Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Human Malaria
2. Clinical Spectrum of...
3. Infant and Childhood Anemia...
4. Disease Burden of Severe...
5. Etiological Basis of Severe...
6. Impaired Erythropoietic...
7. Role of Inflammatory...
8. Role of Parasitic Products in...
9. Hemozoin Formation
10. Hemozoin Accumulation as a...
11. Severe Malarial Anemia is...
12. In vitro Models for...
13. Conclusion
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(9):1427-1442. doi:10.7150/ijbs.7.1427 This issue Cite
Review
Severe Malarial Anemia: Innate Immunity and Pathogenesis
Douglas J. Perkins1, 2 ![]() , Tom Were2,3, Gregory C. Davenport1, 2, Prakasha Kempaiah1, 2, James B. Hittner1, 4, John Michael Ong'echa2
, Tom Were2,3, Gregory C. Davenport1, 2, Prakasha Kempaiah1, 2, James B. Hittner1, 4, John Michael Ong'echa2
1. Center for Global Health, Department of Internal Medicine, University of New Mexico Health Sciences Center, Albuquerque NM, USA
2. University of New Mexico/Kenya Medical Research Institute, Laboratories of Parasitic and Viral Diseases, Centre for Global Health Research, Kisumu, Kenya
3. Department of Pathology, School of Health Sciences, Kenyatta University, Nairobi, Kenya
4. Department of Psychology, College of Charleston, Charleston, SC, USA
Received 2011-9-1; Accepted 2011-10-1; Published 2011-11-2
Abstract
Greater than 80% of malaria-related mortality occurs in sub-Saharan Africa due to infections with Plasmodium falciparum. The majority of P. falciparum-related mortality occurs in immune-naïve infants and young children, accounting for 18% of all deaths before five years of age. Clinical manifestations of severe falciparum malaria vary according to transmission intensity and typically present as one or more life-threatening complications, including: hyperparasitemia; hypoglycemia; cerebral malaria; severe malarial anemia (SMA); and respiratory distress. In holoendemic transmission areas, SMA is the primary clinical manifestation of severe childhood malaria, with cerebral malaria occurring only in rare cases. Mortality rates from SMA can exceed 30% in pediatric populations residing in holoendemic transmission areas. Since the vast majority of the morbidity and mortality occurs in immune-naïve African children less than five years of age, with SMA as the primary manifestation of severe disease, this review will focus primarily on the innate immune mechanisms that govern malaria pathogenesis in this group of individuals. The pathophysiological processes that contribute to SMA involve direct and indirect destruction of parasitized and non-parasitized red blood cells (RBCs), inefficient and/or suppression of erythropoiesis, and dyserythropoiesis. While all of these causal etiologies may contribute to reduced hemoglobin (Hb) concentrations in malaria-infected individuals, data from our laboratory and others suggest that SMA in immune-naïve children is characterized by a reduced erythropoietic response. One important cause of impaired erythroid responses in children with SMA is dysregulation in the innate immune response. Phagocytosis of malarial pigment hemozoin (Hz) by monocytes, macrophages, and neutrophils is a central factor for promoting dysregulation in innate inflammatory mediators. As such, the role of P. falciparum-derived Hz (PfHz) in mediating suppression of erythropoiesis through its ability to cause dysregulation in pro- and anti-inflammatory cytokines, growth factors, chemokines, and effector molecules is discussed in detail. An improved understanding of the etiological basis of suppression of erythropoietic responses in children with SMA may offer the much needed therapeutic alternatives for control of this global disease burden.
Keywords: Malarial Anemia, Innate Immunity, Pathogenesis
1. Human Malaria
Human malaria is caused by unicellular obligate intracellular protozoan parasites of the genus Plasmodium. Although malaria was once prevalent throughout most of the world, malaria is currently endemic in the tropical zones with extensions into the sub-tropical regions of Asia, Africa, South and Central America. However, about half of the world's population (3.3 billion people) is at risk of malaria in more than 100 countries [1]. Four primary species of malaria parasites infect humans: P. falciparum, P. ovale, P. malariae and P. vivax. In addition, studies in Southeast Asia have shown that P. knowlesi, a malaria parasite that typically involves monkeys as the natural reservoir, can also infect humans, and in some cases, result in fatal disease (reviewed in [2]). Malaria due to P. vivax, P. ovale and P. malariae is less severe than that experienced by P. falciparum infections and collectively, these three species account for slightly less than 10% of the worldwide malaria cases [3]. The most virulent of the human malaria parasites is P. falciparum which is responsible for the bulk of the malaria-related morbidity and mortality. P. falciparum accounts for 91% of malaria cases worldwide of which the majority (i.e., 86%) occurs in the African region [3]. Consistent with the high rate of morbidity within Africa, 90% of the P. falciparum-attributable malaria deaths also occur in the African region, primarily sub-Saharan Africa [3]. Approximately 247 million malaria infections are estimated annually, resulting in greater than one million deaths, primarily in African children under the age of five [1].
2. Clinical Spectrum of Plasmodium falciparum Infections
The clinical spectrum of P. falciparum malaria in African children encompasses a wide range of pathophysiological derangements and includes multiple organ involvement and systemic disorders. Falciparum malaria ranges from asymptomatic infections to the classic symptoms of malaria (e.g., fever, chills, sweating, headache and muscle aches), which in a sub-population of the cases, results in severe life-threatening complications such as hyperparasitemia, hypoglycemia, hyperlactatemia, kidney failure, metabolic acidosis, cerebral malaria, severe malarial anemia [SMA, hemoglobin, (Hb)<5.0 g/dL], and respiratory distress (RD) [4, 5]. Although the pathophysiology of malaria is multifactorial and only partially understood, development of a pathogenic versus protective outcome, once an infection occurs, is mediated by host and parasite interactions of which the following appear critically important: endemicity patterns, acquisition of naturally acquired malarial immunity, parasite virulence, multiplication rate, antigenic variation and polymorphic variability in both the host (human) and parasite [6]. The age of the individual when they first acquire a malarial infection also plays a significant role in the clinical outcomes of the disease process. For example, children typically display enhanced susceptibility to severe anemia and hypoglycemia, while non-resident malaria-naïve adults are more likely to present with jaundice and progress to renal failure and respiratory distress due to pulmonary edema [7].
3. Infant and Childhood Anemia in Developing Nations
Although this review will focus primarily on SMA and the innate immune responses that condition the development and outcomes of the disease, it is important to understand the definitions of anemia and the importance of geographic context. A general definition of anemia is a reduction in Hb levels in relation to the age, gender, and physiological status of the individual within a defined geographical context [8]. In western countries, anemia is defined by a Hb concentration <12.0 g/dL, while in developing countries the standard definition of anemia for children <5 years of age is Hb<11.0 g/dL [8]. Throughout much of the developing world, particularly in regions of sub-Saharan Africa with high rates of malaria and human immunodeficiency virus (HIV), the majority of infants and young children suffer from anemia [9]. Anemia in the developing world is largely a product of inadequate feeding practices, frequent infections, and micronutrient deficiencies which culminate in high rates of mortality in infants and young children [9].
4. Disease Burden of Severe Malarial Anemia
The World Health Organization (WHO) defines SMA as Hb concentrations <5.0 g/dL (or a hematocrit <15.0%) in the presence of any density parasitemia [5]. SMA is a major public health problem in developing countries where it contributes 3-46% of inpatient pediatric fatalities in referral care facilities [10]. Despite efforts aimed at ameliorating the anemia burden, SMA remains an important childhood health burden in sub-Saharan Africa [11]. Previous studies in endemic areas of Africa demonstrated that the annual rate of presentation to hospital with SMA was 7.6/1000 in children aged 0-4 years with a case fatality of 9.7% [12]. Additional studies illustrate that the risk for SMA peaks at 1 year of age in high (holoendemic) transmission regions and at approximately 2 years of age in areas with moderate and low transmission intensities, such that (in general) the overall risk of SMA decreases with increasing age [13]. Multicentre studies indicate that SMA affects 7.5-34% of the African children that acquire malaria with an overall prevalence of 21.2% and case fatality rate of 8.4% [14].
5. Etiological Basis of Severe Malarial Anemia
The etiology of SMA can include a number of distinct, as well as overlapping features, including lysis of infected and uninfected RBCs [15-18], splenic sequestration of RBCs [19], dyserythropoiesis and bone marrow suppression [20, 21], co-infections with bacteremia, HIV-1, and hookworm [22-26], and chronic transmission of malaria in holoendemic regions. It is important to note that some or all of these factors can culminate in the chronically low Hb values observed in infants and young children residing in holoendemic regions. As such, the degree of parasitemia is typically a poor indicator of malaria disease severity in these locales, especially considering that peripheral parasitemia is a “snapshot” in time of the complex and continuously evolving disease process. However, it is important to stress that high levels of parasitemia, particularly in non-immune individuals, can certainly result in massive lysis and clearance of RBCs, resulting in profound anemia [21, 27].
6. Impaired Erythropoietic Responses in Severe Malarial Anemia
Although parasite-driven hemolysis will contribute to a reduction in Hb levels in childhood malaria, one of the primary mechanisms responsible for low Hb levels in children with SMA is impaired and/or ineffective erythropoiesis. Loss of appropriate production of erythrocytes translates into a failure in the ability to replenish the reduced pool of erythrocytes due to parasite- and/or anti-malarial-driven hemolysis. Earlier studies demonstrated that there is parenchymal damage of bone marrow, ineffective erythropoiesis, and a reduced rate of erythropoietic proliferation in patients with acute falciparum malaria [28]. Subsequent studies in Gambian children demonstrated that SMA was defined by erythroid hyperplasia with dyserythropoiesis [6]. Examination of bone marrow from children with SMA revealed hypercellularity, mild to normal erythroid hyperplasia, and abnormal features of late erythroid progenitors, but absence of damage to burst forming unit-erythrocyte (BFU-E) or colony forming unit-erythrocyte (CFU-E) early stages [6]. In addition, we have shown that children with SMA have an inefficient reticulocyte production index (RPI) characterized by levels <2.0 [29]. The RPI is a measure of the extent to which the reticulocyte count has risen (or not) in response to the level of anemia [30]. Taken together, these investigations demonstrate that suppression of erythropoiesis is a primary cause of severe anemia in children with P. falciparum infections.
7. Role of Inflammatory Mediators in Impaired Erythropoiesis
Although the precise mechanisms responsible for reduced reticulocyte responses in children with SMA have been somewhat elusive, it is clear that one important cause of reduced erythropoiesis in children with SMA is due to an imbalance in inflammatory mediators. In an attempt to control the parasitemia, the host releases an array of pro- and anti-inflammatory cytokines, chemokines, growth factors, and effector molecules as part of the innate immune response. Depending on the magnitude and timing of inflammatory mediator release, the immune response to malaria can result in either successful control of the parasitemia or alternatively, an inappropriate balance in the inflammatory milieu that can induce damage to the host, including suppression of the erythropoietic response. As such, although malaria is typically viewed as an 'acute' infection, in regions with high levels of falciparum endemicity, SMA is often a more 'chronic' condition in which the immunological response to infection drives an inflammatory milieu that can promote suppression of eythropoiesis. This premise is supported by observations showing that persistent childhood P. falciparum infections are associated with bone marrow suppression [31].
8. Role of Parasitic Products in Stimulating the Innate Immune Response
There are a number of key parasitic products that drive the innate immune response in the malaria-infected human host, including malarial pigment (hemozoin, Hz), glycosylphosphatidylinositols (GPIs), and parasitic antigens. While it is clear that GPIs, which form the connection between the parasite's cellular membrane and external antigens [32], as well as an array of parasitic antigens play an important role in activating the immune response, we will focus our discussion on the role of P. falciparum-derived Hz (PfHz) in promoting the host immune response and the potential implications that this process has on suppression of the erythropoietic response. Excellent reviews on the means by which parasite GPIs [33] and malarial antigens stimulate the innate immune response can be found elsewhere [34].
9. Hemozoin Formation
The importance of hemozoin in malarial infections was recognized over five decades ago following the discovery that accumulation of phagocytosed malarial pigment in bone marrow produces a brown/black appearance in patients with repeated malaria infections [35]. Formation of PfHz occurs during the intraerythrocytic asexual replication cycle in which P. falciparum metabolizes host Hb as a source of amino acids [36, 37], leaving the iron-rich heme portion designated ferriprotoporphyrin IX (FP-IX). P. falciparum then promotes aggregation of the toxic FP-IX molecules, using heme polymerase [38, 39], into an insoluble product known as PfHz [40-42]. Once the schizont has gone through replication within the host RBC, the erythrocyte ruptures [43], releasing PfHz along with merozoites, with the newly-formed merozoites infecting other RBCs, and PfHz being phagocytosed by monocytes/macrophages and neutrophils.
10. Hemozoin Accumulation as a Causal Factor in Suppression of Erythropoiesis
Accumulation of phagocytes containing malaria pigment in the microvasculature of deep tissues such as the bone marrows of patients with acute malaria was recognized in earlier studies [44]. Intraleukocytic malaria pigment is a useful direct diagnostic marker with the amount of phagocytosed pigment being a good indirect measure of the sequestered parasite burden, recent schizogony, and disease severity [45]. Studies by others and our group have also found an association between the presence of PfHz-containing monocytes and suppression of erythropoiesis in children with P. falciparum-induced anemia [46, 47]. In addition, our studies in western Kenya, using multivariate regression analyses, controlling for age, gender, and parasitemia, revealed that elevated levels of pigment-containing monocytes (PCM) were a significant predictor of SMA [47]. A recent multi-site study that included 26,296 hospitalized children with P. falciparum malaria demonstrated that the percentage of PfHz-containing monocytes was negatively correlated with hematocrit [48]. Additional evidence that PfHz-containing cells are an important source for promoting erythroid suppression is supported by histological observations of bone marrow in children with SMA, and those dying from malaria, showing that developing erythroid progenitors located within the vicinity of pigmented macrophages have abnormal cellular development [46]. Consistent with these observations, we recently demonstrated that elevated levels of PfHz in monocytes are associated with inefficient erythropoiesis in Kenyan children with malaria [49].
11. Severe Malarial Anemia is Characterized by Dysregulation in Innate Inflammatory Mediators
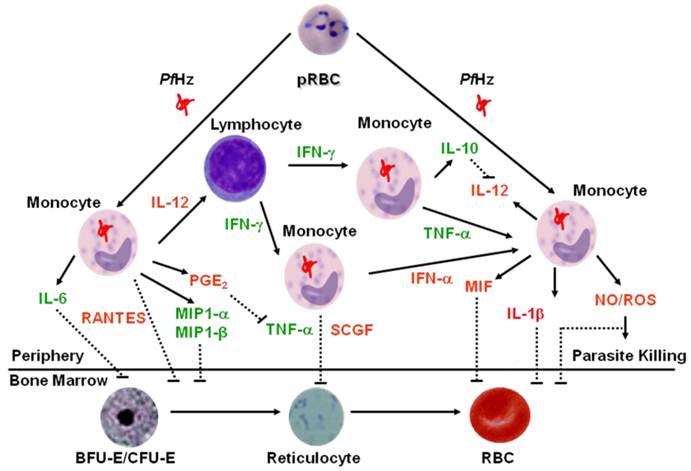
One of the primary factors for stimulating the innate immune response to P. falciparum is phagocytosis of PfHz by circulating monocytes and neutrophils, and resident macrophages. One of the primary means by which PfHz generates an innate immune response is through the toll-like receptors (TLRs). A recent review discussing the role of hemozoin in stimulating the TLRs can be found elsewhere [50]. The studies discussed below support a model in which phagocytosis of PfHz, and the host immune response patterns that ensue following this event, are a critical step in the promotion of the reduced erythroid responses observed in children with SMA (Figure 1). Although it is clear that a wealth of important cellular biology is yet to be elucidated about the mechanisms by which phagocytosis of PfHz shapes the innate immune response, the ability of PfHz to alter inflammatory mediator profiles is central to its action. However, it is important to note that clarification of strict protective versus pathological roles for inflammatory mediators remains poorly defined and extremely difficult to quantify in human malaria in which manipulation of the biological systems is typically not feasible.
Proposed Model of Dysregulation in Innate Immune Responses in Severe Malarial Anemia. Based on concomitant measurement of innate inflammatory mediators (using multiplex technologies) in children with varying severities of malarial anemia, we developed a model to describe how dysregulation in innate inflammatory mediators promotes suppression of erythropoiesis in children with SMA. Central to the model is the fact that phagocytosis of hemozoin (PfHz) by monocytes is one of the primary causes of altered production of innate inflammatory mediators. Elevated inflammatory mediators are shown in green text, while those that are decreased in children with SMA are shown in red text. Solid lines indicate positive signaling (up-regulation), whereas dashed lines indicate suppression (down-regulation). Children with SMA have decreased levels of IL-12 in response to ingestion of parasitized red blood cells (pRBC) and/or hemozoin by monocytes. Suppression of IL-12 in children with SMA is due to PfHz-induced IL-10 over-production. Children with SMA have increased circulating levels of TNF-α, IFN-γ, IL-6, MIP-1α, and MIP-1β. Although TNF-α can induce PGE2 and nitric oxide (NO), these effector molecules are suppressed in children with SMA. Suppression of PGE2 allows over-production of TNF-α, which is associated with enhanced severity of anemia. In addition, MIF is suppressed in children with falciparum malaria, an event associated with phagocytosis of PfHz by monocytes, and enhanced severity of anemia. Circulating levels of IFN-α, IL-1β, RANTES, and SCGF are also decreased in children with SMA. Reduced production of these innate inflammatory mediators, along with increased TNF-α, IL-6, MIP-1α and MIP-β, likely contribute to the development of SMA by suppressing the erythropoietic response. Lastly, although the reduced NO and reactive oxygen species (ROS) generation reported in children with falciparum malaria may promote ineffective parasite killing and, thereby, prolong parasitemia, children with malarial anemia have elevated levels of NO and ROS that can directly inhibit erythropoiesis.
Pro-inflammatory Mediators
A successful type 1 response to malaria requires a well-timed and proportional release of interleukin (IL)-12, interferon (IFN)-γ, and tumor necrosis factor (TNF)-α to minimize parasitemia and preserve erythropoiesis [51, 52]. The pro-inflammatory phase should be followed by an equally timely abrogation of this response by type 2 cytokines such as IL-10, transforming growth factor (TGF)-β, and IL-4, to avoid inflammatory host damage [53].
TNF-α is the molecule typically associated with pathology in malaria and was first hypothesized to be involved in the host immune response to malaria in 1978 [54]. Several studies have shown an association between elevated TNF-α levels and morbidity and mortality in individuals with malaria [55, 56]. However, TNF-α is critical for parasite killing and preventing parasite replication [55-59]. In addition to its direct effects [60, 61], TNF-α also mediates its effects by inducing production of macrophage migration inhibitory factor (MIF) [62, 63] and nitric oxide synthase type 2 (NOS2, inducible nitric oxide synthase, iNOS) [64], which through generation of nitric oxide (NO) also has direct parasite killing effects [65]. TNF-α can also exacerbate inflammation by inducing cyclooxygenase (COX)-2 and subsequently generated effector molecules, such as prostaglandins [66]. Many of the signs and symptoms associated with malaria, such as fever, headache, nausea, vomiting, diarrhea, anorexia, myalgias, and thrombocytopenia can be linked to TNF-α [67].
IFN-γ is produced by natural killer and αβ-T cells, as well as the regulatory γδ-T cells, during the early phases of the immune response to a malaria infection [68-70]. This prototypical type 1 cytokine is a key molecule for protecting against infection in childhood malaria [71] and in non-immune volunteers experimentally infected with malaria [72]. The ability to generate IFN-γ from mononuclear cells exposed to asexual malaria parasites has recently been attributed to the significant resistance to P. falciparum malaria in the Fulani people of West Africa compared to other tribes in the Mali, Burkina Faso, and Sudan regions [73]. Consistent with a protective role, IFN-γ responses to CD8+ T cell epitopes from pre-erythrocytic antigens are associated with higher Hb levels, and reduced prevalence of SMA in Kenyan children [74].
Although both TNF-α and IFN-γ appear to play a protective role in children and adults during the early stages of a P. falciparum infection through their ability to stimulate monocyte/macrophage activation and aid in controlling parasitemia [75], over-production of these innate inflammatory mediators is also associated with anemia [76, 77]. In addition, persistent macrophage activation is significantly greater in children with complicated malaria [78]. Excessive release of IFN-γ and TNF-α, along with NO, also promote enhanced malarial anemia pathogenesis by contributing to bone marrow suppression, dyserythropoiesis, and erythrophagocytosis [79].
IL-1 is a potent endogenous pyrogen that promotes an acute inflammatory response and provides a first line of defense against invading pathogens [80]. IL-1β and IL-1α synergize with TNF-α to enhance the production of NO and IFN-γ in murine models of malaria [81]. However, high levels of sustained IL-1β production in inflammatory diseases can induce a number of hematological abnormalities, including anemia [82, 83]. In murine models, administration of recombinant IL-1 can inhibit development of the pre-erythrocytic stages of malaria [84], protect against the development of cerebral malaria, and aid in controlling parasitemia [85]. Although several studies have reported elevated peripheral blood levels of circulating IL-1β levels in humans with severe malaria [86-88], an additional investigation failed to find significant changes in IL-1β levels in children with SMA [76]. Studies in Gambian children with falciparum malaria illustrated that IL-1α and TNF-α levels, measured upon admission to hospital, were positively correlated with venous blood lactate concentrations, which were approximately two-fold higher in fatal cases compared to survivors [89]. A study by our group investigating the role of IL-1β in the immunopathogenesis of SMA revealed that children with SMA had significantly lower levels of IL-1β than parasitized children without SMA [90]. In addition, haplotypes of IL-1β promoter polymorphisms that were associated with significantly greater risk of developing SMA were also associated with reduced IL-1β production, whereas those haplotypes associated with protection against SMA produced higher levels of IL-1β [90]. Thus, although sustained production of IL-1β can promote anemia [82, 83], it appears that 'high producing' haplotypes of IL-1β provide protection against SMA.
Elevated levels of IL-6 in the peripheral blood of patients with severe P. falciparum malaria was recognized two decades ago [57]. This finding has since been corroborated by a number of studies showing elevated IL-6 levels in children with severe malaria [75, 76, 88]. An investigation in Gabonese children further demonstrated that peripheral blood mononuclear cells (PBMC) are a primary source of increased IL-6 production during acute malaria [91]. However, studies in murine models illustrate that IL-6 mediates protective immunity against the pre-erythrocytic stages of malaria by inducing IL-1β and TNF-α, and during the erythrocytic stage of disease by controlling parasitemia through boosting of specific immunoglobulin (Ig) G antibodies [84]. Experimental infections with P. falciparum in humans support these findings in that early IL-6 production is associated with protective effects [92]. Consistent with the protective effects of IL-6, lower circulating levels of IL-6 are associated with hyperparasitemia in Malian children with falciparum malaria [76]. Taken together, these studies support a protective role for IL-6 during the early stages of disease by controlling parasitemia. However, lack of control over the parasitemia and the resulting progression towards severe disease may explain the association between elevated levels of IL-6 and enhanced pathophysiology.
MIF was the first soluble mediator described in malaria [93], but has only recently been more fully explored in the context of malaria pathophysiology by our group and a number of others [47, 94-98]. MIF is a ubiquitous cytokine produced in response to pro-inflammatory stimuli by T cells [99, 100], monocytes/macrophages [101], and the anterior pituitary gland [102]. However, unlike most cytokines, MIF is constitutively expressed at high levels and stored in preformed vesicles, and as such can therefore be rapidly released without de novo gene expression [102, 103]. MIF has potent pro-inflammatory properties that are important for both innate and adaptive immune responses to bacterial and parasitic infections [100, 101, 104-106]. Murine models of malaria demonstrated that elevated MIF levels were associated with enhanced disease severity [107] with the MIF gene knock-out mice having less anemia and higher survival rates following infection with P. chabaudi compared to wild types [108].
Previous investigations in humans showed that MIF was elevated in intervillous blood during placental malaria [109, 110], thoracic blood vessels of Malawian children with cerebral malaria [111], and peripheral blood from Zambian children with acute malaria [108]. However, results from our laboratory were the first to show that elevated MIF protein (in circulation) and MIF transcripts (in PBMC) were associated with less severe forms of falciparum malaria [94]. We subsequently confirmed these results in a larger study of children (aged <3 years, n=357) with P. falciparum-induced malarial anemia in western Kenya [47]. In the Kenyan cohort, circulating MIF concentrations declined with increasing severity of anemia and significantly correlated with peripheral blood leukocyte MIF transcripts. Interestingly, MIF concentrations in peripheral blood were not significantly correlated with reticulocyte responses in these children. However, multivariate regression modeling, controlling for age, gender, and parasitemia, demonstrated that elevated levels of PCM were significantly associated with both SMA and decreased MIF production. As a complement to the in vivo studies in children with malarial anemia, additional experiments were conducted in PBMC from malaria-naïve individuals which showed that phagocytosis of PfHz caused dysregulation in MIF production in a apoptosis-independent manner [47]. Taken together, investigations in Gabonese and Kenyan children with malarial anemia demonstrate that elevated levels of monocytic PfHz are associated with suppression of peripheral blood MIF production and enhanced severity of anemia.
Another pro-inflammatory mediator that appears important in conditioning the pathogenesis of SMA is IL-23. Although largely unexplored in the context of SMA, IL-23 is important in mediating the development of anemia in autoimmune diseases [112] and chronic inflammation [113]. IL-23 is composed of two subunits, p19 and p40 [114]. IL-23 shares a number of common properties with IL-12, including the p40 subunit [115], the ability to bind to the IL-12Rβ1 receptor [116], release from activated myeloid antigen presenting cells, promotion of a type 1 immune response [114-118], and suppression by both IL-10 [119, 120] and IL-12p40 homodimers [115, 121]. In addition to the common properties IL-23 shares with IL-12, there are also distinct immunological roles in that IL-23 acts on activated memory CD4+ T cells, while IL-12 promotes Th1 differentiation of naïve CD4+ T cells [114, 122]. Based on the common and distinct roles of IL-23 and IL-12, along with the well established importance of IL-12 in the pathogenesis of malarial anemia (discussed below in detail), we explored the relationships among these cytokines in Kenyan children with varying severities of malarial anemia [123]. Children with malarial anemia had increased peripheral blood levels of IL-23 and suppressed IL-12 relative to healthy controls. Additional experiments in cultured PBMC revealed that hemozoin caused a sustained induction of IL-23p19 transcripts over 72 h, while IL-12p40 and IL-10 transcripts peaked at 24 h, and rapidly declined thereafter. This line of investigation suggests that elevated IL-23 levels may be important in the pathogenesis of SMA, and that both IL-10 and IL-12 may regulate IL-23 production during an infection with P. falciparum.
Perhaps one of the most important innate inflammatory mediators in the pathogenesis of SMA is IL-12, a heterodimeric protein composed of 35 and 40 kDa subunits, and a prototypical cytokine of the type 1 immune response [124, 125]. IL-12 is secreted from dendritic cells, monocytes, and B-cells in response to bacterial cell wall components, intracellular pathogens, and CD40 ligation [124-126]. IL-12 stimulates production of IFN-γ and TNF-α from T-cells and natural killer (NK) cells [124, 125], thereby further augmenting type 1 responses. A number of cytokines and chemokines can promote IL-12 [e.g., granulocyte macrophage-colony stimulating factor (GM-CSF) and IFN-γ], while others decrease IL-12 production [e.g., IL-4, IL-10, IL-11, IL-13, monocyte chemotactic protein (MCP)-1/CCL2, and TGF-β] [125, 126]. As such, the overall ability of the innate immune response to generate IL-12 is an important event that mediates the development of malarial anemia.
Previous studies in the murine models showed that administration of recombinant IL-12 and chloroquine ameliorated blood-stage malaria and severe anemia, and induced immunity against re-infection [127]. Additional murine malaria studies demonstrated that deficient IL-12 production is associated with severe anemia and dyserythropoiesis [128]. The protective responses associated with IL-12 against blood-stage malaria appear to be due to increased IL-12 production from splenic macrophages and NK cells [129, 130] and the ability of IL-12 to stimulate antibody production [131]. Central to the role of IL-12 in malaria is its ability to act as a hematopoietic growth factor [132, 133]. In concert with IL-3, IL-12 along with IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-11 can bolster colony formation in dormant hematopoietic progenitors during times of cytopenic crisis [132, 133]. Consistent with this role, our previous studies in children with falciparum malaria showed that suppression of circulating IL-12 is associated with enhanced malarial anemia [77, 134]. Suppression of IL-12 in these children was also associated with increasing concentrations of PfHz-containing leukocytes [134]. Additional studies from our laboratories demonstrated that children with SMA have reduced IL-12 levels through a mechanism that involves, at least in part, phagocytosis of PfHz which promotes up-regulation of monocyte-derived IL-10 that, in turn, suppresses IL-12p40 subunits [135].
Anti-inflammatory Mediators
High levels of anti-inflammatory cytokines, such as IL-10, appear to provide protective effects against SMA by preventing the over-production of pro-inflammatory mediators. Anti-inflammatory cytokines are typically produced during the later stages of the innate immune response to P. falciparum in which they down-regulate the potentially pathogenic pro-inflammatory responses that are important for controlling parasitemia [136]. Previous investigations show that a low IL-10 to TNF-α ratio is associated with enhanced severity of malarial anemia [77, 137], suggesting that the timing and magnitude of pro-inflammatory cytokine production, relative to the anti-inflammatory cytokine response, is an important determinant of the clinical outcomes of malaria. Although plasma IL-10 levels are elevated in Malian children with SMA relative to healthy controls [76], studies in Ghana showed that plasma IL-10 levels were significantly lower in children with SMA compared to children with cerebral malaria, uncomplicated malaria, or moderate malarial anemia [138]. Additional studies have shown significant associations between circulating IL-10 levels and pigment-containing phagocytes in the peripheral blood [134], suggesting that malarial pigment plays an important role in governing the systemic pathology of malaria through up-regulation of IL-10 production.
TGF-β1 is an anti-inflammatory cytokine (and growth factor), which down-regulates the production of TNF-α and IL-10 and protects against severe murine malaria [139]. TGF-β appears to be important in malaria pathogenesis [86, 110, 140], and is attributed to both positive [141] and negative [141-143] effects on erythropoiesis. Previous studies from our group showed that TGF-β1 levels in peripheral blood are significantly reduced in Gabonese children with severe malaria [77]. However, studies in Burkina Faso revealed opposite effects in which severe childhood malaria was associated with increased plasma concentrations of TGF-β1 [144]. The reason for differing results may be due to the differences in malaria endemicity since the rural site in Lambaréné, Gabon, has a high level of P. falciparum transmission, whereas the urban region of Ouagadougou, Burkina Faso, is a mesoendemic area for P. falciparum. Alternatively, it is unclear whether or not 'platelet poor' samples were used to measure TGF-β1 in the Burkina Faso study, an important consideration since platelets contain high levels of TGF-β1. A more recent study further supports the importance of the TGF-β family in malaria pathogenesis by showing that serum levels of the soluble form of the TGF-β co-receptor, endoglin (sEng or CD105/ TGF-βRIII), was significantly elevated in children with severe falciparum malaria [145]. In addition to TGF-β1, a recent investigation in Angolan children illustrates that polymorphic variability in TGF-β2 conditions susceptibility to the risk of progressing to cerebral malaria [146]. It remains to be definitively determined if TGF-β2 is also an important mediator of SMA pathogenesis.
Chemokines
Chemotactic cytokines, or chemokines, are primarily known for their chemotactic properties, but also play important roles in immune activation, hematopoiesis, angiogenesis, and antimicrobial activities [147]. Burgmann et al. were the first to investigate chemokines in the context of malaria in 1995 by measuring the C-C chemokine, macrophage inflammatory protein (MIP)-1α/CCL3, and the C-X-C chemokine, IL-8/CXCL8, in the serum of acutely infected adult patients with P. falciparum infections in which they found a positive correlation between parasitemia and IL-8/CXCL8 [148]. A subsequent investigation revealed that Malian children with severe malaria had ten-fold higher concentrations of IL-8/CXCL8 compared to either healthy controls or individuals with uncomplicated malaria [76]. IL-8 is an important neutrophil activating chemokine that was also previously shown to be elevated in Thai patients with severe, non-fatal malaria [149]. Additional studies in Gabonese children and adults illustrated that higher plasma IL-8 levels were associated with acute malaria and a slow rate of cure after malaria chemotherapy [75].
It appears that phagocytosis of PfHz is an important signal for promoting chemokine production and/or suppression. For example, PfHz treatment of a bone marrow-derived murine cell line increased transcript levels of MIP-1α/CCL3, MIP-1β/CCL4, MIP-2/CXCL2, and MCP-1/CCL2 [150]. Concomitant studies from our group demonstrated that Gabonese children with severe malaria had elevated levels of MIP-1α/CCL3 and MIP-1β/CCL4 protein (measured in circulation) and transcripts (determined in ex vivo PBMC) [151]. Additional experiments in cultured PBMC from healthy, malaria-naïve donors revealed that PfHz promoted increased MIP-1α/CCL3 and MIP-1β/CCL4 production [151].
Regulated on activation, normal T-cell expressed and secreted (RANTES, CCL5) also appears to play a critically important role in the pathogenesis of SMA. RANTES is secreted by a number of cell types including monocytes, macrophages, fibroblasts, NK and T cells, and CD34+ hematopoietic progenitors [152-155]. RANTES protein is sequestered in the α-granules of platelets and is released by thrombin stimulated platelets [156], indicating that RANTES is involved in both innate and adaptive immune response. In addition, RANTES stimulates hematopoiesis, angiogenesis, cell proliferation, and development [157]. An earlier study by our group, which was the first to examine RANTES in the context of malaria, demonstrated that RANTES was suppressed in Gabonese children with severe malaria, at least in part, through PfHz-induced down-regulation of RANTES transcripts in PBMC [151]. The inherent ability to produce RANTES/CCL5 also appears important in conditioning susceptibility to severe malaria. For example, our investigation in Gabon revealed that healthy children with prior mild malaria produced significantly higher RANTES transcripts and protein than children with a history of severe malaria [151]. These investigations were then confirmed in Kenya where we showed that RANTES was significantly suppressed in children with SMA [29]. Suppression of RANTES in these children was also significantly associated with inefficient erythropoiesis and malaria-induced thrombocytopenia [29]. Subsequent studies from our laboratories determined that naturally acquired PfHz by monocytes promotes suppression of RANTES in children with malarial anemia through an IL-10-dependent mechanism [49]. Taken together, these findings suggest that thrombocytopenia may be an important source of reduced RANTES which appears to contribute to suppression of erythropoiesis in children with SMA.
Growth Factors
Since a number of growth factors influence the erythropoietic cascade, and SMA is clearly a disease process in which phagocytosis and lysis of RBCs necessitates the production of new RBCs to recover from anemia, growth factors will clearly emerge as critical determinants of clinical outcomes. However, the literature is largely lacking with respect to the importance of growth factors in conditioning the development of SMA. A time-course study in patients with P. falciparum malaria in Thailand showed that serum levels of granulocyte-colony stimulating factor (G-CSF) were significantly elevated in individuals with complicated disease on day 0 that then declined to within the normal range by day 7; G-CSF at day 0 was correlated with procalcitonin, parasite density, and erythropoietin [158]. Although not specifically examined in children with SMA, the potential negative consequences associated with over-production could certainly emerge since G-CSF has a negative impact on erythropoiesis [159-161].
GM-CSF is important for promoting erythropoiesis [162] and synergizes with TNF-α to increase the killing capabilities of neutrophils on blood-stage malaria parasites [163]. In addition, enhanced pathology in a murine model of malaria (characterized by parasitemia and anemia) was associated with elevated levels of erythropoietin (EPO), which were strongly correlated with the level of anemia, and negatively correlated with GM-CSF concentrations [164]. The influence that GM-CSF has on anemia outcomes in children with SMA remains to be determined.
As part of our investigations aimed at identifying genes/gene pathways that could play a role in the pathogenesis of SMA, we utilized gene expression profiling from pooled fractions of human PBMCs stimulated with PfHz. These experiments revealed that human stem cell growth factor [SCGF, C-type lectin domain family member 11A, (CLEC11A)] was up-regulated following treatment with PfHz [165]. SCGF is a hematopoietic growth factor, expressed primarily by myeloid cells and fibroblasts that possess burst-promoting activity for human bone marrow erythroid progenitors [166]. Human SCGF-α is a 323-amino acid protein, while SCGF-β is a 245-amino acid protein that results from cleavage of the conserved carbohydrate domain [167]. After determining the in vitro kinetics of SCGF expression in response to PfHz, we then examined circulating SCGF levels in Kenyan children with malarial anemia. SCGF levels in circulation and in cultured peripheral blood were significantly suppressed in children with SMA, with circulating SCGF levels being positively correlated with Hb concentration and the RPI [165]. SCGF was significantly lower in children with a suppressed erythropoietic response and in children with high levels of naturally acquired monocytic PfHz [165]. Our additional investigation showed that a novel SCGF promoter variant (-539C/T, rs7246355) was significantly associated with susceptibility to SMA and reduced erythropoietic responses with the 'high producing' TT genotype protecting against development of SMA and suppression of erythropoiesis in parasitized children [168]. Taken together, these results illustrate that SCGF is an important mediator of SMA pathogenesis that may offer the potential for immunotherapy in future clinical trials.
Effector Molecules
As described above, the clinical outcomes of malaria are largely conditioned by the relative expression of inflammatory mediators. The relative timing and magnitude of pro- and anti-inflammatory cytokines, chemokines, and growth factors released into the inflammatory milieu have direct actions on the cellular response as well as the 'down-stream' effector molecules that ultimately get produced. As such, effector molecules play a critical role in the pathogenesis of SMA. One important effector molecule in malaria is the toxic free radical, NO. NO and equimolar amounts of L-citrulline are generated by catalysis of L-arginine by the NO synthases (NOS) [169]. In the context of an acute inflammatory disease, such as malaria, much of the NO produced comes from the cytokine inducible isoform, nitric oxide synthase type 2 [NOS2 or inducible NO synthase (iNOS)] present in monocytes, macrophages, and neutrophils [170]. In general, pro-inflammatory cytokines (e.g. IL-12, IFN-γ, and TNF-α) increase NOS2-generated NO production, whereas anti-inflammatory cytokines (e.g. IL-10 and TGF-β) down-regulate NOS2 expression (for review see [171]). Although the role of NO in the pathogenesis of malaria has been debated for nearly a decade, it is apparent that NO is both protective and pathogenic. For example, NO is protective in that it has potent parasiticidal properties against P. falciparum [65] and can thereby limit parasitemia [172]. A protective effect is also illustrated by our previous investigation showing that healthy, malaria-exposed Gabonese children with a history of mild malaria have significantly higher levels of ex vivo PBMC NO production and NOS enzymatic activity than their age-matched cohort with a history of severe malaria [173]. However, our follow-up investigation in the same population revealed that ex vivo and in vitro NOS activity in PBMCs was significantly higher in children with malarial anemia in which there was an inverse association between NOS enzyme activity and hemoglobin levels [174]. Additional experiments confirmed that PfHz was an important source of NOS2 transcripts and NO production [174]. Thus, although NO serves an important role in controlling parasitemia, it is likely that sustained, high levels of NO production also promotes anemia. This premise is supported by the fact that generation of NO during a malarial infection can promote severe anemia through bone marrow suppression, dyserythropoiesis, and erythrophagocytosis (for review see [79]).
In addition to NO, reactive oxygen species (ROS) also appear to be both protective and pathogenic in human malaria. High levels of oxygen radical production are associated with accelerated clearance of parasitemia in Gabonese children with falciparum malaria [175]. In addition, ROS are important for controlling peripheral parasitemia in children with severe malaria [176]. A pathogenic role for ROS is illustrated by a study in Kenyan children showing that ROS cause damage to the erythrocytic membrane (demonstrated by measurement of α-tocopherol and polyunsaturated fatty acid levels in the erythrocyte membrane) [177]. A recent investigation in Indian children (<15 years of age) infected with P. falciparum (in which the primary disease manifestation of severe malaria was SMA) revealed that severe malaria cases had significantly elevated markers of oxidant stress, including malondialdehyde, protein carbonyl, nitrite, ascorbic acid, and copper levels [178]. Although a wealth of data exists on the topic of free radicals in malaria, such a discussion is beyond the scope of this review. However, studies outlined here highlight both the protective and pathogenic roles of reactive nitrogen and oxygen intermediates.
Prostaglandin (PG)E2 is synthesized from arachidonic acid (AA) through the catalytic activity of cyclooxygenase (COX) enzymes also known as prostaglandin-H2 (PGH2) synthase, which exists in two isozymes: COX-1 (PGH synthase-1) and COX-2 (PGH synthase-2). Constitutively expressed COX-1 catalyses immediate biosynthesis of PGE2 and other prostanoids involved in physiological homeostasis, whereas inducible COX-2 catalyses delayed formation of PGE2 and prostanoids involved in regulating the inflammatory response and immunity to invading pathogens [179]. Formation of PGH2, the committed step in prostanoid biosynthesis, promotes generation of primary prostanoids [i.e., PGE2, PGH2, thromboxane A2, PGD2, PGF2α, and prostacyclin (PGI2) through the action of respective terminal prostanoid synthases [179]. Our previous study illustrates that intervillous blood mononuclear cell (IVBMC) PGE2 production is reduced in parasitemic women of all gravidae due, at least in part, to acquisition of intraleukocytic PfHz [180]. Additional studies from our group have shown that plasma bicyclo-PGE2 (a stable end metabolite of PGE2) and ex vivo PBMC COX-2 gene expression are significantly reduced in Gabonese children with severe malaria [181]. Studies in Tanzanian children also showed that suppression of systemic bicyclo-PGE2 production (measured in urine) was suppressed in children with cerebral malaria [182]. In addition, in vitro experiments in our laboratories revealed that reduced PGE2 biosynthesis in children with falciparum malaria was largely due to inhibition of de novo COX-2 transcripts following phagocytosis of PfHz by monocytes [183, 184]. Further investigation of the role of prostaglandins in childhood malaria showed that suppression of PGE2 by PfHz and commonly used antipyretics to treat the malarial fever promoted over-production of TNF-α, an event associated with enhanced malaria pathogenesis [183, 184].
12. In vitro Models for Investigating Suppression of Erythropoiesis in Malarial Anemia
In vitro studies have shown that PfHz directly (or in synergy with TNF-α) inhibits erythroid cell development [46]. In addition to inducing erythropoiesis-inhibiting cytokines, PfHz has direct effects on the erythropoietic cascade through its ability to cause apoptosis of erythroid precursor cells via oxidative stress [185]. We recently developed an in vitro model of erythropoiesis using CD34+ stem cells isolated from human peripheral blood to investigate the effects of inflammatory mediators on erythroid development [186]. This model showed that PfHz only slightly suppressed erythroid cell proliferation and maturation, marked by decreased expression of glycophorin A (GPA) [186]. However, the addition of PfHz-stimulated PBMC-conditioned media (PfHz-CM), recombinant TNF-α, and NO donors significantly inhibited erythroid cell proliferation [186]. The decreased proliferation witnessed in cells treated with PfHz-CM and NO was accompanied by increased apoptosis of erythropoietin-stimulated CD34+ cells [186]. Interestingly, the addition of NO donors significantly inhibited erythroid cell maturation, whereas TNF-α failed to impact on maturation. These results demonstrate that PfHz suppresses erythropoiesis by acting directly on erythroid cells, and to a greater extent, through indirect effects in which phagocytosis of PfHz generates inflammatory mediators which have adverse effects on erythroid development.
13. Conclusion
Studies outlined here support a model in which the pathogenesis of SMA is largely driven by dysregulation in pro- and anti-inflammatory cytokines, chemokines, growth factors, and effector molecules. Altered patterns of these innate inflammatory mediators is due, at least in part, to the phagocytosis of PfHz by monocytes, resident macrophages (including those in bone marrow), and neutrophils. The mechanisms that lead to the profoundly low Hb concentrations witnessed in children with SMA are due to hemolysis and phagocytosis of parasitized and non-parasitized RBCs, and to a large extent, by suppression of erythropoiesis that is driven by PfHz-generated dysregulation in innate inflammatory mediators. While it is clear that overcoming the global burden of SMA will continue to present a serious challenge, it is our hope that gaining an improved understanding of pathogenic events that cause suppression of erythropoiesis, particularly dysregulation in innate immunity, may offer future treatment strategies to combat the unacceptable rates of morbidity and mortality associated with SMA.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. WHO. World Malaria Report 2008. Geneva, Switzerland: WHO. 2008
2. Sabbatani S, Fiorino S, Manfredi R. The emerging of the fifth malaria parasite (Plasmodium knowlesi). A public health concern? Braz J Infect Dis. 2010;14(3):299-309
3. WHO UNICEF. World malaria report 2008. Geneva: World Health Organization. 2008
4. Marsh K. et al. Indicators of life-threatening malaria in African children. N Engl Med J. 1995;332(21):1399-404
5. WHO. Severe falciparum malaria; World Health Organization, Communicable Diseases Cluster. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2000;94(Suppl 1):S1-90
6. Abdalla S.H, Pasvol G. Malaria: a hematological perspective. Tropical medicine; v4 (xvi, 429 p). London, River Edge, NJ: Imperial College Press, Distributed by World Scientific Pub. 2004
7. White N.J. Not much progress in treatment of cerebral malaria. Lancet. 1998;352(9128):594-5
8. Murray C.J.L. et al. The global burden of disease: a comprehensive assessment of mortality and disability from diseases, injuries, and risk factors in 1990 and projected to 2020. Cambridge, Mass: Harvard University Press. 1996:43
9. Lartey A. Maternal and child nutrition in Sub-Saharan Africa: challenges and interventions. Proc Nutr Soc. 2008;67(1):105-8
10. English M. et al. Assessment of inpatient paediatric care in first referral level hospitals in 13 districts in Kenya. Lancet. 2004;363(9425):1948-53
11. Brabin B.J, Premji Z, Verhoeff F. An analysis of anemia and child mortality. Nutr J. 2001;131(2S-2):636S-645S
12. Snow R.W. et al. A preliminary continental risk map for malaria mortality among African children. Parasitol Today. 1999;15(3):99-104
13. Reyburn H. et al. Association of transmission intensity and age with clinical manifestations and case fatality of severe Plasmodium falciparum malaria. JAMA. 2005;293(12):1461-70
14. Taylor T. et al. Standardized data collection for multi-center clinical studies of severe malaria in African children: establishing the SMAC network. Trans R Soc Trop Med Hyg. 2006;100(7):615-22
15. Dondorp A.M. et al. Red blood cell deformability as a predictor of anemia in severe falciparum malaria. Am J Trop Med Hyg. 1999;60(5):733-7
16. Dondorp A.M. et al. Red cell deformability, splenic function and anaemia in thalassaemia. Br Haematol J. 1999;105(2):505-8
17. Price R.N. et al. Factors contributing to anemia after uncomplicated falciparum malaria. Am J Trop Med Hyg. 2001;65(5):614-22
18. Egan A.F. et al. Aotus New World monkeys: model for studying malaria-induced anemia. Blood. 2002;99(10):3863-6
19. Buffet P.A. et al. Retention of erythrocytes in the spleen: a double-edged process in human malaria. Curr Opin Hematol. 2009;16(3):157-64
20. Abdalla S. et al. The anaemia of P. falciparum malaria. Br Haematol J. 1980;46(2):171-83
21. Phillips R.E. et al. The importance of anaemia in cerebral and uncomplicated falciparum malaria: role of complications, dyserythropoiesis and iron sequestration. Q Med J. 1986;58(227):305-23
22. Berkley J.A. et al. Bacteremia among children admitted to a rural hospital in Kenya. N Engl Med J. 2005;352(1):39-47
23. Otieno R.O. et al. Increased severe anemia in HIV-1-exposed and HIV-1-positive infants and children during acute malaria. AIDS. 2006;20(2):275-80
24. Bassat Q. et al. Severe malaria and concomitant bacteraemia in children admitted to a rural Mozambican hospital. Trop Med Int Health. 2009;14(9):1011-9
25. Davenport G.C. et al. Hematological predictors of increased severe anemia in Kenyan children coinfected with Plasmodium falciparum and HIV-1. Am Hematol J. 2010;85(4):227-33
26. Were T. et al. Bacteremia in Kenyan children presenting with malaria. J Clin Microbiol. 2011;49(2):671-6
27. Molyneux M.E. et al. Clinical features and prognostic indicators in paediatric cerebral malaria: a study of 131 comatose Malawian children. Q Med J. 1989;71(265):441-59
28. Dormer P. et al. Ineffective erythropoiesis in acute human P. falciparum malaria. Blut. 1983;46(5):279-88
29. Were T. et al. Suppression of RANTES in children with Plasmodium falciparum malaria. Haematologica. 2006;91(10):1396-9
30. Turgeon M.L. Clinical hematology: theory and procedures; 4th ed; ix, 570 p. Philadelphia: Lippincott Williams & Wilkins. 2005
31. Helleberg M. et al. Bone marrow suppression and severe anaemia associated with persistent Plasmodium falciparum infection in African children with microscopically undetectable parasitaemia. Malaria Journal. 2005;4:56
32. Krishnegowda G. et al. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J Biol Chem. 2005;280(9):8606-16
33. Nebl T, De Veer M.J, Schofield L. Stimulation of innate immune responses by malarial glycosylphosphatidylinositol via pattern recognition receptors. Parasitology. 2005;130(Suppl):S45-62
34. McKenzie F.E. et al. Strain theory of malaria: the first 50 years. Adv Parasitol. 2008;66:1-46
35. Deegan T, Maegraith B.G. Studies on the nature of malarial pigment (haemozoin). II. The pigment of the human species, Plasmodium falciparum and malariae P. Ann Trop Med Parasitol. 1956;50(2):212-22
36. Krugliak M, Zhang J, Ginsburg H. Intraerythrocytic Plasmodium falciparum utilizes only a fraction of the amino acids derived from the digestion of host cell cytosol for the biosynthesis of its proteins. Mol Biochem Parasitol. 2002;119(2):249-56
37. Liu J. et al. Plasmodium falciparum ensures its amino acid supply with multiple acquisition pathways and redundant proteolytic enzyme systems. Proc Natl Acad Sci U S A. 2006;103(23):8840-5
38. Chou A.C, Fitch C.D. Heme polymerase: modulation by chloroquine treatment of a rodent malaria. Life Sci. 1992;51(26):2073-8
39. Slater A.F, Cerami A. Inhibition by chloroquine of a novel haem polymerase enzyme activity in malaria trophozoites. Nature. 1992;355(6356):167-9
40. Slater A.F. et al. An iron-carboxylate bond links the heme units of malaria pigment. Proc Natl Acad Sci U S A. 1991;88(2):325-9
41. Egan T.J. Physico-chemical aspects of hemozoin (malaria pigment) structure and formation. J Inorg Biochem. 2002;91(1):19-26
42. Pandey A.V. et al. Hemozoin formation in malaria: a two-step process involving histidine-rich proteins and lipids. Biochem Biophys Res Commun. 2003;308(4):736-43
43. Omodeo-Sale F. et al. Destabilisation and subsequent lysis of human erythrocytes induced by Plasmodium falciparum haem products. Eur Haematol J. 2005;74(4):324-32
44. Srichaikul T, Panikbutr N, Jeumtrakul P. Bone-marrow changes in human malaria. Ann Trop Med Parasitol. 1967;61(1):40-51
45. Nguyen P.H. et al. Intraleucocytic malaria pigment and prognosis in severe malaria. Trans R Soc Trop Med Hyg. 1995;89(2):200-4
46. Casals-Pascual C. et al. Suppression of erythropoiesis in malarial anemia is associated with hemozoin in vitro and in vivo. Blood. 2006;108(8):2569-77
47. Awandare G.A. et al. Role of monocyte-acquired hemozoin in suppression of macrophage migration inhibitory factor in children with severe malarial anemia. Infect Immun. 2007;75(1):201-10
48. Kremsner P.G. et al. Prognostic value of circulating pigmented cells in African children with malaria. J Infect Dis. 2009;199(1):142-50
49. Were T. et al. Naturally acquired hemozoin by monocytes promotes suppression of RANTES in children with malarial anemia through an IL-10-dependent mechanism. Microbes Infect. 2009;11(8-9):811-9
50. Shio M. et al. Innate inflammatory response to the malarial pigment hemozoin. Microbes and Infection. 2010;12(12-13):889-899
51. Crutcher J.M. et al. Interleukin-12 and malaria. Research in Immunology. 1995;146:552-559
52. Stevenson M.M. et al. IL-12-induced protection against blood-stage Plasmodium chabaudi AS requires IFN-gamma and TNF-alpha and occurs via a nitric oxide-dependent mechanism. Immunol J. 1995;155(5):2545-56
53. Clark I.A. et al. Human malarial disease: a consequence of inflammatory cytokine release. Malar J. 2006;5:85
54. Clark I.A. Does endotoxin cause both the disease and parasite death in acute malaria and babesiosis? Lancet. 1978;2(8080):75-7
55. Grau G.E. et al. Tumor necrosis factor and disease severity in children with falciparum malaria. N Engl Med J. 1989;320(24):1586-91
56. Kwiatkowski D. et al. TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet. 1990;336(8725):1201-4
57. Kern P. et al. Elevated tumor necrosis factor alpha and interleukin-6 serum levels as markers for complicated Plasmodium falciparum malaria. Am Med J. 1989;87(2):139-43
58. Kwiatkowski D. et al. Tumour necrosis factor production in Falciparum malaria and its association with schizont rupture. Clin Exp Immunol. 1989;77(3):361-6
59. Clark I.A. et al. TNF and Plasmodium berghei ANKA-induced cerebral malaria. Immunol Lett. 1990;25(1-3):195-8
60. Clark I.A. et al. Possible roles of tumor necrosis factor in the pathology of malaria. Am Pathol J. 1987;129(1):192-9
61. van Hensbroek M.B. et al. The effect of a monoclonal antibody to tumor necrosis factor on survival from childhood cerebral malaria. J Infect Dis. 1996;174(5):1091-7
62. Calandra T, Bucala R. Macrophage migration inhibitory factor: a counter-regulator of glucocorticoid action and critical mediator of septic shock. Inflamm J. 1995;47(1-2):39-51
63. Lan H.Y. et al. TNF-alpha up-regulates renal MIF expression in rat crescentic glomerulonephritis. Mol Med. 1997;3(2):136-44
64. Rockett K.A. et al. In vivo induction of nitrite and nitrate by tumor necrosis factor, lymphotoxin, and interleukin-1: possible roles in malaria. Infect Immun. 1992;60(9):3725-30
65. Rockett K. et al. Killing of Plasmodium falciparum in vitro by nitric oxide derivatives. Infection and Immunity. 1991;59:3280
66. Perkins D.J, Kniss D.A. Tumor necrosis factor-alpha promotes sustained cyclooxygenase-2 expression: attenuation by dexamethasone and NSAIDs. Prostaglandins. 1997;54(4):727-43
67. Schwartz J.E. et al. A phase I trial of recombinant tumor necrosis factor (rTNF) administered by continuous intravenous infusion in patients with disseminated malignancy. Biotherapy. 1989;1(3):207-14
68. Hensmann M, Kwiatkowski D. Cellular basis of early cytokine response to Plasmodium falciparum. Infect Immun. 2001;69(4):2364-71
69. Artavanis-Tsakonas K, Riley E.M. Innate immune response to malaria: rapid induction of IFN-gamma from human NK cells by live Plasmodium falciparum-infected erythrocytes. Immunol J. 2002;169(6):2956-63
70. D'Ombrain M.C. et al. gammadelta-T cells expressing NK receptors predominate over cells NK and conventional T cells in the innate IFN-gamma response to Plasmodium falciparum malaria. Eur Immunol J. 2007;37(7):1864-73
71. D'Ombrain M.C. et al. Association of early interferon-gamma production with immunity to clinical malaria: a longitudinal study among Papua New Guinean children. Clin Infect Dis. 2008;47(11):1380-7
72. Pombo D.J. et al. Immunity to malaria after administration of ultra-low doses of red cells infected with Plasmodium falciparum. Lancet. 2002;360(9333):610-7
73. McCall M.B. et al. Early interferon-gamma response against Plasmodium falciparum correlates with interethnic differences in susceptibility to parasitemia between sympatric Fulani and Dogon in Mali. J Infect Dis. 2010;201(1):142-52
74. Ong'echa J.M. et al. Association of interferon-gamma responses to pre-erythrocytic stage vaccine candidate antigens of Plasmodium falciparum in young Kenyan children with improved hemoglobin levels: XV. Asembo Bay Cohort Project. Am J Trop Med Hyg. 2003;68(5):590-7
75. Kremsner P.G. et al. Prediction of accelerated cure in Plasmodium falciparum malaria by the elevated capacity of tumor necrosis factor production. Am J Trop Med Hyg. 1995;53(5):532-8
76. Lyke K.E. et al. Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1beta), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect Immun. 2004;72(10):5630-7
77. Perkins D.J, Weinberg J.B, Kremsner P.G. Reduced interleukin-12 and transforming growth factor-beta1 in severe childhood malaria: relationship of cytokine balance with disease severity. J Infect Dis. 2000;182(3):988-92
78. Biemba G. et al. Prolonged macrophage activation and persistent anaemia in children with complicated malaria. Trop Med Int Health. 1998;3(1):60-5
79. Clark I.A, Cowden W.B. The pathophysiology of falciparum malaria. Pharmacol Ther. 2003;99(2):221-60
80. Dinarello C.A. Infection, fever, and exogenous and endogenous pyrogens: some concepts have changed. J Endotoxin Res. 2004;10(4):201-22
81. Rockett K.A. et al. Tumor necrosis factor and interleukin-1 synergy in the context of malaria pathology. Am J Trop Med Hyg. 1994;50(6):735-42
82. Pascual V. et al. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201(9):1479-86
83. Dinarello C.A. Blocking IL-1 in systemic inflammation. J Exp Med. 2005;201(9):1355-9
84. Pied S. et al. IL-6 induced by IL-1 inhibits malaria pre-erythrocytic stages but its secretion is down-regulated by the parasite. Immunol J. 1992;148(1):197-201
85. Curfs J.H. et al. Low dosages of interleukin 1 protect mice against lethal cerebral malaria. J Exp Med. 1990;172(5):1287-91
86. Prakash D. et al. Clusters of cytokines determine malaria severity in Plasmodium falciparum-infected patients from endemic areas of Central India. J Infect Dis. 2006;194(2):198-207
87. Vogetseder A. et al. Time course of coagulation parameters, cytokines and adhesion molecules in Plasmodium falciparum malaria. Trop Med Int Health. 2004;9(7):767-73
88. John C.C. et al. Low levels of RANTES are associated with mortality in children with cerebral malaria. J Infect Dis. 2006;194(6):837-45
89. Krishna S. et al. Lactic acidosis and hypoglycaemia in children with severe malaria: pathophysiological and prognostic significance. Trans R Soc Trop Med Hyg. 1994;88(1):67-73
90. Ouma C. et al. Polymorphic variability in the interleukin (IL)-1beta promoter conditions susceptibility to severe malarial anemia and functional changes in IL-1beta production. J Infect Dis. 2008;198(8):1219-26
91. Aubouy A, Deloron P, Migot-Nabias F. Plasma and in vitro levels of cytokines during and after a Plasmodium falciparum malaria attack in Gabon. Acta Trop. 2002;83(3):195-203
92. Harpaz R. et al. Serum cytokine profiles in experimental human malaria. Relationship to protection and disease course after challenge. J Clin Invest. 1992;90(2):515-23
93. Coleman R.M, Bruce A, Rencricca N.J. Malaria: macrophage migration inhibition factor (MIF). Parasitol J. 1976;62(1):137-8
94. Awandare G.A. et al. Decreased circulating macrophage migration inhibitory factor (MIF) protein and blood mononuclear cell MIF transcripts in children with Plasmodium falciparum malaria. Clin Immunol. 2006;119(2):219-25
95. Awandare G.A. et al. A macrophage migration inhibitory factor promoter polymorphism is associated with high-density parasitemia in children with malaria. Genes Immun. 2006;7(7):568-75
96. De Mast Q. et al. A decrease of plasma macrophage migration inhibitory factor concentration is associated with lower numbers of circulating lymphocytes in experimental Plasmodium falciparum malaria. Parasite Immunol. 2008;30(3):133-8
97. Awandare G.A. et al. MIF (macrophage migration inhibitory factor) promoter polymorphisms and susceptibility to severe malarial anemia. J Infect Dis. 2009;200(4):629-37
98. Jain V. et al. Macrophage migration inhibitory factor is associated with mortality in cerebral malaria patients in India. BMC Res Notes. 2009;2:36
99. David J.R, Delayed hypersensitivity in vitro. its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc Natl Acad Sci U S A. 1966;56(1):72-7
100. Bacher M. et al. An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc Natl Acad Sci U S A. 1996;93(15):7849-54
101. Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3(10):791-800
102. Bernhagen J, Calandra T, Bucala R. Regulation of the immune response by macrophage migration inhibitory factor: biological and structural features. J Mol Med (Berl). 1998;76(3-4):151-61
103. Bernhagen J. et al. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature. 1993;365(6448):756-9
104. Calandra T. et al. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med. 2000;6(2):164-70
105. Koebernick H. et al. Macrophage migration inhibitory factor (MIF) plays a pivotal role in immunity against Salmonella typhimurium. Proc Natl Acad Sci U S A. 2002;99(21):13681-6
106. Juttner S. et al. Migration inhibitory factor induces killing of Leishmania major by macrophages: dependence on reactive nitrogen intermediates and endogenous TNF-alpha. Immunol J. 1998;161(5):2383-90
107. Martiney J. et al. Macrophage migration inhibitory factory release by macrophages after ingestion of Plasmodium chaubaudi-Infected erythrocytes: possible role in the pathogenesis of malarial anemia. Infection and Immunity. 2000;68(4):2259-2267
108. McDevitt M.A. et al. A critical role for the host mediator macrophage migration inhibitory factor in the pathogenesis of malarial anemia. J Exp Med. 2006;203(5):1185-96
109. Chaisavaneeyakorn S. et al. Levels of macrophage inflammatory protein 1 alpha (MIP-1 alpha) and MIP-1 beta in intervillous blood plasma samples from women with placental malaria and human immunodeficiency virus infection. Clin Diagn Lab Immunol. 2003;10(4):631-6
110. Chaiyaroj S.C. et al. Reduced levels of transforming growth factor-beta1, interleukin-12 and increased migration inhibitory factor are associated with severe malaria. Acta Trop. 2004;89(3):319-27
111. Clark I.A. et al. Tissue distribution of migration inhibitory factor and inducible nitric oxide synthase in falciparum malaria and sepsis in African children. Malar J. 2003;2:6
112. Cua D. et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:774-748
113. Wiekowski M.T. et al. Ubiquitous transgenic expression of the IL-23 subunit p19 induces multiorgan inflammation, runting, infertility, and premature death. Immunol J. 2001;166(12):7563-70
114. Oppmann B. et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13(5):715-25
115. Shimozato O. et al. The secreted form of the p40 subunit of interleukin (IL)-12 inhibits IL-23 functions and abrogates IL-23-mediated antitumour effects. Immunology. 2006;117(1):22-8
116. Parham C. et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. Immunol J. 2002;168(11):5699-708
117. Pirhonen J, Matikainen S, Julkunen I. Regulation of virus-induced IL-12 and IL-23 expression in human macrophages. Immunol J. 2002;169(10):5673-8
118. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3(2):133-46
119. Aste-Amezaga M. et al. Molecular mechanisms of the induction of IL-12 and its inhibition by IL-10. Immunol J. 1998;160(12):5936-44
120. Schuetze N. et al. IL-12 family members: differential kinetics of their TLR4-mediated induction by Salmonella enteritidis and the impact of IL-10 in bone marrow-derived macrophages. Int Immunol. 2005;17(5):649-59
121. Gately M.K. et al. Interleukin-12 antagonist activity of mouse interleukin-12 p40 homodimer in vitro and in vivo. Ann N Y Acad Sci. 1996;795:1-12
122. Aggarwal S. et al. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278(3):1910-4
123. Ong'echa J.M. et al. Increased circulating interleukin (IL)-23 in children with malarial anemia: In vivo and in vitro relationship with co-regulatory cytokines IL-12 and IL-10. Clin Immunol. 2008Feb;126(2):211-21
124. Gately M.K. et al. The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu Rev Immunol. 1998;16:495-521
125. Trinchieri G. Interleukin-12: a cytokine at the interface of inflammation and immunity. Adv Immunol. 1998;70:83-243
126. Mosser D.M, Karp C.L. Receptor mediated subversion of macrophage cytokine production by intracellular pathogens. Curr Opin Immunol. 1999;11(4):406-11
127. Mohan K, Sam H, Stevenson M.M. Therapy with a combination of low doses of interleukin 12 and chloroquine completely cures blood-stage malaria, prevents severe anemia, and induces immunity to reinfection. Infect Immun. 1999;67(2):513-9
128. Mohan K, Stevenson M.M. Dyserythropoiesis and severe anaemia associated with malaria correlate with deficient interleukin-12 production. Br Haematol J. 1998;103(4):942-9
129. Mohan K, Moulin P, Stevenson M.M. Natural killer cell cytokine production, not cytotoxicity, contributes to resistance against blood-stage Plasmodium chabaudi AS infection. Immunol J. 1997;159(10):4990-8
130. Sam H, Stevenson M.M. Early IL-12 p70, but not p40, production by splenic macrophages correlates with host resistance to blood-stage Plasmodium chabaudi AS malaria. Clin Exp Immunol. 1999;117(2):343-9
131. Su Z, Stevenson M.M. IL-12 is required for antibody-mediated protective immunity against blood-stage Plasmodium chabaudi AS malaria infection in mice. Immunol J. 2002;168(3):1348-55
132. Bellone G, Trinchieri G. Dual stimulatory and inhibitory effect of NK cell stimulatory factor/IL-12 on human hematopoiesis. Immunol J. 1994;153(3):930-7
133. Dybedal I, Larsen S, Jacobsen S.E. IL-12 directly enhances in vitro murine erythropoiesis in combination with IL-4 and stem cell factor. Immunol J. 1995;154(10):4950-5
134. Luty A.J. et al. Low interleukin-12 activity in severe Plasmodium falciparum malaria. Infect Immun. 2000;68(7):3909-15
135. Keller C.C. et al. Acquisition of hemozoin by monocytes down-regulates interleukin-12 p40 (IL-12p40) transcripts and circulating IL-12p70 through an IL-10-dependent mechanism: in vivo and in vitro findings in severe malarial anemia. Infect Immun. 2006;74(9):5249-60
136. Ho M. et al. Endogenous interleukin-10 modulates proinflammatory response in Plasmodium falciparum malaria. J Infect Dis. 1998;178(2):520-5
137. Othoro C. et al. A low interleukin-10 tumor necrosis factor-alpha ratio is associated with malaria anemia in children residing in a holoendemic malaria region in western Kenya. J Infect Dis. 1999;179(1):279-82
138. Kurtzhals J.A. et al. Low plasma concentrations of interleukin 10 in severe malarial anaemia compared with cerebral and uncomplicated malaria. Lancet. 1998;351(9118):1768-72
139. Omer F.M, Riley E.M. Transforming growth factor beta production is inversely correlated with severity of murine malaria infection. J Exp Med. 1998;188(1):39-48
140. Gourley I.S. et al. Profound bias in interferon-gamma and interleukin-6 allele frequencies in western Kenya, where severe malarial anemia is common in children. J Infect Dis. 2002;186(7):1007-12
141. Zermati Y. et al. Transforming growth factor inhibits erythropoiesis by blocking proliferation and accelerating differentiation of erythroid progenitors. Exp Hematol. 2000;28(8):885-94
142. Hino M. et al. Effects of type beta transforming growth factors on haematopoietic progenitor cells. Br Haematol J. 1988;70(2):143-7
143. Sing G.K. et al. Transforming growth factor beta selectively inhibits normal and leukemic human bone marrow cell growth in vitro. Blood. 1988;72(5):1504-11
144. Malaguarnera L. et al. Plasma levels of interleukin-12 (IL-12), interleukin-18 (IL-18) and transforming growth factor beta (TGF-beta) in Plasmodium falciparum malaria. Eur Cytokine Netw. 2002;13(4):425-30
145. Dietmann A. et al. Endoglin in African children with Plasmodium falciparum malaria: a novel player in severe malaria pathogenesis? J Infect Dis. 2009;200(12):1842-8
146. Sambo M.R. et al. Transforming growth factor beta 2 and heme oxygenase 1 genes are risk factors for the cerebral malaria syndrome in Angolan children. PLoS One. 2010;5(6):e11141
147. Rollins B.J. Chemokines. Blood. 1997;90(3):909-28
148. Burgmann H. et al. Serum concentrations of MIP-1 alpha and interleukin-8 in patients suffering from acute Plasmodium falciparum malaria. Clin Immunol Immunopathol. 1995;76(1 Pt 1):32-6
149. Friedland J.S. et al. Interleukin-8 and Plasmodium falciparum malaria in Thailand. Trans R Soc Trop Med Hyg. 1993;87(1):54-5
150. Jaramillo M, Godbout M, Olivier M. Hemozoin induces macrophage chemokine expression through oxidative stress-dependent and -independent mechanisms. Immunol J. 2005;174(1):475-84
151. Ochiel D.O. et al. Differential regulation of beta-chemokines in children with Plasmodium falciparum malaria. Infect Immun. 2005;73(7):4190-7
152. Conlon K. et al. CD8+ and CD45RA+ human peripheral blood lymphocytes are potent sources of macrophage inflammatory protein 1 alpha, interleukin-8 and RANTES. Eur Immunol J. 1995;25(3):751-6
153. Marfaing-Koka A. et al. Contrasting effects of IL-4, IL-10 and corticosteroids on RANTES production by human monocytes. Int Immunol. 1996;8(10):1587-94
154. Mariani E. et al. RANTES and MIP-1alpha production by lymphocytes T, monocytes and NK cells from nonagenarian subjects. Exp Gerontol. 2002;37(2-3):219-26
155. Umland O. et al. Induction of various immune modulatory molecules in CD34(+) hematopoietic cells. J Leukoc Biol. 2004;75(4):671-9
156. Tang Y.Q, Yeaman M.R, Selsted M.E. Antimicrobial peptides from human platelets. Infect Immun. 2002;70(12):6524-33
157. Luster A.D. The role of chemokines in linking innate and adaptive immunity. Current Opinion in Immunology. 2002;14(1):129-135
158. Stoiser B. et al. Serum concentrations of granulocyte-colony stimulating factor in complicated Plasmodium falciparum malaria. Eur Cytokine Netw. 2000;11(1):75-80
159. Van Zant G, Goldwasser E. Simultaneous effects of erythropoietin and colony-stimulating factor on bone marrow cells. Science. 1977;198(4318):733-5
160. Kojima S. et al. Treatment of aplastic anemia in children with recombinant human granulocyte colony-stimulating factor. Blood. 1991;77(5):937-41
161. Papaldo P. et al. Does granulocyte colony-stimulating factor worsen anemia in early breast cancer patients treated with epirubicin and cyclophosphamide? J Clin Oncol. 2006;24(19):3048-55
162. Liehl E. et al. Prediction of the role of granulocyte-macrophage colony-stimulating factor in animals and man from in vitro results. Eur J Clin Microbiol Infect Dis. 1994;13(Suppl 2):S9-17
163. Kumaratilake L.M. et al. GM-CSF-induced priming of human neutrophils for enhanced phagocytosis and killing of asexual blood stages of Plasmodium falciparum: synergistic effects of GM-CSF and TNF. Parasite Immunol. 1996;18(3):115-23
164. Chang K.H, Stevenson M.M. Effect of anemia and renal cytokine production on erythropoietin production during blood-stage malaria. Kidney Int. 2004;65(5):1640-6
165. Keller C.C. et al. Suppression of a novel hematopoietic mediator in children with severe malarial anemia. Infect Immun. 2009;77(9):3864-71
166. Hiraoka A. et al. Stem cell growth factor: in situ hybridization analysis on the gene expression, molecular characterization and in vitro proliferative activity of a recombinant preparation on primitive hematopoietic progenitor cells. Hematol J. 2001;2(5):307-15
167. Mio H. et al. Isolation and characterization of a cDNA for human mouse, and rat full-length stem cell growth factor, a new member of C-type lectin superfamily. Biochem Biophys Res Commun. 1998;249(1):124-30
168. Ouma C. et al. A novel functional variant in the stem cell growth factor promoter protects against severe malarial anemia. Infect Immun. 2010;78(1):453-60
169. Nathan C, Xie Q.W. Nitric oxide synthases—Roles, tolls, and controls [Review]. Cell. 1994;78(6):915-918
170. Perkins D. et al. Blood monocuclear cell nitric oxide production and plasma cytokine levels in healthy Gabonese children with prior mild or severe malaria. Infection and Immunity. 1999;67:4977-4981
171. Geller D.A, Billiar T.R. Molecular biology of nitric oxide synthases. Cancer & Metastasis Reviews. 1998;17(1):7-23
172. Kremsner P.G. et al. High plasma levels of nitrogen oxides are associated with severe disease and correlate with rapid parasitological and clinical cure in Plasmodium falciparum malaria. Trans R Soc Trop Med Hyg. 1996;90(1):44-7
173. Perkins D.J. et al. Blood mononuclear cell nitric oxide production and plasma cytokine levels in healthy gabonese children with prior mild or severe malaria. Infect Immun. 1999;67(9):4977-81
174. Keller C.C. et al. Elevated nitric oxide production in children with malarial anemia: hemozoin-induced nitric oxide synthase type 2 transcripts and nitric oxide in blood mononuclear cells. Infect Immun. 2004;72(8):4868-73
175. Greve B. et al. High oxygen radical production is associated with fast parasite clearance in children with Plasmodium falciparum malaria. J Infect Dis. 1999;179(6):1584-6
176. Postma N.S. et al. Oxidative stress in malaria; implications for prevention and therapy. Pharm World Sci. 1996;18(4):121-9
177. Griffiths M.J. et al. Oxidative stress and erythrocyte damage in Kenyan children with severe Plasmodium falciparum malaria. Br Haematol J. 2001;113(2):486-91
178. Narsaria N. et al. Oxidative stress in children with severe malaria. J Trop Pediatr. 2011 epub
179. Vane J.R, Bakhle Y.S, Botting R.M. Cyclooxygenases 1 and 2. Annual Review of Pharmacology and Toxicology. 1998;38:97-120
180. Perkins D.J. et al. In vivo acquisition of hemozoin by placental blood mononuclear cells suppresses PGE2, TNF-alpha, and IL-10. Biochem Biophys Res Commun. 2003;311(4):839-46
181. Perkins D.J, Kremsner P.G, Weinberg J.B. Inverse relationship of plasma prostaglandin E2 and blood mononuclear cell cyclooxygenase-2 with disease severity in children with Plasmodium falciparum malaria. J Infect Dis. 2001;183(1):113-8
182. Perkins D.J. et al. Impaired systemic production of prostaglandin E2 in children with cerebral malaria. J Infect Dis. 2005;191(9):1548-57
183. Keller C.C. et al. Reduced peripheral PGE2 biosynthesis in Plasmodium falciparum malaria occurs through hemozoin-induced suppression of blood mononuclear cell cyclooxygenase-2 gene expression via an interleukin-10-independent mechanism. Mol Med. 2004;10(1-6):45-54
184. Keller C.C. et al. Suppression of prostaglandin E2 by malaria parasite products and antipyretics promotes overproduction of tumor necrosis factor-alpha: association with the pathogenesis of childhood malarial anemia. J Infect Dis. 2006;193(10):1384-93
185. Lamikanra A.A. et al. Hemozoin (malarial pigment) directly promotes apoptosis of erythroid precursors. PLoS One. 2009;4(12):e8446
186. Awandare G.A. et al. Mechanisms of erythropoiesis inhibition by malarial pigment and malaria-induced proinflammatory mediators in an in vitro model. Am Hematol J. 2011;86(2):155-62
Author contact
![]() Corresponding author: Douglas J. Perkins, dperkinsunm.edu
Corresponding author: Douglas J. Perkins, dperkinsunm.edu