional study [117]. Therefore, MIF, together with other chemokines, cytokines and IEL subpopulations, mediates host defense mechanisms against Eimeria infection. However, the role of MIF as a chemokine or a potent inducer of proinflammatory cytokines will need further clarification.
Concluding remarks
Since the discovery of MIF, we have witnessed slow progress in our understanding of the MIF-mediated immune mechanisms in infectious diseases, such as the protozoan parasites that cause major human diseases in the developing world, including malaria, human African trypanosomiasis, Chagas disease, leishmaniasis and toxoplasmosis. Although the life cycles of these parasites were defined years ago, specific therapeutic or preventive interventions are not yet available, and better understanding of the immune response to these protozoans is required. Recently, MIF has been considered a key player in protozoan infections, where it plays a pivotal role in early innate immunity rather than favoring the adaptive immune response. However, the role MIF plays in the response to protozoan infection is complex. For example, high levels of MIF can be dangerous for the host during malaria infections (Fig 2), on the other hand, MIF appears to be necessary to successfully control leishmaniasis, trypanosomiasis, toxoplasmosis, neosporosis, coccidiosis and meningoencephalitis (Fig 3). Together, the studies described in this review suggest that MIF participates in regulating the expression of some important molecules of the early immune response to different protozoa infections. However, the exact mechanism by which MIF acts during the immune response against parasites has not yet been defined. MIF signaling pathways have not been demonstrated for the activation of innate immunity during protozoa infections. Understanding the complex nature of these innate immunity responses will be necessary for the development of strategies to target MIF therapeutically, or for the development of a novel adjuvant or vaccine against protozoan infections.
Overview of MIF in Protozoan infections. Infected mice with intracellular protozoan (exception plasmodium) parasites (1) have high levels of MIF in serum (3) produced mainly by the immune cells (2). MIF acts in paracrine and autocrine manner (4) favoring the expression of proinflammatory cytokines and its receptors, chemokines, parasites recognition receptors and costimulatory molecules on antigen presenting cells (5). These conditions favor the microbicidal activity by macrophages, cellular chemiotaxis, parasite recognition and efficient antigen presentation (6). All together indicate that MIF has a protective role against protozoan infections (7).
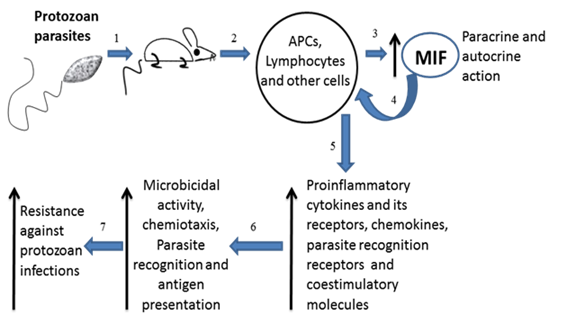
Acknowledgements
We thank Imelda Juarez by collecting the items required for this article. J. D. R.. is fellow of the National Council of Science and Technology (CONACYT). The author's research is supported by the grants ICYT-DF and UNAM-PAPIIT IN213009-3.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. David J.R. Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc Natl Acad Sci U S A. 1966;56(1):72-7
2. David J.R, Paterson P.Y. In vitro demonstration of cellular sensitivity in allergic encephalomyelitis. J Exp Med. 1965;122(6):1161-71
3. Bozza F.A, Gomes RN, Japiassú AM. et al. Macrophage migration inhibitory factor levels correlate with fatal outcome in sepsis. Shock. 2004;22(4):309-13
4. Yang N, Nikolic-Paterson DJ, Ng YY. et al. Reversal of established rat crescentic glomerulonephritis by blockade of macrophage migration inhibitory factor (MIF): potential role of MIF in regulating glucocorticoid production. Mol Med. 1998;4(6):413-24
5. Leech M, Lacey D, Xue JR. et al. Regulation of p53 by macrophage migration inhibitory factor in inflammatory arthritis. Arthritis Rheum. 2003;48(7):1881-9
6. Cvetkovic I. et al. Critical role of macrophage migration inhibitory factor activity in experimental autoimmune diabetes. Endocrinology. 2005;146(7):2942-51
7. Powell N.D, Papenfuss TL, McClain MA. et al. Cutting edge: macrophage migration inhibitory factor is necessary for progression of experimental autoimmune encephalomyelitis. J Immunol. 2005;175(9):5611-4
8. Mizue Y, Ghani S, Leng L. et al. Role for macrophage migration inhibitory factor in asthma. Proc Natl Acad Sci U S A. 2005;102(40):14410-5
9. Fukuyama S, Yoshino I, Yamaguchi M. et al. Blockage of the macrophage migration inhibitory factor expression by short interference RNA inhibited the rejection of an allogeneic tracheal graft. Transpl Int. 2005;18(10):1203-9
10. de Jong Y.P, Abadia-Molina AC, Satoskar AR. et al. Development of chronic colitis is dependent on the cytokine MIF. Nat Immunol. 2001;2(11):1061-6
11. Lin S.G, Yu XY, Chen YX. et al. De novo expression of macrophage migration inhibitory factor in atherogenesis in rabbits. Circ Res. 2000;87(12):1202-8
12. Lan H.Y, Bacher M, Yang N. et al. The pathogenic role of macrophage migration inhibitory factor in immunologically induced kidney disease in the rat. J Exp Med. 1997;185(8):1455-65
13. Boyce N.W, Tipping PG, Holdsworth SR. Lymphokine (MIF) production by glomerular T-lymphocytes in experimental glomerulonephritis. Kidney Int. 1986;30(5):673-7
14. Mikulowska A, Metz CN, Bucala R. et al. Macrophage migration inhibitory factor is involved in the pathogenesis of collagen type II-induced arthritis in mice. J Immunol. 1997;158(11):5514-7
15. Denkinger C.M, Denkinger M, Kort JJ. et al. In vivo blockade of macrophage migration inhibitory factor ameliorates acute experimental autoimmune encephalomyelitis by impairing the homing of encephalitogenic T cells to the central nervous system. J Immunol. 2003;170(3):1274-82
16. Matsui Y, Okamoto H, Jia N. et al. Blockade of macrophage migration inhibitory factor ameliorates experimental autoimmune myocarditis. J Mol Cell Cardiol. 2004;37(2):557-66
17. Bernhagen J, Calandra T, Mitchell RA. et al. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365(6448):756-9
18. Bacher M, Meinhardt A, Lan HY. et al. Migration inhibitory factor expression in experimentally induced endotoxemia. Am J Pathol. 1997;150(1):235-46
19. Calandra T, Echtenacher B, Roy DL. et al. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med. 2000;6(2):164-70
20. Bozza M, Satoskar AR, Lin G. et al. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J Exp Med. 1999;189(2):341-6
21. Bernhagen J, Calandra T, Cerami A. et al. Macrophage migration inhibitory factor is a neuroendocrine mediator of endotoxaemia. Trends Microbiol. 1994;2(6):198-201
22. Bernhagen J, Calandra T, Bucala R. The emerging role of MIF in septic shock and infection. Biotherapy. 1994;8(2):123-7
23. Pollak N, Sterns T, Echtenacher B. et al. Improved resistance to bacterial superinfection in mice by treatment with macrophage migration inhibitory factor. Infect Immun. 2005;73(10):6488-92
24. Calandra T, Spiegel LA, Metz CN. et al. Macrophage migration inhibitory factor is a critical mediator of the activation of immune cells by exotoxins of Gram-positive bacteria. Proc Natl Acad Sci U S A. 1998;95(19):11383-8
25. Ohkawara T, Miyashita K, Nishihira J. et al. Transgenic over-expression of macrophage migration inhibitory factor renders mice markedly more susceptible to experimental colitis. Clin Exp Immunol. 2005;140(2):241-8
26. Waeber G, Calandra T, Roduit R. et al. Insulin secretion is regulated by the glucose-dependent production of islet beta cell macrophage migration inhibitory factor. Proc Natl Acad Sci U S A. 1997;94(9):4782-7
27. Lan H.Y, Mu W, Yang N. et al. De Novo renal expression of macrophage migration inhibitory factor during the development of rat crescentic glomerulonephritis. Am J Pathol. 1996;149(4):1119-27
28. Yabunaka N, Nishihira J, Mizue Y. et al. Elevated serum content of macrophage migration inhibitory factor in patients with type 2 diabetes. Diabetes Care. 2000;23(2):256-8
29. Bacher M, Meinhardt A, Lan HY. et al. MIF expression in experimentally-induced endotoxemia. Am J Phatol. 1997;150:235-246
30. Baugh JA, Bucala R. Macrophage migration inhibitory factor. Crit Care Med. 2002;30:27-35
31. Bernhagen J, Calandra T, Bucala R. Regulation of the immune response by macrophage migration inhibitory factor: biological and structural features. J Mol Med. 1998;76(3-4):151-61
32. Calandra T, Bernhagen J, Metz CN. et al. MIF as a glucocorticoid-induced modulator of cytokine production. Nature. 1995;377(6544):68-71
33. Sprong T. et al. Macrophage migration inhibitory factor (MIF) in meningococcal septic shock and experimental human endotoxemia. Shock. 2007;27(5):482-7
34. Calandra T, Bernhagen J, Mitchell RA. et al. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J Exp Med. 1994;179(6):1895-902
35. Bacher M, Metz CN, Calandra T. et al. An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc Natl Acad Sci U S A. 1996;93(15):7849-54
36. Donnelly S.C, Haslett C, Reid PT. et al. Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nat Med. 1997;3(3):320-3
37. Makita H, Nishimura M, Miyamoto K. et al. Effect of anti-macrophage migration inhibitory factor antibody on lipopolysaccharide-induced pulmonary neutrophil accumulation. Am J Respir Crit Care Med. 1998;158(2):573-9
38. Bernhagen J, Mitchell RA, Calandra T. et al. Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF). Biochemistry. 1994;33(47):14144-55
39. Mitchell RA, Metz CN, Peng T. et al. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999;274(25):18100-6
40. Mitchell RA, Liao H, Chesney J. et al. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci U S A. 2002;99(1):345-50
41. Roger T, David J, Glauser MP. et al. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature. 2001;414(6866):920-4
42. Roger T, Froidevaux C, Martin C. et al. Macrophage migration inhibitory factor (MIF) regulates host responses to endotoxin through modulation of Toll-like receptor 4 (TLR4). J Endotoxin Res. 2003;9(2):119-23
43. Roger T, Miconnet I, Schiesser AL. et al. Critical role for Ets, AP-1 and GATA-like transcription factors in regulating mouse Toll-like receptor 4 (Tlr4) gene expression. Biochem J. 2005;387(Pt 2):355-65
44. Petrovsky N, Socha L, Silva D. et al. Macrophage migration inhibitory factor exhibits a pronounced circadian rhythm relevant to its role as a glucocorticoid counter-regulator. Immunol Cell Biol. 2003;81(2):137-43
45. Isidori A.M, Kaltsas GA, Korbonits M. et al. Response of serum macrophage migration inhibitory factor levels to stimulation or suppression of the hypothalamo-pituitary-adrenal axis in normal subjects and patients with Cushing's disease. J Clin Endocrinol Metab. 2002;87(4):1834-40
46. Leech M, Metz C, Hall P. et al. Macrophage migration inhibitory factor in rheumatoid arthritis: evidence of proinflammatory function and regulation by glucocorticoids. Arthritis Rheum. 1999;42(8):1601-8
47. Hudson J.D, Shoaibi MA, Maestro R. et al. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999;190(10):1375-82
48. Hou G, Valujskikh A, Bayer J. et al. In vivo blockade of macrophage migration inhibitory factor prevents skin graft destruction after indirect allorecognition. Transplantation. 2001;72(12):1890-7
49. Demir Y, Chen Y, Metz C. et al. Cardiac allograft rejection in the absence of macrophage migration inhibitory factor. Transplantation. 2003;76(1):244-7
50. Huang X.R, Chun Hui CW, Chen YX. et al. Macrophage migration inhibitory factor is an important mediator in the pathogenesis of gastric inflammation in rats. Gastroenterology. 2001;121(3):619-30
51. Ferro E.A, Mineo JR, Ietta F. et al. Macrophage migration inhibitory factor is up-regulated in human first-trimester placenta stimulated by soluble antigen of Toxoplasma gondii, resulting in increased monocyte adhesion on villous explants. Am J Pathol. 2008;172(1):50-8
52. Miller L.H, Good M.F, Milon G. Malaria pathogenesis. Science. 1994;264(5167):1878-83
53. Biemba G, Gordeuk VR, Thuma PE. et al. Prolonged macrophage activation and persistent anaemia in children with complicated malaria. Trop Med Int Health. 1998;3(1):60-5
54. Biemba G, Gordeuk VR, Thuma P. et al. Markers of inflammation in children with severe malarial anaemia. Trop Med Int Health. 2000;5(4):256-62
55. Camargo L.M, Ferreira MU, Krieger H. et al. Unstable hypoendemic malaria in Rondonia (western Amazon region, Brazil): epidemic outbreaks and work-associated incidence in an agro-industrial rural settlement. Am J Trop Med Hyg. 1994;51(1):16-25
56. Walther M, Woodruff J, Edele F. et al. Innate immune responses to human malaria: heterogeneous cytokine responses to blood-stage Plasmodium falciparum correlate with parasitological and clinical outcomes. J Immunol. 2006;177(8):5736-45
57. Dodoo D, Omer FM, Todd J. et al. Absolute levels and ratios of proinflammatory and anti-inflammatory cytokine production in vitro predict clinical immunity to Plasmodium falciparum malaria. J Infect Dis. 2002;185(7):971-9
58. Martiney J.A, Sherry B, Metz CN. et al. Macrophage migration inhibitory factor release by macrophages after ingestion of Plasmodium chabaudi-infected erythrocytes: possible role in the pathogenesis of malarial anemia. Infect Immun. 2000;68(4):2259-67
59. Malu D.T, Bélanger B, Desautels F. et al. Macrophage migration inhibitory factor: a downregulator of early T cell-dependent IFN-gamma responses in Plasmodium chabaudi adami (556 KA)-infected mice. J Immunol. 2011;186(11):6271-9
60. McDevitt M.A, Xie J, Shanmugasundaram G. et al. A critical role for the host mediator macrophage migration inhibitory factor in the pathogenesis of malarial anemia. J Exp Med. 2006;203(5):1185-96
61. Jain V, McClintock S, Nagpal AC. et al. Macrophage migration inhibitory factor is associated with mortality in cerebral malaria patients in India. BMC Res Notes. 2009;2:36
62. Fernandes A.A, Carvalho LJ, Zanini GM. et al. Similar cytokine responses and degrees of anemia in patients with Plasmodium falciparum and Plasmodium vivax infections in the Brazilian Amazon region. Clin Vaccine Immunol. 2008;15(4):650-8
63. Chaisavaneeyakorn S, Moore JM, Othoro C. et al. Immunity to placental malaria. IV. Placental malaria is associated with up-regulation of macrophage migration inhibitory factor in intervillous blood. J Infect Dis. 2002;186(9):1371-5
64. Chaiyaroj S.C, Rutta AS, Muenthaisong K. et al. Reduced levels of transforming growth factor-beta1, interleukin-12 and increased migration inhibitory factor are associated with severe malaria. Acta Trop. 2004;89(3):319-27
65. Chaisavaneeyakorn S, Lucchi N, Abramowsky C. et al. Immunohistological characterization of macrophage migration inhibitory factor expression in Plasmodium falciparum-infected placentas. Infect Immun. 2005;73(6):3287-93
66. Clark I.A, Awburn MM, Whitten RO. et al. Tissue distribution of migration inhibitory factor and inducible nitric oxide synthase in falciparum malaria and sepsis in African children. Malar J. 2003;2:6
67. Awandare G.A, Hittner JB, Kremsner PG. et al. Decreased circulating macrophage migration inhibitory factor (MIF) protein and blood mononuclear cell MIF transcripts in children with Plasmodium falciparum malaria. Clin Immunol. 2006;119(2):219-25
68. Awandare G.A, Ouma Y, Ouma C. et al. Role of monocyte-acquired hemozoin in suppression of macrophage migration inhibitory factor in children with severe malarial anemia. Infect Immun. 2007;75(1):201-10
69. De Mast Q, Sweep FC, McCall M. et al. A decrease of plasma macrophage migration inhibitory factor concentration is associated with lower numbers of circulating lymphocytes in experimental Plasmodium falciparum malaria. Parasite Immunol. 2008;30(3):133-8
70. Kwiatkowski D.P. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. 2005;77(2):171-92
71. Donn R, Alourfi Z, De Benedetti F. et al. Mutation screening of the macrophage migration inhibitory factor gene: positive association of a functional polymorphism of macrophage migration inhibitory factor with juvenile idiopathic arthritis. Arthritis Rheum. 2002;46(9):2402-9
72. Donn R, Alourfi Z, Zeggini E. et al. A functional promoter haplotype of macrophage migration inhibitory factor is linked and associated with juvenile idiopathic arthritis. Arthritis Rheum. 2004;50(5):1604-10
73. Donn R.P, Shelley E, Ollier WE. et al. A novel 5'-flanking region polymorphism of macrophage migration inhibitory factor is associated with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2001;44(8):1782-5
74. Barton A. et al. Macrophage migration inhibitory factor (MIF) gene polymorphism is associated with susceptibility to but not severity of inflammatory polyarthritis. Genes Immun. 2003;4(7):487-91
75. Baugh J.A, Chitnis S, Donnelly SC. et al. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun. 2002;3(3):170-6
76. Renner P, Roger T, Calandra T. Macrophage migration inhibitory factor: gene polymorphisms and susceptibility to inflammatory diseases. Clin Infect Dis. 2005;41(Suppl 7):S513-9
77. Amoli M.M, Donn RP, Thomson W. et al. Macrophage migration inhibitory factor gene polymorphism is associated with sarcoidosis in biopsy proven erythema nodosum. J Rheumatol. 2002;29(8):1671-3
78. Nohara H, Okayama N, Inoue N. et al. Association of the -173 G/C polymorphism of the macrophage migration inhibitory factor gene with ulcerative colitis. J Gastroenterol. 2004;39(3):242-6
79. Hizawa N, Yamaguchi E, Takahashi D. et al. Functional polymorphisms in the promoter region of macrophage migration inhibitory factor and atopy. Am J Respir Crit Care Med. 2004;169(9):1014-8
80. Miterski B, Drynda S, Böschow G. et al. Complex genetic predisposition in adult and juvenile rheumatoid arthritis. BMC Genet. 2004;5:2
81. Zhong X.B, Leng L, Beitin A. et al. Simultaneous detection of microsatellite repeats and SNPs in the macrophage migration inhibitory factor (MIF) gene by thin-film biosensor chips and application to rural field studies. Nucleic Acids Res. 2005;33(13):e121
82. Awandare G.A, Ouma C, Keller CC. et al. A macrophage migration inhibitory factor promoter polymorphism is associated with high-density parasitemia in children with malaria. Genes Immun. 2006;7(7):568-75
83. Awandare G.A, Martinson JJ, Were T. et al. MIF (macrophage migration inhibitory factor) promoter polymorphisms and susceptibility to severe malarial anemia. J Infect Dis. 2009;200(4):629-37
84. WHO. The world Health Report. Geneva: WHO. 1996
85. Tonui W.K. Situational analysis of leishmaniases research in Kenya. Afr J Health Sci. 2006;13(1-2):7-21
86. Juttner S, Bernhagen J, Metz CN. et al. Migration inhibitory factor induces killing of Leishmania major by macrophages: dependence on reactive nitrogen intermediates and endogenous TNF-alpha. J Immunol. 1998;161(5):2383-90
87. Satoskar A.R, Bozza M, Rodriguez SM. et al. Migration-inhibitory factor gene-deficient mice are susceptible to cutaneous Leishmania major infection. Infect Immun. 2001;69(2):906-11
88. Xu D, McSorley SJ, Tetley L. et al. Protective effect on Leishmania major infection of migration inhibitory factor, TNF-alpha, and IFN-gamma administered orally via attenuated Salmonella typhimurium. J Immunol. 1998;160(3):1285-9
89. Kozaci D.L, Ertuğ S, Kavak T. et al. [Investigation of Serum Macrophage Migration Inhibitor Factor (MIF) levels in patients with cutaneous leishmaniasis.]. Turkiye Parazitol Derg. 2005;29(3):145-148
90. Santos-Oliveira J.R, Regis EG, Leal CR. et al. Evidence that lipopolisaccharide may contribute to the cytokine storm and cellular activation in patients with visceral leishmaniasis. PLoS Negl Trop Dis. 2011;5(7):e1198
91. Bimal S, Singh SK, Das VN. et al. Leishmania donovani: effect of therapy on expression of CD2 antigen and secretion of macrophage migration inhibition factor by T-cells in patients with visceral leishmaniasis. Exp Parasitol. 2005;111(2):130-2
92. Kar S, Metz C, McMahon-Pratt D. CD4+ T cells play a dominant role in protection against New World leishmaniasis induced by vaccination with the P-4 amastigote antigen. Infect Immun. 2005;73(6):3823-7
93. The world health report, Trypanosomiasis; 1996: 53; Revised 19 May 2011. WHO. http://www.who.int/whr/1996/media_centre/en/index.html
94. The worl Health Report, Trypanosomiasis; 1996: 51; Revised 19 May 2011. WHO. http://www.who.int/whr/1996/media_centre/en/index.html
95. Reyes J.L, Terrazas LI, Espinoza B. et al. Macrophage migration inhibitory factor contributes to host defense against acute Trypanosoma cruzi infection. Infect Immun. 2006;74(6):3170-9
96. Nishimura K, Nakaya H, Nakagawa H. et al. Differential effects of Trypanosoma brucei gambiense and Trypanosoma brucei brucei on rat macrophages. J Parasitol. 2011;97(1):48-54
97. Paiva C.N, Arras RH, Lessa LP. et al. Unraveling the lethal synergism between Trypanosoma cruzi infection and LPS: a role for increased macrophage reactivity. Eur J Immunol. 2007;37(5):1355-64
98. Cutrullis R.A, Postan M, Petray PB. et al. Timing of expression of inflammatory mediators in skeletal muscles from mice acutely infected with the RA strain of Trypanosoma cruzi. Pathobiology. 2009;76(4):170-80
99. Bosschaerts T, Guilliams M, Stijlemans B. et al. Tip-DC development during parasitic infection is regulated by IL-10 and requires CCL2/CCR2, IFN-gamma and MyD88 signaling. PLoS Pathog. 2010;6(8):e1001045
100. Lieberman LA, Hunter CA. The role of cytokines and their signaling pathways in the regulation of immunity to Toxoplasma gondii. Int Rev Immunol. 2002;21(4-5):373-403
101. Desmonts G, Couvreur J. Toxoplasmosis in pregnancy and its transmission to the fetus. Bull N Y Acad Med. 1974;50(2):146-59
102. Williams K.A, Scott JM, Macfarlane DE. et al. Congenital toxoplasmosis: a prospective survey in the West of Scotland. J Infect. 1981;3(3):219-29
103. Guerina N.G. Congenital infection with Toxoplasma gondii. Pediatr Ann. 1994;23(3):138-42
104. Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363(9425):1965-76
105. Terrazas C.A, Juarez I, Terrazas LI. et al. Toxoplasma gondii: impaired maturation and pro-inflammatory response of dendritic cells in MIF-deficient mice favors susceptibility to infection. Exp Parasitol. 2010;126(3):348-58
106. de Oliveira Gomes A, de Oliveira Silva DA, Silva NM. et al. Effect of macrophage migration inhibitory factor (MIF) in human placental explants infected with Toxoplasma gondii depends on gestational age. Am J Pathol. 2011;178(6):2792-801
107. Marciano-Cabral F. Biology of Naegleria spp. Microbiol Rev. 1988;52(1):114-33
108. Rodriguez-Zaragoza S. Ecology of free-living amoebae. Crit Rev Microbiol. 1994;20(3):225-41
109. Szenasi Z, Endo T, Yagita K. et al. Isolation, identification and increasing importance of 'free-living' amoebae causing human disease. J Med Microbiol. 1998;47(1):5-16
110. Marciano-Cabral F, Cabral GA. The immune response to Naegleria fowleri amebae and pathogenesis of infection. FEMS Immunol Med Microbiol. 2007;51(2):243-59
111. Cursons R.T, Brown TJ, Keys EA. et al. Immunity to pathogenic free-living amoebae: role of cell-mediated immunity. Infect Immun. 1980;29(2):408-10
112. Haq A, Sharma A, Ahmad S. Increased macrophage migration inhibition factor production in hamsters sensitized by amoebic antigen and glucan. Parasite Immunol. 1984;6(4):391-6
113. Carvalho JV, Alves CM, Cardoso MR. et al. Differential susceptibility of human trophoblastic (BeWo) and uterine cervical (HeLa) cells to Neospora caninum infection. Int J Parasitol. 2010;40(14):1629-37
114. Dubey J.P, Schares G, Ortega-Mora LM. Epidemiology and control of neosporosis and Neospora caninum. Clin Microbiol Rev. 2007;20(2):323-67
115. Tranas J, Heinzen RA, Weiss LM. et al. Serological evidence of human infection with the protozoan Neospora caninum. Clin Diagn Lab Immunol. 1999;6(5):765-7
116. Lobato J, Silva DA, Mineo TW. et al. Detection of immunoglobulin G antibodies to Neospora caninum in humans: high seropositivity rates in patients who are infected by human immunodeficiency virus or have neurological disorders. Clin Vaccine Immunol. 2006;13(1):84-9
117. Hong Y.H, Lillehoj HS, Lee SH. et al. Analysis of chicken cytokine and chemokine gene expression following Eimeria acervulina and Eimeria tenella infections. Vet Immunol Immunopathol. 2006;114(3-4):209-23
118. Novilla MN, Carpenter JW. Pathology and pathogenesis of disseminated visceral coccidiosis in cranes. Avian Pathol. 2004;33(3):275-80
119. Williams R.B. A compartmentalised model for the estimation of the cost of coccidiosis to the world's chicken production industry. Int J Parasitol. 1999;29(8):1209-29
120. Hong Y.H, Lillehoj HS, Lillehoj EP. et al. Changes in immune-related gene expression and intestinal lymphocyte subpopulations following Eimeria maxima infection of chickens. Vet Immunol Immunopathol. 2006;114(3-4):259-72
121. Flores M, Saavedra R, Bautista R. et al. Macrophage migration inhibitory factor (MIF) is critical for the host resistance against Toxoplasma gondii. FASEB J. 2008;22(10):3661-71
Author contact
![]() Corresponding author: Miriam Rodriguez-Sosa, Unidad de Biomedicina, FES-Iztacala, UNAM. Av. de los Barrios # 1, Los Reyes Iztacala, 54090 Tlalnepantla, Edo. de México. Mexico. Phone: (52-55) 5623-1333 ext. 39789. Fax: (52-55) 5623-1138. Email: rodriguezmiztacala.unam.mx
Corresponding author: Miriam Rodriguez-Sosa, Unidad de Biomedicina, FES-Iztacala, UNAM. Av. de los Barrios # 1, Los Reyes Iztacala, 54090 Tlalnepantla, Edo. de México. Mexico. Phone: (52-55) 5623-1333 ext. 39789. Fax: (52-55) 5623-1138. Email: rodriguezmiztacala.unam.mx