ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2012; 8(1):1-14. doi:10.7150/ijbs.8.1 This issue Cite
Research Paper
Enhanced Antitumor Efficacy of Gemcitabine by Evodiamine on Pancreatic Cancer via Regulating PI3K/Akt Pathway
Wei-Tian Wei2,3,#, Hui Chen3, #, Zhao-Hong Wang3, Zhong-Lin Ni3, Hai-Bin Liu3, Hong-Fei Tong3, Hong-Chun Guo3, Dian-Lei Liu3, Sheng-Zhang Lin1,3, ![]()
1. Department of Hepato-biliary-pancreatic Surgery, First Affiliated Hospital of Medical College, Zhejiang University, Hangzhou, China.
2. Department of Oncological Surgery, Zhejiang Cancer Hospital, Hangzhou, China.
3. Department of Hepato-biliary-pancreatic Surgery, Second Affiliated Hospital of Wenzhou Medical College, Zhejiang, China.
# These authors contributed equally to this work.
Abstract
Evodiamine has therapeutic potential against cancers. This study was designed to investigate whether combination therapy with gemcitabine and evodiamine enhanced antitumor efficacy in pancreatic cancer. In vitro application of the combination therapy triggered significantly higher frequency of pancreatic cancer cells apoptosis, inhibited the activities of PI3K, Akt, PKA, mTOR and PTEN, and decreased the activation of NF-κB and expression of NF-κB-regulated products. In vivo application of the combination therapy induced significant enhancement of tumor cell apoptosis, reductions in tumor volume, and inhibited activation of mTOR and PTEN. In conclusion, evodiamine can augment the therapeutic effect of gemcitabine in pancreatic cancer through direct or indirect negative regulation of the PI3K/Akt pathway.
Keywords: evodiamine, gemcitabine, pancreatic cancer SW1990 cells, apoptosis, PTEN, mTOR
Introduction
Pancreatic cancer has a high incidence of local recurrence and develops distant metastasis, leading to extremely poor prognosis (1). Many patients with locally advanced or metastatic pancreatic cancer are stable on chemotherapy with gemcitabine. Although gemcitabine is the most potent and standard treatment of pancreatic cancer (2, 3), the tumor response rate of gemcitabine is below 10%. Previous studies have shown that treatment with gemcitabine results in the median survival time of about five to six months (4, 5). Furthermore, although treatment with gemcitabine and erlotinib or capecitabine benefits patients with pancreatic cancer, these therapeutic strategies fail to significantly prolong the survival time of pancreatic cancer patients (6, 7). Therefore, development of new chemotherapeutic approaches to enhance the therapeutic effect and reduce the development of drug-resistance will be of great significance in the clinical management of pancreatic cancer.
Constitutive activation of nuclear factor-κB (NF-κB) can promote cell proliferation, inhibit cell apoptosis and regulate the expression of genes associated with the tumor-related invasion (8) and angiogenesis (9, 10), reflecting the aggressive behavior of pancreatic cancer (11). Notably, gemcitabine can up-regulate NF-κB expression in pancreatic cancer cells, which is associated with the development of chemoresistance and poor outcome in cancer patients, including patients with pancreatic cancer (12-16). PI3K and Akt are kinases that play a critical role in human cancer. Asano et al. (17) has reported that PI3K and Akt are activated due to aberrant PTEN expression and essential for the function of constitutively activated NF-κB in pancreatic cancer. Another report has shown that PI3K/Akt pathway is constitutively activated in a majority of human pancreatic cancer cell lines and PI3K/Akt has emerged as a promising target for therapeutic intervention (18). Arlt et al. (16) suggested that PI3K/Akt was not involved in gemcitabine resistance, but another report (19) demonstrated that Akt activity is necessary for the induction of NF-κB after gemcitabine treatment, though the mechanism does not involve activation of Akt.
Evodiamine is one of the main constituents of Evodiae fructus (20) and has been shown to exhibit anti-tumor properties (21). The apoptotic activity of evodiamine was shown to be due to its inhibition of NF-κB activation via suppression of IκBα kinase activity, which leads to inhibition of NF-κB-regulated gene products such as XIAP, Bcl-2, and Bcl-xL (8). Previous study has also revealed the molecular mechanism by which evodiamine increases the expression of proapoptotic Bax and decreases that of anti-apoptotic Bcl-2, which amplified the activation of the caspase cascade, triggering consequent responses and cell death (21-23). PI3K was also found to exhibit essential regulatory effects on functions of SIRT1, p53 and other signaling proteins involved in evodiamine-induced A375-S2 cell death (24, 25). Based on these reports, we hypothesized that evodiamine may augment the gemcitabine-induced anti-tumor effect on pancreatic cancer via direct or possibly indirect inhibition of the PI3K/Akt pathway targeting NF-κB.
This study showed that evodiamine inhibited the spontaneous and gemcitabine-induced NF-κB activation, and the expression of NF-κB-regulated proteins in SW1990 cells. Importantly, evodiamine inhibited the activation of PI3K/Akt pathway, phosphorylation of PTEN and mammalian target of rapamycin (mTOR), and activity of cAMP-dependent protein kinase A (PKA) that was not influenced by gemcitabine. Our data suggest that evodiamine may augment the therapeutic effect of gemcitabine in pancreatic cancer through direct or indirect negative regulation of the PI3K/Akt pathway that targets NF-κB.
Materials and Methods
Reagents
Evodiamine (purity: >99%) was purchased from Sigma (St. Louis, MO, USA), and dissolved in dimethylsulfoxide (DMSO) at 0.2 mmol/L to make the stock solution. Gemcitabine was purchased from Ely Lilly (Bad Homburg, Germany) and dissolved in sterile saline at 50 g/L for stock solution, with a final concentration of DMSO at <0.1%. Rabbit polyclonal antibodies against phospho-PTEN(Ser380/Thr382/383) and PI3K(Tyr458) were purchased from Cell Signaling Technology (Beverly, MA, USA). Rabbit polyclonal antibodies against Bax and Bcl-2, phospho-mTOR(Ser2448), Rictor-mTOR, phospho-Akt(Ser473), rabbit monoclonal antibody against NF-κB(p65), survivin and active caspase 3 were purchased from Abcam (Cambridge, UK). The in Situ Cell Death Detection Kit was purchased from Roche (Basel, Switzerland).
Cell culture
Human pancreatic tumor cell line, SW1990, was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in RPMI-1640 supplemented with 10% heat-incubated fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA), 100 units/mL of penicillin, and 100 μg/mL of streptomycin and incubated at 37˚C in a humidified 5% CO2 atmosphere. Some SW1990 cells were stably transfected with luciferase, as previously described for Panc-1 cells (26). Luciferase-transfected SW1990 cells were routinely cultured in the same condition as SW1990 cells.
MTT assay
The cytotoxicity of individual drugs against SW1990 cells was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (MTT; Sigma), as previously described (27). Briefly, SW1990 cells at 5×103 cells/well were cultured in 96-well microtiter plates overnight and treated in triplicate with 20 μmol/L of gemcitabine for 24 h, and/or 1.0 μmol/L of evodiamine for 48 h. Untreated cells in medium alone were used as controls. The viability of control cells was designated as 100% and the viability of other experimental groups was calculated relative to controls.
In vitro treatment protocol and apoptosis assay
SW1990 cells at 2×105/mL were cultured overnight in six-well plates and treated in triplicate with 20 μmol/L of gemcitabine for 24 h, and/or 1.0 μmol/L of evodiamine for 48 h. Untreated cells in medium alone were used as controls. Subsequently, the cells were harvested, washed and stained with 10 µL of Annexin V and 5 µL of propidium iodide (PI) in the dark for 15 min at room temperature, according to the manufacturer's instructions (Biosea, Beijing, China). The apoptotic cells were detected and measured by flow cytometer (Epics AltraII; Beckman Coulter, Fullerton, CA, USA) and examined under an inverse fluorescent microscope.
Electrophoretic mobility shift assay (EMSA)
NF-κB activity was evaluated by EMSA analysis, as described elsewhere (28). Nuclear proteins were extracted from treated or untreated cells and protein concentrations were determined by BCA assay. The Biotin end-labeled DNA duplex of sequence 5′-AGT TGA GGG GAC TTT CCC AGG C-3′ and 3′-TCA ACT CCC CTG AAA GGG TCC G-5′ containing a putative binding site for NF-κB was incubated with the nuclear extracts. Subsequently, the DNA-protein complexes were subjected to a 6% native polyacrylamide gel electrophoresis (PAGE) and transferred to a nylon membrane (Pierce, Rockford, IL, USA), and cross-linked for 15 min on a UV transilluminator, followed by detection using the LightShiftTM Chemiluminescent EMSA kit (Pierce), according to the manufacturers' instructions. The membranes were exposed to X-ray films and the relative intensities were analyzed using the NIH Image 1.62 package. The nuclear extracts from unstimulated gastric cancer SGC7901 cells were used as negative control and SGC7901cells stimulated with 50 ng/mL TNFα were used as positive controls.
Western blot analysis
SW1990 cells (2×106/plate) were treated with drug(s) as described above and harvested. Total proteins were extracted and concentrations were determined by the BCA assay. The lysates (20 μg/lane) were separated by 8-12% sodium dodecyl sulfate-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes. After being blocked with 5% fat-free milk, the membranes were probed with individual primary antibodies overnight at 4ºC and the bound antibodies were detected with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG. The formed immunocomplex was visualized by enhanced chemiluminescence reagent (ECL, Pierce) and exposed to X-ray films. Quantitative data were expressed as mean%±SD of the relative levels of the objective protein and control β-actin of each group of cells from three independent experiments.
Measurement of intracellular cAMP concentration
SW1990 cells (2×106/plate) were treated with drug(s) as described above and harvested. cAMP levels were determined by cAMP enzyme immunoassay kit following the manufacturer's instructions (Cayman Chemical Co., Ann Arbor, MI, USA). The protein concentration of total cell lysate was used as loading control, and the results were expressed as picomoles of cAMP per microgram of total protein.
Measurement of intracellular PKA activity
SW1990 cells were treated drug(s) as described above and collected with PKA extraction buffer before breakdown under 4˚C. Individual supernatants were collected by centrifugation (11,000×g, 10 min at 4˚C) to detect PKA activity using a PKA activity assay kit (Upstate, Lake Placid, NY, USA).
Experimental animals and protocols
Female BALB/c nude mice at four to six weeks of age were purchased from the Shanghai Laboratory Animals Center (China). All animals were housed in a specific pathogen-free facility. The experimental protocols involving animals were approved by the Animal Research and Ethical Committee of Wenzhou Medical College.
To establish the SW1990 pancreatic cancer cells xenograft tumor model, individual mice were implanted subcutaneously with 2×106 SW1990 cells in 200 μL of PBS on the back of each animal and monitored for the development of tumors for up to three weeks post-implantation. When tumors reached ~5 mm in one dimension (length, width, or height measured by Vernier calipers), the mice were randomized and treated intraperitoneally (IP) with saline, gemcitabine alone (125 mg/kg) (29), evodiamine alone (10 mg/kg) (30), or combined gemcitabine (80 mg/kg) with evodiamine (10 mg/kg) every three days for up to 30 days post-implantation (n=12 per group). The experimental protocols are illustrated in Figure 1. One week after the last treatment, animals were weighed, sacrificed and the tumors were excised. The final tumor volume was estimated by the following formula: V=2/3πr3, where r is the mean of the three dimensions (length, width, and height). A portion of the tumor tissue was formalin-fixed and paraffin-embedded for subsequent apoptosis assay and immunohistochemistry analysis.
Luciferase-transfected SW1990 cells xenograft tumor models were established by subcutaneously inoculating the cells into the bilateral abdominal flanks (three places on each side) of nude mice. After three weeks of implantation, a total of 48 nude mice were randomized into four treatment groups (n=12) based on the bioluminescence measured after the first IVIS imaging. The treatment protocol of each group was the same as described above. One week after the last treatment, the numbers of tumors that had grown in each mouse were monitored by the bioluminescence IVIS Imaging System 200 (Caliper Life Sciences, Hopkinton, MA, USA) using a cryogenically cooled imaging system coupled to a data acquisition computer running Living Image software (Xenogen Corp., Alameda, CA, USA). Before imaging, animals were anesthetized by asphyxiation in an acrylic chamber filled with 2.5% isoflurane/air mixture. Immediately afterwards, mice were intraperitoneally (i.p.) injected with 40 mg/mL D-luciferin potassium salt in PBS at a dose of 150 mg/kg. After 10 min of incubation with luciferin, mice were placed in a prone position and a digital grayscale animal image was acquired followed by acquisition and overlay of a pseudocolor image representing the spatial distribution of detected photons emerging from active luciferase within the animal. Signal intensity was quantified as the sum of all detected photons within the region of interest per second.
In situ detection of apoptotic cells in tumor tissue
The apoptotic tumor cells in the tumor tissues were characterized by the terminal deoxynucleotidyl transferase-mediated deoxyuridine tri-phosphate nick-end labeling (TUNEL) method using the In Situ Cell Death Detection Kit, according to the manufacturers' instruction. The tumor tissue sections were subjected to TUNEL analysis and apoptotic cells were examined under a laser scanning confocal microscope with 400x observation camera. Images were captured and a total of 10 fields with strongest fluorescence from individual tissue samples were examined. The integrated optical density (IOD) was analyzed by using Image Pro Plus software (MediaCybernetics, Bethesda, MD, USA). Statistical differences between different groups were then analyzed.
Immunohistochemistry detection of phospho-mTOR(Ser2448) and phospho-PTEN(Ser380) by
Sections of paraffin-embedded pancreatic cancer tissues were deparaffinized and rehydrated prior to non-specific antigen blocking with goat serum. Immunostaining was performed using primary antibodies specific for phospho-mTOR(Ser2448) or phospho-PTEN(Ser380), followed by staining with the appropriate HRP-conjugated secondary antibodies. The immunostained sections were developed in diaminobenzidine (DAB) and counterstained with a weak solution of haematoxylin solution. The stained slides were dehydrated and mounted in Permount (Fisher Scientific, Pittsburg, PA, USA) and visualized on a light microscope (Olympus, Tokyo, Japan). Images were captured with an attached camera linked to a computer. For data quantification, IOD level was analyzed by using the Image Pro Plus 6.0 software. Twelve nude mice were included for each experimental group. One section was obtained from each animal. At least six fields were randomly selected from each section. The average IOD levels in different groups were then compared by statistical analysis.
Statistical analysis
Data shown are representative images or expressed as mean%±SD of each group. The difference among groups of cells or mice was analyzed by Chi-square (X2) test or ANOVA, followed by post hoc Student's t-test using the SPSS v17.0 software (Chicago, IL, USA). A P-value of less than 0.05 was considered statistically significant.
Schematic illustration of the experimental protocol.

Results
Cytotoxicity of evodiamine plus gemcitabine against SW1990 cells in vitro
As shown by MTT assays (Fig. 2A), treatment with evodiamine or gemcitabine alone reduced the cell viability by nearly 42% and 57%, respectively. Treatment with combination of evodiamine and gemcitabine significantly reduced the cell viability by 80%. Further analysis by cell apoptosis assay indicated that single evodiamine increased the apoptotic rate from 12.6% to 28.6% and single gemcitabine increased the apoptotic rate from 12.6% to 34.2%, while treatment with evodiamine plus gemcitabine induced nearly 57.6% of cell apoptosis (Fig. 2B and C). These data are consistent with results from cell growth inhibition studies using MTT, suggesting that the loss of viable cells by evodiamine and/or gemcitabine is partly due to the induction of cell apoptosis.
Effect of evodiamine on the gemcitabine-induced NF-κB activity in vitro
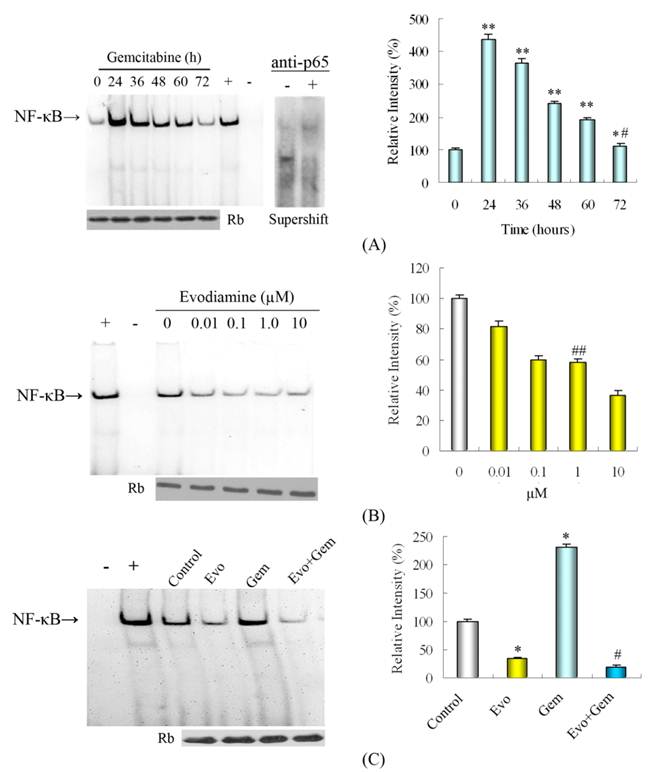
SW1990 cells were treated with 20 μM of gemcitabine for varying periods and the DNA binding activity of NF-κB in the nuclear extracts was characterized by EMSA. As shown in Fig. 3A, SW1990 cells in the absence of gemcitabine displayed low levels of DNA binding activity, indicating low levels of spontaneous NF-κB activation. Treatment of SW1990 cells with gemcitabine for 24 h increased the DNA binding activity of NF-κB by 436% in vitro, then activity of NF-κB gradually decreased to nearly a normal level at 72 h after treatment (Fig. 3A). Treatment with different doses of evodiamine inhibited the DNA binding activity of NF-κB in SW1990 cells (Fig. 3B). More importantly, treatment with gemcitabine plus evodiamine significantly reduced the DNA binding of NF-κB, as compared with that of gemcitabine treatment or control cells (Fig. 3C). Therefore, treatment with evodiamine effectively inhibited the gemcitabine-induced NF-κB activation in vitro, which may contribute to inducing more SW1990 cells apoptosis.
Treatment with evodiamine plus gemcitabine inhibits the proliferation and induces apoptosis of SW1990 cells in vitro. SW1990 cells at 2×105/mL were cultured overnight in six-well plates and treated in triplicate with 20 μmol/L of gemcitabine for 24 h, and/or 1.0 μmol/L of evodiamine for 48 h. Untreated cells in medium alone were used as controls. (A) Proliferation of SW1990 cells. (B) Representative dot-plots illustrating apoptotic status in SW1990 cells. (C) The percentage of SW1990 cell apoptosis. Data are expressed as mean%±SD of each group of cells from three separate experiments. Evo: Evodiamine; Gem: Gemcitabine. *P<0.05 vs. control. **P<0.05 vs. cells treated with single drug or control.
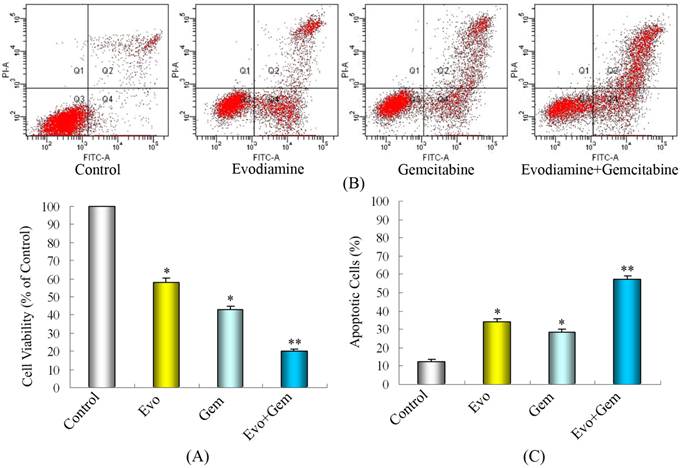
Effect of evodiamine on the spontaneous and gemcitabine-induced NF-κB activation in vitro. (A) Gemcitabine (20 μmol/L) induces NF-κB activation in SW1990 cells, as evidenced by EMSA supershift experiment. (B) Treatment with evodiamine for 48 h inhibits NF-κB activation in SW1990 cells. (C) SW1990 cells at 2×105/mL were cultured overnight in six-well plates and treated in triplicate with 20 μmol/L of gemcitabine for 24 h, and/or 1.0 μmol/L of evodiamine for 48 h. Untreated cells in medium alone were used as controls. Treatment with evodiamine attenuates the gemcitabine-induced NF-κB activation in SW1990 cells. Equal protein loading was ensured by immunoblotting 10 μg of nuclear protein with anti-retinoblastoma antibody. Data are representative images (left column) and expressed as mean%±SD of the relative levels of NF-κB activation in SW1990 cells from tree separate experiments (right column). The nuclear extracts from unstimulated gastric cancer SGC7901 cells were used as negative control and SGC7901 cells stimulated with 50 ng/mL TNFα were used as positive controls. +: positive control; -: negative control; Evo: Evodiamine; Gem: Gemcitabine. *P<0.05 vs. control. #P<0.05 vs. untreated cells or cells treated with gemcitabine. **P<0.05 vs. untreated cells; *# P>0.05 vs. untreated cells; ##P<0.05 vs. untreated cells.
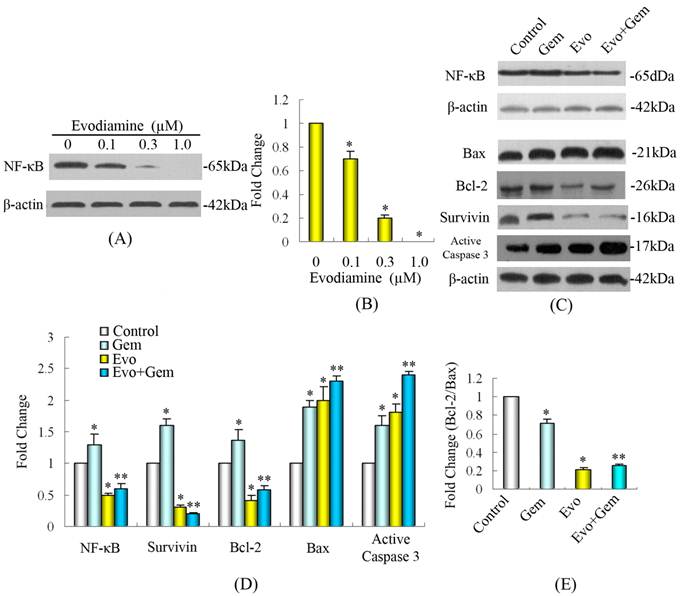
Evodiamine inhibits gemcitabine-induced expression of NF-κB and modulates the expression of apoptosis-related proteins in SW1990 cells
Western blot analysis showed that treatment with evodiamine significantly down-regulated the expression of NF-κB/p65 in SW1990 cells and its inhibitory effects were dose-dependent (Fig. 4A and B). Furthermore, while treatment with gemcitabine significantly up-regulated the expression of NF-κB/p65, as compared with that in control, treatment with evodiamine significantly mitigated the spontaneous and gemcitabine-induced expression of NF-κB/p65 (Fig. 4C and D). Evodiamine significantly decreased the expression of Bcl-2 and survivin, while active caspase-3 and Bax were up-regulated (Fig. 4C and D). Reduced ratio of Bcl-2/Bax was seen in combination treatment group (Fig. 4E).
Treatment with evodiamine modulates the expression of NF-κB and the NF-κB-targeted proteins in vitro. (A) Treatment with evodiamine for 48 h inhibits the expression of NF-κB/p65 in a dose-dependent manner. (B) Quantification of NF-κB/p65 was performed by assigning a value of 1 to the group without evodiamine treatment. (C) SW1990 cells at 2×105/mL were cultured overnight in six-well plates and treated in triplicate with 20 μmol/L of gemcitabine for 24 h, and/or 1.0 μmol/L of evodiamine for 48 h. Untreated cells in medium alone were used as controls. The relative levels of NF-κB/p65, Bcl-2 and Bax expression and active caspase-3 in SW1990 cells. (D) Quantification was performed by assigning a value of 1 to the control group. (E) Bcl-2/Bax ratio was quantified by assigning a value of 1 to the control group. Evo: Evodiamine; Gem: Gemcitabine. *P<0.05 vs. controls; **P<0.05 vs. control or gemcitabine alone group.
Western blot analysis illustrating the effect of evodiamine on phospho-mTOR(Ser2448), Rictor-mTOR, phospho-PTEN(Ser380/Thr382/383), and their downstream substrates PI3K(Tyr458), phospho-Akt(Ser473) protein expression in SW1990 cells. SW1990 cells at 2×105/mL were cultured overnight in six-well plates and treated in triplicate with 20 μmol/L of gemcitabine for 24 h, and/or 1.0 μmol/L of evodiamine for 48 h. Untreated cells in medium alone were used as controls. (A) Treatment with evodiamine or evodiamine plus gemcitabine inhibits the expression of phospho-mTOR(Ser2448), Rictor-mTOR, phospho-PTEN(Ser380/Thr382/383), PI3K(Tyr458), and phospho-Akt(Ser473) protein. (B) The relative levels of phospho-mTOR(Ser2448), Rictor-mTOR, phospho-PTEN(Ser380/Thr382/383), PI3K(Tyr458), and phospho-Akt(Ser473) in SW1990 cells. Quantification was performed by assigning a value of 1 to the control group. (C) Effect of evodiamine or evodiamine plus gemcitabine on cAMP concentration in SW1990 cells. (D) Effect of evodiamine or evodiamine plus gemcitabine on PKA activity in SW1990 cells. Evo: Evodiamine; Gem: Gemcitabine. #P>0.05 vs. control; *P<0.05 vs. control; **P<0.05 vs. control or cells treated with gemcitabine alone.
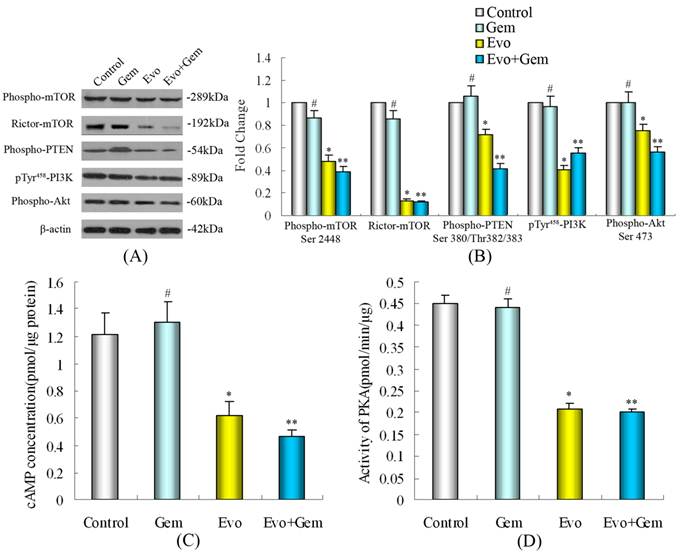
Evodiamine inhibits the phosphorylation of PI3K, Akt, PTEN and mTOR in SW1990 cells
Evodiamine significantly down-regulated the expression of phospho-Akt(Ser473), which was not influenced by gemcitabine, and treatment with evodiamine plus gemcitabine also significantly inhibited the phosphorylation of Akt. Evodiamine or evodiamine plus gemcitabine markedly reduced the expression of PI3K(Tyr458), phospho-PTEN and phospho-mTOR, which were not significantly influenced by gemcitabine. Evodiamine or evodiamine combined with gemcitabine down-regulated the expression of Rictor-mTOR, which was not modulated by gemcitabine.
Evodiamine inhibits cAMP concentration and PKA activity
Evodiamine or evodiamine plus gemcitabine decreased cAMP concentration in SW1990 cells (Fig. 5C). PKA is the primary mediator of cAMP activity and a key regulatory enzyme responsible for many normal cellular processes, such as cell growth and metabolism. We found that evodiamine or evodiamine plus gemcitabine caused significant reduction in PKA activity (Fig. 5D), which was similar to the drug-induced effects observed for phospho-Akt(Ser473) and PI3K(Tyr458).
Antitumor effect of evodiamine plus gemcitabine on the growth of implanted pancreatic tumors in vivo
We examined the effects of evodiamine and gemcitabine, alone or in combination, on the growth of subcutaneously implanted pancreatic tumors (Fig. 6A). Analysis of the final tumor volumes measured on day 37 after the start of treatment revealed that treatment with gemcitabine or evodiamine alone led to significantly slower growth than treatment with saline alone (Fig. 6B) (P<0.05 vs. controls). The final tumor volumes on day 37 of treatment in the mice treated with combination of evodiamine plus gemcitabine were further significantly minimized (Fig. 6B) (P<0.05 vs. control or gemcitabine-treated mice). A similar pattern of inhibitory effect was observed in the dissected tumor weights (Fig. 6C).
Evodiamine potentiates the effect of gemcitabine in blocking the growth of pancreatic cancer in nude mice. After three weeks of implantation, a total of 48 nude mice were randomized into four treatment groups (n=12 per group) based on the bioluminescence measured after the first IVIS imaging. (A) Photographs of subcutaneously implanted pancreatic tumors on day 37 of treatment. Combination of evodiamine with gemcitabine significantly inhibited tumor growth. (B) Tumor volumes in mice measured on the last day of the experiment (Day 37) (n=12). (C) The tumor weights from individual groups of mice. (D) Bioluminescence IVIS images of subcutaneously implanted pancreatic tumors in live, anesthetized mice. (E) The amount of tumor in each nude mouse, as measured by live IVIS imaging on day 37 of treatment (n=12). Evo: Evodiamine; Gem: Gemcitabine. Points are and columns represent mean values; bars represent SD. *P<0.05 vs. control; **P<0.05 vs. control or mice treated with single agent.
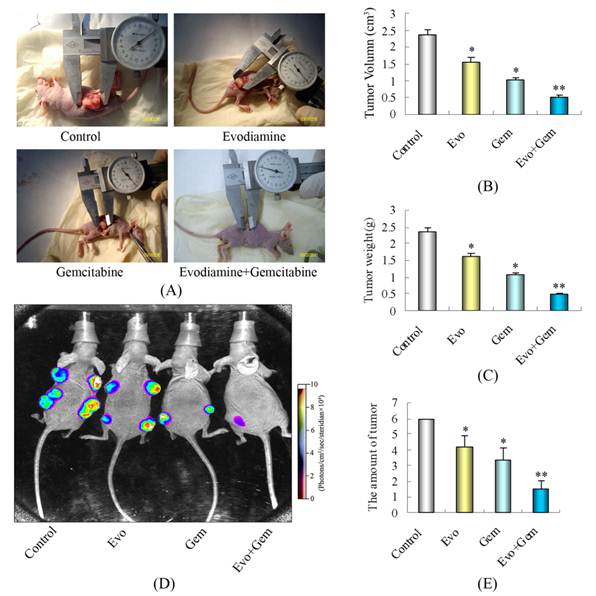
The enhanced antitumor effect caused by combination of evodiamine plus gemcitabine was further demonstrated in the luciferase-transfected SW1990 pancreatic cancer cells xenograft tumor model. Measurements of bioluminescence by IVIS imaging (Fig. 6D and E) indicated that the amount of tumors in the combined evodiamine plus gemcitabine treatment group was lower than in any other group.
Before treatment, the average body weight of mice was not significantly different among the four experimental groups. However, measurement of mouse body weights on day 37 of treatments revealed that the body weight in the control group, evodiamine therapy group, gemcitabine therapy group and combination therapy group was 16.3±1.53 g, 17.2±1.67 g, 13.2±1.54 g, and 15.7±1.54 g, respectively. The mean body weight in the mice treated with gemcitabine alone was significantly less than other groups of mice. The potent inhibition on the growth of implanted tumors and the maintained body weight in the evodiamine-treated mice suggested that evodiamine improved the tumor- and gemcitabine-related deterioration in mice.
Treatment with evodiamine plus gemcitabine triggers pancreatic tumor cell apoptosis in vivo
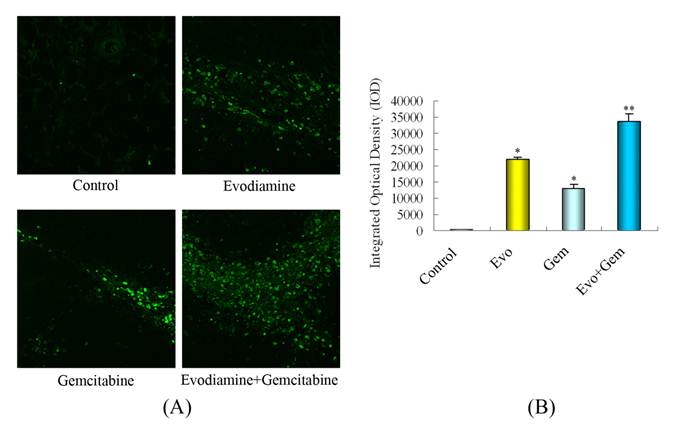
As shown by TUNEL assays (Fig. 7A and B), few cells underwent apoptosis in the control group, while significantly more apoptotic cells were observed in the tumors from the combination treatment group (P<0.05 vs. mice treated with gemcitabine alone or controls).
Evodiamine modulates the activation of phospho-mTOR(Ser2448) and phospho-PTEN(Ser380) in tumor cells
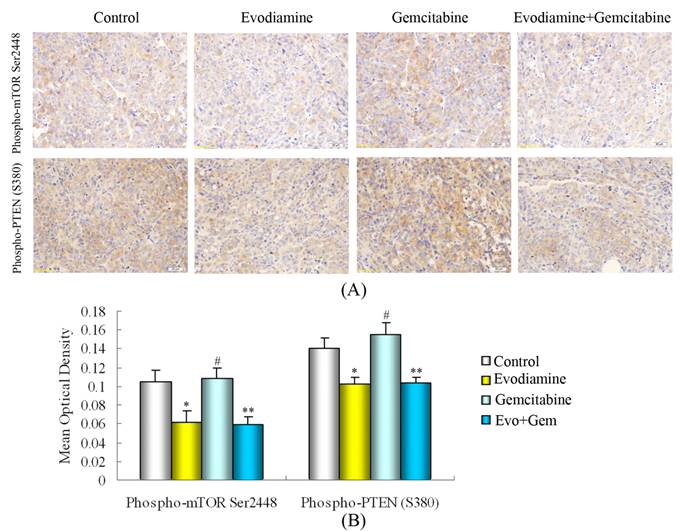
Consistent with the in vitro results, evodiamine alone or evodiamine plus gemcitabine significantly reduced the expression of phospho-PTEN(Ser380) and phospho-mTOR(Ser2448) in transplanted pancreatic cancer (Fig. 8A and B).
Treatment with evodiamine enhances the gemcitabine-induced pancreatic cancer cell apoptosis in vivo. (A) TUNEL analysis of apoptotic cells (400x). (B) Quantitative analysis of apoptotic cells. Evo: Evodiamine; Gem: Gemcitabine. *P<0.05 vs. control; **P<0.05 vs. control or mice treated with single agent.
Immunohistochemistry detection of phospho-mTOR(Ser2448) and phospho-PTEN(Ser380). (A) Evodiamine or evodiamine plus gemcitabine inhibit the activation of phospho-PTEN(Ser380) and phospho-mTOR(Ser2448). (B) Quantified data are presented. Evo: Evodiamine; Gem: Gemcitabine. #P>0.05 vs. control; *P<0.05 vs. control; **P<0.05 vs. control or mice treated with gemcitabine.
Discussion
Gemcitabine may activate NF-κB, and activation of NF-κB is believed to be one of the reasons for the development of chemoresistance during cancer therapy. In fact, spontaneous activation of NF-κB has been detected in human pancreatic cancer tissues, and activation of NF-κB is believed to result in the chemoresistant phenotype in pancreatic cancer cells (31-33). On the other hand, inactivation of NF-κB sensitizes cancer cells to conventional chemotherapies (34-36). Evodiamine is widely believed to inhibit constitutive and inducible NF-κB activation (8) in several kinds of tumors, such as lung adenocarcinoma, T-cell lymphoma, and multiple myeloma. In this study, we found that evodiamine potentiated anti-tumor effects of gemcitabine by inhibiting pancreatic cancer cell proliferation and inducing SW1990 cell apoptosis in vitro and in vivo. In addition, evodiamine was also found to inhibit spontaneous and gemcitabine-induced NF-κB expression and activation in SW1990 cells. Taken together, these findings suggest that evodiamine potentiates the cytotoxicity of gemcitabine against pancreatic cancer cells by inhibiting the expression and activation of NF-κB.
Previous studies have demonstrated that inhibition of NF-κB can potentiate the anti-cancer effect of multiple chemotherapeutic agents (36, 37). Similarly, siRNA-mediated knockdown of survivin expression was reported to enhance the chemosensitivity of pancreatic cancer cells to gemcitabine (41). In this study, we found that gemcitabine-induced activation of NF-κB and up-regulation of Bcl-2 and survivin practically undermined its proapoptotic effect on pancreatic cancer cells. However, evodiamine significantly down-regulated the gemcitabine-induced NF-κB activation and altered expression of Bcl-2 and survivin. Moreover, the combination therapy with gemcitabine and evodiamine also significantly up-regulated Bax, as compared to single-agent treatment, resulting in down-regulation of the Bcl-2/Bax ratio, and then increased the activation of caspase-3, which induced apoptosis.
Earlier reports have shown that treatment of human melanoma A375-S2 cells with evodiamine negatively affects the PI3K/Akt signaling pathway (25, 38). PI3K and Akt are both considered as viable and effective targets for pancreatic cancer therapy (38, 39). Previous studies have also indicated that inhibition of Akt can enhance the activity of gemcitabine chemotherapy in pancreatic cancer (40-42). Here, we found that evodiamine alone or combined with gemcitabine decreased the phospho-Akt(Ser473) levels in SW1990 cells, implying that evodiamine may also potentiate the anti-tumor activity of gemcitabine through inhibiting Akt activation. Fahy et al. (43) showed that Akt inhibition sensitized pancreatic cancer cells to the apoptotic effect of gemcitabine by suppressing the activity of NF-κB and reducing the Bcl-2/Bax ratio in the pancreatic cancer cell line MIA-PaCa-2. Madrid et al. (44) demonstrated that the PI3K/Akt pathway is involved in development of chemoresistance, at least in part, by the activation of NF-κB. In our study, a remarkable deactivation of NF-κB and decrease in the Bcl-2/Bax ratio in SW1990 cells was detected in the evodiamine therapy group and in the evodiamine plus gemcitabine combination group. The PI3K signaling pathway has been shown to be activated in clinical samples of pancreatic cancer (17), and activation of PI3K is known to ultimately lead to Akt phosphorylation at Thr308 and Ser473 (45). In the present study, we found that PI3K(Tyr458) in SW1990 cells was down-regulated in response to evodiamine or evodiamine plus gemcitabine treatments. PKA is the primary mediator of cAMP action and a key regulatory enzyme responsible for many normal cellular processes, such as cell growth and metabolism. Activation of PI3K/Akt can be achieved by cAMP-dependent PKA (46). Akt activation in human coronary artery endothelial cells was found to be inhibited by application of PI3K, Akt, or PKA inhibitors (46). Here, for the first time, we demonstrated that evodiamine or evodiamine plus gemcitabine down-regulated the activity of PKA in SW1990 cells, suggesting that inhibition of PI3K/Akt by evodiamine is partly due to suppressing PKA activity. It has been reported that cAMP formation up-regulates PI3K/Akt and PKA activities, leading to NF-κB activation (47). Here, we also found that evodiamine or evodiamine plus gemcitabine down-regulated the cAMP concentration in SW1990 cells, suggesting that inhibition of PI3K/Akt by evodiamine is partly due to inhibition of cAMP/PKA. PI3K/Akt kinases phosphorylate multiple downstream substrates, including the serine/threonine protein kinase mTOR (48). A study by Sarbassov et al. (49) demonstrated that mTOR in complex with Rictor:G_L targets AKT for phosphorylation at Ser473. Therefore, interplay between mTOR and PI3K/Akt may exist. Since PTEN is known to be able to negatively affect the PI3K pathway in vivo (45), it is possible that dephosphorylation of Ser380/Thr382/383 might indicate the up-regulation of PTEN phosphatase activity, a critical event that leads to destabilization and down-regulation of the PI3K pathway (45). Our results showed that treatment with evodiamine alone or combined with gemcitabine decreased the expression of phospho-PTEN(Ser380/Thr382/383), phospho-mTOR(Ser2448) and Rictor-mTOR. In general, our study suggested that evodiamine might directly or indirectly inhibit the PI3K/Akt pathway targeting NF-κB and inhibit the phosphorylation of PTEN and mTOR, thereby sensitizing pancreatic cancer cells to gemcitabine-induced apoptosis.
We found that evodiamine significantly augmented the antitumor efficacy of gemcitabine in subcutaneously implanted tumors. Experiments based on the luciferase-transfected SW1990 cells xenograft tumor model also showed that evodiamine plus gemcitabine were more efficacious for treating pancreatic cancer. Furthermore, the study also showed that evodiamine and evodiamine plus gemcitabine down-regulated the expression of phospho-PTEN(Ser380) and phospho-mTOR(Ser2448), but gemcitabine had no remarked effect on their expression in tumor tissue, consistent with the in vitro results of Western blot analysis.
Chemotherapeutic agents often cause various adverse effects in patients. Gastrointestinal adverse effects, such as anorexia, constipation, or diarrhea often result in under nourishment and loss of body weight. We found that the body weight of mice treated with gemcitabine alone was significantly less than that of other groups. However, the body weight of the mice treated with both evodiamine and gemcitabine was similar to that of controls. These data suggest that treatment with a reduced dose of gemcitabine, combined with evodiamine, not only enhanced the antitumor effect, but also reduced the gemcitabine-induced body weight loss in mice. A similar approach may also prevent and mitigate the side effect of gemcitabine treatment in patients with pancreatic cancer to improve the life quality of patients.
In conclusion, our data indicated that evodiamine may augment the therapeutic effect of gemcitabine in pancreatic cancer by direct or indirect inhibition of the PI3K/Akt pathway that was not influenced by gemcitabine and targeted NF-κB, thereby leading to sensitization of pancreatic cancer cells to gemcitabine.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010Mar;7(3):163-72
2. Colucci G, Giuliani F, Gebbia V, Biglietto M, Rabitti P, Uomo G, Cigolari S, Testa A, Maiello E, Lopez M. Gemcitabine alone or with cisplatin for the treatment of patients with locally advanced and/or metastatic pancreatic carcinoma: a prospective, randomized phase III study of the Gruppo Oncologia dell'Italia Meridionale. Cancer. 2002Feb15;94(4):902-10
3. Louvet C, Labianca R, Hammel P, Lledo G, Zampino MG, Andre T, Zaniboni A, Ducreux M, Aitini E, Taieb J, Faroux R, Lepere C, de Gramont A. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. J Clin Oncol. 2005May20;23(15):3509-16
4. Abou-Alfa GK, Letourneau R, Harker G, Modiano M, Hurwitz H, Tchekmedyian NS, Feit K, Ackerman J, De Jager RL, Eckhardt SG, O'Reilly EM. Randomized phase III study of exatecan and gemcitabine compared with gemcitabine alone in untreated advanced pancreatic cancer. J Clin Oncol. 2006Sep20;24(27):4441-7
5. Berlin JD, Catalano P, Thomas JP, Kugler JW, Haller DG, Benson AB 3rd. Phase III study of gemcitabine in combination with fluorouracil versus gemcitabine alone in patients with advanced pancreatic carcinoma: Eastern Cooperative Oncology Group Trial E2297. J Clin Oncol. 2002Aug1;20(15):3270-5
6. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007May20;25(15):1960-6
7. Herrmann R, Bodoky G, Ruhstaller T, Glimelius B, Bajetta E, Schuller J, Saletti P, Bauer J, Figer A, Pestalozzi B, Kohne CH, Mingrone W, Stemmer SM, Tamas K, Kornek GV, Koeberle D, Cina S, Bernhard J, Dietrich D, Scheithauer W. Gemcitabine plus capecitabine compared with gemcitabine alone in advanced pancreatic cancer: a randomized, multicenter, phase III trial of the Swiss Group for Clinical Cancer Research and the Central European Cooperative Oncology Group. J Clin Oncol. 2007Jun1;25(16):2212-7
8. Takada Y, Kobayashi Y, Aggarwal BB. Evodiamine abolishes constitutive and inducible NF-kappaB activation by inhibiting IkappaBalpha kinase activation, thereby suppressing NF-kappaB-regulated antiapoptotic and metastatic gene expression, up-regulating apoptosis, and inhibiting invasion. J Biol Chem. 2005Apr29;280(17):17203-12
9. Gately S, Kerbel R. Therapeutic potential of selective cyclooxygenase-2 inhibitors in the management of tumor angiogenesis. Prog Exp Tumor Res. 2003;37:179-92
10. North S, Moenner M, Bikfalvi A. Recent developments in the regulation of the angiogenic switch by cellular stress factors in tumors. Cancer Lett. 2005Jan31;218(1):1-14
11. Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999Jan;5(1):119-27
12. Wang CY, Cusack JC Jr, Liu R, Baldwin AS Jr. Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kappaB. Nat Med. 1999Apr;5(4):412-7
13. Yeh PY, Chuang SE, Yeh KH, Song YC, Ea CK, Cheng AL. Increase of the resistance of human cervical carcinoma cells to cisplatin by inhibition of the MEK to ERK signaling pathway partly via enhancement of anticancer drug-induced NF kappa B activation. Biochem Pharmacol. 2002Apr15;63(8):1423-30
14. Wang W, McLeod HL, Cassidy J. Disulfiram-mediated inhibition of NF-kappaB activity enhances cytotoxicity of 5-fluorouracil in human colorectal cancer cell lines. Int J Cancer. 2003Apr20;104(4):504-11
15. Patel NM, Nozaki S, Shortle NH, Bhat-Nakshatri P, Newton TR, Rice S, Gelfanov V, Boswell SH, Goulet RJ Jr, Sledge GW Jr, Nakshatri H. Paclitaxel sensitivity of breast cancer cells with constitutively active NF-kappaB is enhanced by IkappaBalpha super-repressor and parthenolide. Oncogene. 2000Aug24;19(36):4159-69
16. Arlt A, Gehrz A, Muerkoster S, Vorndamm J, Kruse ML, Folsch UR, Schafer H. Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene. 2003May22;22(21):3243-51
17. Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, Reddy SA. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene. 2004Nov11;23(53):8571-80
18. Bondar VM, Sweeney-Gotsch B, Andreeff M, Mills GB, McConkey DJ. Inhibition of the phosphatidylinositol 3'-kinase-AKT pathway induces apoptosis in pancreatic carcinoma cells in vitro and in vivo. Mol Cancer Ther. 2002Oct;1(12):989-97
19. Fahy BN, Schlieman MG, Virudachalam S, Bold RJ. Inhibition of AKT abrogates chemotherapy-induced NF-kappaB survival mechanisms: implications for therapy in pancreatic cancer. J Am Coll Surg. 2004Apr;198(4):591-9
20. Shoji N, Umeyama A, Takemoto T, Kajiwara A, Ohizumi Y. Isolation of evodiamine, a powerful cardiotonic principle, from Evodia rutaecarpa Bentham (Rutaceae). J Pharm Sci. 1986Jun;75(6):612-3
21. Fei XF, Wang BX, Li TJ, Tashiro S, Minami M, Xing DJ, Ikejima T. Evodiamine, a constituent of Evodiae Fructus, induces anti-proliferating effects in tumor cells. Cancer Sci. 2003Jan;94(1):92-8
22. Zhang Y, Wu LJ, Tashiro S, Onodera S, Ikejima T. Intracellular regulation of evodiamine-induced A375-S2 cell death. Biol Pharm Bull. 2003Nov;26(11):1543-7
23. Zhang Y, Wu LJ, Tashiro S, Onodera S, Ikejima T. Evodiamine induces tumor cell death through different pathways: apoptosis and necrosis. Acta Pharmacol Sin. 2004Jan;25(1):83-9
24. Wang C, Wang MW, Tashiro S, Onodera S, Ikejima T. Roles of SIRT1 and phosphoinositide 3-OH kinase/protein kinase C pathways in evodiamine-induced human melanoma A375-S2 cell death. J Pharmacol Sci. 2005Apr;97(4):494-500
25. Wang C, Li S, Wang MW. Evodiamine-induced human melanoma A375-S2 cell death was mediated by PI3K/Akt/caspase and Fas-L/NF-kappaB signaling pathways and augmented by ubiquitin-proteasome inhibition. Toxicol In Vitro. 2010Apr;24(3):898-904
26. Khanbolooki S, Nawrocki ST, Arumugam T, Andtbacka R, Pino MS, Kurzrock R, Logsdon CD, Abbruzzese JL, McConkey DJ. Nuclear factor-kappaB maintains TRAIL resistance in human pancreatic cancer cells. Mol Cancer Ther. 2006Sep;5(9):2251-60
27. Gerlier D, Thomasset N. Use of MTT colorimetric assay to measure cell activation. J Immunol Methods. 1986Nov20;94(1-2):57-63
28. Matata BM, Galinanes M. Peroxynitrite is an essential component of cytokines production mechanism in human monocytes through modulation of nuclear factor-kappa B DNA binding activity. J Biol Chem. 2002Jan18;277(3):2330-5
29. Tang ZY, Wu YL, Gao SL, Shen HW. Effects of the proteasome inhibitor bortezomib on gene expression profiles of pancreatic cancer cells. J Surg Res. 2008Mar;145(1):111-23
30. Ogasawara M, Matsubara T, Suzuki H. Inhibitory effects of evodiamine on in vitro invasion and experimental lung metastasis of murine colon cancer cells. Biol Pharm Bull. 2001Aug;24(8):917-20
31. Arlt A, Schafer H. NFkappaB-dependent chemoresistance in solid tumors. Int J Clin Pharmacol Ther. 2002Aug;40(8):336-47
32. Liptay S, Weber CK, Ludwig L, Wagner M, Adler G, Schmid RM. Mitogenic and antiapoptotic role of constitutive NF-kappaB/Rel activity in pancreatic cancer. Int J Cancer. 2003Jul20;105(6):735-46
33. Holcomb B, Yip-Schneider M, Schmidt CM. The role of nuclear factor kappaB in pancreatic cancer and the clinical applications of targeted therapy. Pancreas. 2008Apr;36(3):225-35
34. Sarkar FH, Li Y. Using chemopreventive agents to enhance the efficacy of cancer therapy. Cancer Res. 2006Apr1;66(7):3347-50
35. Banerjee S, Zhang Y, Ali S, Bhuiyan M, Wang Z, Chiao PJ, Philip PA, Abbruzzese J, Sarkar FH. Molecular evidence for increased antitumor activity of gemcitabine by genistein in vitro and in vivo using an orthotopic model of pancreatic cancer. Cancer Res. 2005Oct1;65(19):9064-72
36. Banerjee S, Zhang Y, Wang Z, Che M, Chiao PJ, Abbruzzese JL, Sarkar FH. In vitro and in vivo molecular evidence of genistein action in augmenting the efficacy of cisplatin in pancreatic cancer. Int J Cancer. 2007Feb15;120(4):906-17
37. Ali S, Varghese L, Pereira L, Tulunay-Ugur OE, Kucuk O, Carey TE, Wolf GT, Sarkar FH. Sensitization of squamous cell carcinoma to cisplatin induced killing by natural agents. Cancer Lett. 2009Jun18;278(2):201-9
38. Yotsumoto F, Fukami T, Yagi H, Funakoshi A, Yoshizato T, Kuroki M, Miyamoto S. Amphiregulin regulates the activation of ERK and Akt through epidermal growth factor receptor and HER3 signals involved in the progression of pancreatic cancer. Cancer Sci. 2010Nov;101(11):2351-60
39. He L, Wu Y, Lin L, Wang J, Chen Y, Yi Z, Liu M, Pang X. Hispidulin, a small flavonoid molecule, suppresses the angiogenesis and growth of human pancreatic cancer by targeting vascular endothelial growth factor receptor 2-mediated PI3K/Akt/mTOR signaling pathway. Cancer Sci. 2011Jan;102(1):219-25
40. Liu D, Zhang Y, Dang C, Ma Q, Lee W, Chen W. siRNA directed against TrkA sensitizes human pancreatic cancer cells to apoptosis induced by gemcitabine through an inactivation of PI3K/Akt-dependent pathway. Oncol Rep. 2007Sep;18(3):673-7
41. Yao J, Qian C. Inhibition of Notch3 enhances sensitivity to gemcitabine in pancreatic cancer through an inactivation of PI3K/Akt-dependent pathway. Med Oncol. 2010Sep;27(3):1017-22
42. Zhang B, Shi ZL, Liu B, Yan XB, Feng J, Tao HM. Enhanced anticancer effect of gemcitabine by genistein in osteosarcoma: the role of Akt and nuclear factor-kappaB. Anticancer Drugs. 2010Mar;21(3):288-96
43. Fahy BN, Schlieman M, Virudachalam S, Bold RJ. AKT inhibition is associated with chemosensitisation in the pancreatic cancer cell line MIA-PaCa-2. Br J Cancer. 2003Jul21;89(2):391-7
44. Madrid LV, Wang CY, Guttridge DC, Schottelius AJ, Baldwin AS Jr, Mayo MW. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-kappaB. Mol Cell Biol. 2000Mar;20(5):1626-38
45. Olsen BB, Bjorling-Poulsen M, Guerra B. Emodin negatively affects the phosphoinositide 3-kinase/AKT signalling pathway: a study on its mechanism of action. Int J Biochem Cell Biol. 2007;39(1):227-37
46. Erdogdu O, Nathanson D, Sjoholm A, Nystrom T, Zhang Q. Exendin-4 stimulates proliferation of human coronary artery endothelial cells through eNOS-, PKA- and PI3K/Akt-dependent pathways and requires GLP-1 receptor. Mol Cell Endocrinol. 2010;325(1-2):26-35
47. Wang P, Zhu F, Konstantopoulos K. Prostaglandin E2 induces interleukin-6 expression in human chondrocytes via cAMP/protein kinase A- and phosphatidylinositol 3-kinase-dependent NF-kappaB activation. Am J Physiol Cell Physiol. 2010;298(6):C1445-56
48. Cho DH, Choi YJ, Jo SA, Ryou J, Kim JY, Chung J. et al. Troglitazone acutely inhibits protein synthesis in endothelial cells via a novel mechanism involving protein phosphatase 2A-dependent p70 S6 kinase inhibition. Am J Physiol Cell Physiol. 2006;291(2):C317-26
49. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098-101
Author contact
![]() Corresponding author: Sheng-Zhang Lin, Tel: 86-577-88879016; Fax: 86-577-88816191; Email: zysxswzszedu.cn.
Corresponding author: Sheng-Zhang Lin, Tel: 86-577-88879016; Fax: 86-577-88816191; Email: zysxswzszedu.cn.
Received 2011-6-29
Accepted 2011-10-19
Published 2011-11-5