Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(1):93-107. doi:10.7150/ijbs.8.93 This issue Cite
Research Paper
The Complete Mitochondrial Genome of the Damsel Bug Alloeorhynchus bakeri (Hemiptera: Nabidae)
Hu Li1,*, Haiyu Liu1,*, Liangming Cao2, Aimin Shi1, Hailin Yang1, Wanzhi Cai1 ![]()
1. Department of Entomology, China Agricultural University, Beijing 100193, China
2. The Key Laboratory of Forest Protection of China State Forestry Administration, Research Institute of Forest Ecology, Environment and Protection, Chinese Academy of Forestry, Beijing 100091, China
* These authors contributed equally to this work.
Received 2011-9-17; Accepted 2011-11-10; Published 2011-11-24
Abstract
The complete sequence of the mitochondrial DNA (mtDNA) of the damsel bug, Alloeorhynchus bakeri, has been completed and annotated in this study. It represents the first sequenced mitochondrial genome of heteropteran family Nabidae. The circular genome is 15, 851 bp in length with an A+T content of 73.5%, contains the typical 37 genes that are arranged in the same order as that of the putative ancestor of hexapods. Nucleotide composition and codon usage are similar to other known heteropteran mitochondrial genomes. All protein-coding genes (PCGs) use standard initiation codons (methionine and isoleucine), except COI, which started with TTG. Canonical TAA and TAG termination codons are found in eight protein-coding genes, the remaining five (COI, COII, COIII, ND5, ND1) have incomplete termination codons (T or TA). PCGs of two strands present opposite CG skew which is also reflected by the nucleotide composition and codon usage. All tRNAs have the typical clover-leaf structure, except the dihydrouridine (DHU) arm of tRNASer (AGN) which forms a simple loop as known in many other metazoa. Secondary structure models of the ribosomal RNA genes of A. bakeri are presented, similar to those proposed for other insect orders. There are six domains and 45 helices and three domains and 27 helices in the secondary structures of rrnL and rrnS, respectively. The major non-coding region (also called control region) between the small ribosomal subunit and the tRNAIle gene includes two special regions. The first region includes four 133 bp tandem repeat units plus a partial copy of the repeat (28 bp of the beginning), and the second region at the end of control region contains 4 potential stem-loop structures. Finally, PCGs sequences were used to perform a phylogenetic study. Both maximum likelihood and Bayesian inference analyses highly support Nabidae as the sister group to Anthocoridae and Miridae.
Keywords: Mitochondrial genome, Alloeorhynchus bakeri, Nabidae, RNA secondary structure, phylogenetic relationship, Cimicomorpha
Introduction
Mitochondrial (mt) genome sequence and structure is widely used to provide information on comparative and evolutionary genomics, on molecular evolution and patterns of gene flow, on phylogenetics and population genetics [1, 2]. Several analyses have demonstrated recently that complete mt genomes provide higher levels of support than those based on individual or partial mt genes [3-5]. Mt genome of insect is typically a double-stranded, circular molecule of 14-20 kb in length, which usually encodes 13 protein-coding genes (PCGs), two ribosomal RNA (rRNA) genes, and 22 transfer RNA (tRNA) genes [6, 7]. Additionally, insect mt genome contains a major non-coding region known as the A+T-rich region or the control region (CR) that plays a role in initiation of transcription and replication [6]. The CRs of different insect taxa have turned out to be very divergent, showing differences in primary sequence, organization, as well as in their location relative to flanking genes, raising the question of whether CRs are homologous across different taxa [7]. Moreover, the length of this region is also highly variable due to its high rates of nucleotide substitution, insertions/deletions, and the presence of varying copy numbers of tandem repeats [8, 9].
The reconstruction of the phylogeny of insects has been a focus of studies for more than a century [10, 11]. The growing interest in phylogenetic reconstruction of the mt genome has triggered a rapid increase in the number of published complete mt genome sequence [12]. To date, the complete or nearly complete mt genomes of 32 species of true bugs are available at NCBI (status April 25, 2011).
Nabidae is a relatively small family of Heteroptera with 20 genera and approximately 500 species [13]. The members of this family are important natural enemies of pests and are distributed throughout the world. Nabidae is proposed to be one of the most primitive families in the infraorder Cimicomorpha and hence it is of major importance for the classification and phylogeny of this infraorder [14]. No complete mt genome has been sequenced from members of this family prior to this study. Here, we present the complete mt genome of Alloeorhynchus bakeri, a representative of Prostemmatinae, and provide analyses of the nucleotide composition, codon usage, compositional biases, RNA secondary structure, and evaluate the phylogenetic position of Nabidae in Heteroptera based on the sequences of PCGs.
Materials and Methods
Samples and DNA extraction
Adult specimens of A. bakeri were collected from Mengla (21°43.474N, 101°32.635E), Yunnan Province, China in April 2007. All specimens were preserved in 95% ethanol in the field. After being transported to the laboratory, they were stored at -20℃ until DNA extraction. Total genomic DNA was extracted from thorax muscle tissue using a CTAB-based method [15]. Voucher specimens (Nos. VHem-00101), preserved in alcohol, are deposited at the Entomological Museum of China Agricultural University (Beijing).
PCR amplification, cloning and sequencing
The genome was amplified in overlapping PCR fragments (Supplementary Material: Table S1). Initially, 13 fragments were amplified using the universal primers from previous work [16] (Fig. 1). Seven perfectly matching primers were designed on the basis of these short fragments for secondary PCRs.
Map of the mt genome of A. bakeri. The tRNAs are denoted by the color blocks and are labeled according to the IUPACIUB single-letter amino acid codes. Gene name without underline indicates the direction of transcription from left to right, and with underline indicates right to left. PCGs are denoted by the grey blocks indicate the direction of transcription from right to left, and the sky-blue indicate the direction of transcription from left to right. Overlapping lines within the circle denote PCR fragments used for cloning and sequencing.
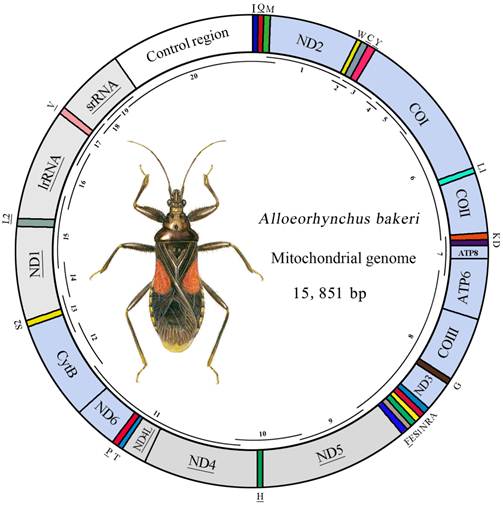
Short PCRs were conducted using Qiagen Taq DNA polymerase (Qiagen, Beijing, China) with the following cycling conditions: 5 min at 94℃, followed by 35 cycles of 50 s at 94℃, 50 s at 48-55℃, and 1-2 min at 72℃. The final elongation step was continued for 10 min at 72℃. Long PCRs were performed using NEB Long Taq DNA polymerase (New England Biolabs) under the following cycling conditions: 30 s at 95℃, followed by 45 cycles of 10 s at 95℃, 50 s at 48-55℃, and 3-6 min at 65℃. The final elongation was continued for 10 min at 65℃. These PCR products were analyzed by 1.0% agarose gel electrophoresis.
The fragments were ligated into pGEM-T Easy Vector (Promega) and the resultant plasmid DNA was isolated using the TIANprp Midi Plasmid Kit (Qiagen). All fragments were sequenced in both directions using the BigDye Terminator Sequencing Kit (Applied Bio Systems) and the ABI 3730XL Genetic Analyzer (PE Applied Biosystems, San Francisco, CA, USA) with two vector-specific primers and internal primers for primer walking.
Sequence analysis and inferences of secondary structures
Raw sequence files were proof-read and aligned into contigs in BioEdit version 7.0.5.3 [17]. Protein-coding regions and ribosomal RNA genes were identified by sequence comparison with published insect mt sequences.
The tRNAs were identified by tRNAscan-SE Search Server v.1.21 [18] with default setting. Some tRNA genes that could not be found by tRNAscan-SE were identified by comparing to other hemipterans. Secondary structures of the small and large ribosomal RNAs were inferred using alignment to the models predicted for Drosophila melanogaster and D. virilis [19], Apis mellifera [20], Manduca sexta [21] and Ruspolia dubia [22]. Stem-loops were named using both the conventions of A. mellifera [20] and M. sexta [21].
Protein-coding gene sequences were aligned using Clustal X [23]. The aligned data were further analyzed by MEGA version 4.0 [24] for the codon usage. The putative control region was examined for regions of potential inverted repeats or palindromes with the aid of the mfold web server (http://www.bioinfo.rpi.edu/applications/mfold/) [25]. Strand asymmetry was calculated using the formulae: AT skew= [A−T]/ [A+T] and GC skew= [G−C]/ [G+C] [26], for the strand encoding the majority of the protein-coding genes.
Phylogenetic analysis
Phylogenetic analysis was carried out based on the 32 complete or nearly complete mt genomes of true bugs from GenBank. Four species from Sternorrhyncha and Auchenorrhyncha were selected as outgroups (Table 1). Based on an analysis of mt genomes of nine Nepomorpha and five other hemipterans, Pleidae were suggested to be raised from a superfamily to the infraorder Plemorpha [27]. Since we didn't add samples to solve this problem, Paraplea frontalis was treated as incertae sedis, and was not included in the phylogenetic analysis to ensure the stability of the topology.
Summary of sample information used in present study
| Order/suborder | Infraorder/superfamily | Family | Species | Accession Number | Reference |
|---|---|---|---|---|---|
| Sternorrhyncha | |||||
| Psylloidea | Psyllidae | Pachypsylla venusta | NC_006157 | [34] | |
| Aphidoidea | Aphididae | Acyrthosiphon pisum | NC_011594 | [35] | |
| Auchenorrhyncha | |||||
| Fulgoroidea | Fulgoridae | Lycorma delicatula | NC_012835 | [27] | |
| Issidae | Sivaloka damnosa | NC_014286 | [36] | ||
| Heteroptera | |||||
| Gerromorpha | |||||
| Hydrometroidea | Hydrometridae | Hydrometra sp. | NC_012842 | [27] | |
| Gerroidea | Gerridae | Gerris sp. | NC_012841 | [27] | |
| Nepomorpha | |||||
| Corixoidea | Corixidae | Sigara septemlineata | FJ456941 | [27] | |
| Ochteroidea | Gelastocoridae | Nerthra sp. | NC_012838 | [27] | |
| Ochteridae | Ochterus marginatus | NC_012820* | [27] | ||
| Notonectoidea | Notonectidae | Enithares tibialis | NC_012819 | [27] | |
| Pleidae | Paraplea frontalis | NC_012822 | [27] | ||
| Nepoidea | Nepidae | Laccotrephes robustus | NC_012817 | [27] | |
| Belostomatidae | Diplonychus rusticus | FJ456939* | [27] | ||
| Naucoroidea | Naucoridae | Ilyocoris cimicoides | NC_012845 | [27] | |
| Aphelocheiridae | Aphelocheirus ellipsoideus | FJ456940* | [27] | ||
| Leptopodomorpha | |||||
| Saldoidea | Saldidae | Saldula arsenjevi | NC_012463 | [49] | |
| Leptopodoidea | Leptopodidae | Leptopus sp. | FJ456946 | [27] | |
| Cimicomorpha | |||||
| Naboidea | Nabidae | Alloeorhynchus bakeri | HM 235722 | ||
| Cimicoidea | Anthocoridae | Orius niger | NC_012429* | [49] | |
| Reduvioidea | Reduviidae | Triatoma dimidiata | NC_002609 | [33] | |
| Valentia hoffmanni | NC_012823 | [27] | |||
| Miroidea | Miridae | Lygus lineolaris | EU401991* | Roehrdanz, unpublished | |
| Pentatomomorpha | |||||
| Aradoidea | Aradidae | Neuroctenus parus | NC_012459 | [49] | |
| Pentatomoidea | Pentatomidae | Nezara viridula | NC_011755 | [49] | |
| Halyomorpha halys | NC_013272 | [37] | |||
| Cydnidae | Macroscytus subaeneus | NC_012457* | [49] | ||
| Plataspidae | Coptosoma bifaria | NC_012449 | [49] | ||
| Lygaeoidea | Berytidae | Yemmalysus parallelus | NC_012464 | [49] | |
| Colobathristidae | Phaenacantha marcida | NC_012460* | [49] | ||
| Malcidae | Malcus inconspicuus | NC_012458 | [49] | ||
| Geocoridae | Geocoris pallidipennis | NC_012424* | [49] | ||
| Pyrrhocoroidea | Largidae | Physopelta gutta | NC_012432 | [49] | |
| Pyrrhocoridae | Dysdercus cingulatus | NC_012421 | [49] | ||
| Coreoidea | Alydidae | Riptortus pedestris | NC_012462 | [49] | |
| Coreidae | Hydaropsis longirostris | NC_012456 | [49] | ||
| Rhopalidae | Aeschyntelus notatus | NC_012446* | [49] | ||
| Stictopleurus subviridis | NC_012888 | [49] |
* Mt genome sequence was incomplete.
A DNA alignment was inferred from the amino acid alignment of the 13 protein-coding genes using Clustal X [23]. Alignments of individual genes were then concatenated excluding the stop codon.
Model selection was done with MrModeltest 2.3 [28] and Modeltest 3.7 [29] for Bayesian inference and ML analysis, respectively. According to the Akaike information criterion, the GTR+I+G model was optimal for analysis with nucleotide alignments. MrBayes Version 3.1.1 [30] and a PHYML online web server [31] were employed to analyze this data set under the GTR+I+G model. In Bayesian inference, two simultaneous runs of 3, 000, 000 generations were conducted for the matrix. Each set was sampled every 200 generations with a burnin of 25%. Trees inferred prior to stationarity were discarded as burnin, and the remaining trees were used to construct a 50% majority-rule consensus tree. In ML analysis, the parameters were estimated during analysis and the node support values were assessed by bootstrap resampling (BP) [32] calculated using 100 replicates.
Results
Genome organization and structure
The mt genome of A. bakeri was a double-stranded circular molecule of 15, 851 bp in length (GenBank: HM 235722; Fig.1), and it contained the entire set of 37 genes usually present in most insect mtDNAs (13 PCGs, 22 tRNA genes, and two rRNA genes), and a large non-coding region (control region) (Table 2).
Organization of the A. bakeri mt genome
| Gene | Direction | Location | Size | Anticodon | Codon | Intergenic nucleotides a | |
|---|---|---|---|---|---|---|---|
| Start | Stop | ||||||
| tRNAIle | F | 1-63 | 63 | 30-32 GAT | |||
| tRNAGln | R | 67-133 | 67 | 102-104 TTG | 3 | ||
| tRNAMet | F | 133-198 | 66 | 164-166 CAT | -1 | ||
| ND2 | F | 199-1197 | 999 | ATT | TAA | 0 | |
| tRNATrp | F | 1196-1258 | 63 | 1227-1229 TCA | -2 | ||
| tRNACys | R | 1251-1316 | 66 | 1281-1283 GCA | -8 | ||
| tRNATyr | R | 1319-1381 | 63 | 1347-1349 GTA | 2 | ||
| COI | F | 1383-2916 | 1534 | TTG | T- | 1 | |
| tRNALeu(UUR) | F | 2917-2981 | 65 | 2946-2948 TAA | 0 | ||
| COII | F | 2982-3660 | 679 | ATT | T- | 0 | |
| tRNALys | F | 3661-3730 | 70 | 3691-3693 CTT | 0 | ||
| tRNAAsp | F | 3730-3794 | 65 | 3761-3763 GTC | -1 | ||
| ATP8 | F | 3795-3953 | 159 | ATA | TAA | 0 | |
| ATP6 | F | 3947-4630 | 684 | ATG | TAA | -7 | |
| COIII | F | 4617-5404 | 788 | ATG | TA- | -14 | |
| tRNAGly | F | 5404-5463 | 60 | 5433-5435 TCC | -1 | ||
| ND3 | F | 5464-5817 | 354 | ATA | TAA | 0 | |
| tRNAAla | F | 5821-5880 | 60 | 5850-5852 TGC | 3 | ||
| tRNAArg | F | 5884-5946 | 63 | 5914-5916 TCG | 3 | ||
| tRNAAsn | F | 5945-6010 | 66 | 5976-5978 GTT | -2 | ||
| tRNASer(AGN) | F | 6010-6078 | 69 | 6037-6039 GCT | -1 | ||
| tRNAGlu | F | 6078-6141 | 64 | 6109-6111 TTC | -1 | ||
| tRNAPhe | R | 6140-6202 | 63 | 6167-6169 GAA | -2 | ||
| ND5 | R | 6202-7907 | 1706 | ATT | TA- | -1 | |
| tRNAHis | R | 7905-7966 | 62 | 7933-7935 GTG | -3 | ||
| ND4 | R | 7966-9294 | 1329 | ATG | TAA | -1 | |
| ND4L | R | 9288-9581 | 294 | ATT | TAG | -7 | |
| tRNAThr | F | 9593-9655 | 63 | 9624-9626 TGT | 11 | ||
| tRNAPro | R | 9656-9718 | 63 | 9687-9689TGG | 0 | ||
| ND6 | F | 9721-10218 | 498 | ATA | TAA | 2 | |
| CytB | F | 10218-11354 | 1137 | ATG | TAG | -1 | |
| tRNASer(UCN) | F | 11353-11420 | 68 | 11384-11386TGA | -2 | ||
| ND1 | R | 11441-12362 | 922 | ATA | T- | 20 | |
| tRNALeu(CUN) | R | 12363-12428 | 66 | 12397-12399TAG | 0 | ||
| lrRNA | R | 12429-13680 | 1252 | 0 | |||
| tRNAVal | R | 13681-13749 | 69 | 13716-13718 TAC | 0 | ||
| srRNA | R | 13750-14539 | 790 | 0 | |||
| Control region | 14540-15851 | 1312 |
a Negative numbers indicate that adjacent genes overlap.
Twenty-three genes were transcribed on the majority strand (J-strand), whereas the others were oriented on the minority strand (N-strand). Gene overlaps were found at 17 gene junctions and involved a total of 54 bp; the longest overlap (14 bp) existed between ATP6 and COIII. In addition to the control region, there were 45 nucleotides dispersed in 8 intergenic spacers, ranging in size from 1 to 20 bp. The longest spacer sequence was located between tRNASer (UCN) and ND1.
Transfer RNAs
The entire complement of 22 tRNAs was found in A. bakeri, and 20 of them were determined using tRNAscane-SE [18]. The tRNAArg and tRNASer (AGN) genes were not detected by software, and were determined through comparison with previously published hemipteran mt genomes [27, 33]. All tRNAs could fold into the typical clover-leaf structure except for tRNASer (AGN), in which its dihydrouridine (DHU) arm simply formed a loop (Fig. 2).
Inferred secondary structure of 22 tRNAs of the A. bakeri mt genome. The tRNAs are labeled with the abbreviations of their corresponding amino acids. Dashed (-) indicate Watson-Crick base pairing and (+) indicate G-U base pairing.
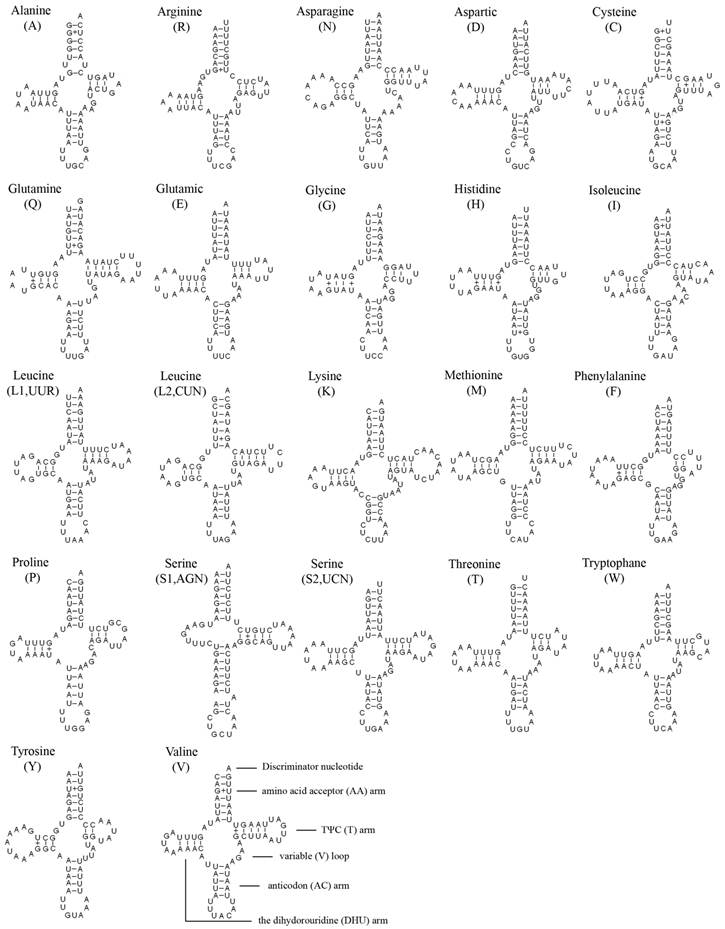
The length of tRNAs ranged from 60 to 70 bp. The aminoacyl (AA) stem (7 bp) and the AC loop (7 nucleotides) were invariable, and most of the size variation was the DHU and TΨC (T) arms, within which the loop size (3-9 bp) was more variable than the stem size (2-5 bp). The size of the anticodon stems was conservative, with the exception of tRNASer (AGN) which possessed a long optimal base pairing (9 bp in contrast to the normal 5) and a bulged nucleotide in the middle for the AC stem.
Based on the secondary structure, a total of 28 unmatched base pairs were found in the A. bakeri tRNAs. Twenty-three of them were G-U pairs, which form a weak bond, located in the AA stem (8 bp), the DHU stem (9 bp), the AC stem (2 bp), the T stem (4 bp), the remaining 5 included C-U (2 bp) mismatches in the AA stem and the T stem of tRNAArg, respectively; A-A (2 bp) mismatches in the AA stem of tRNAArg; U-U mismatches (1 bp) in the AA stem of tRNAAla.
Ribosomal RNAs
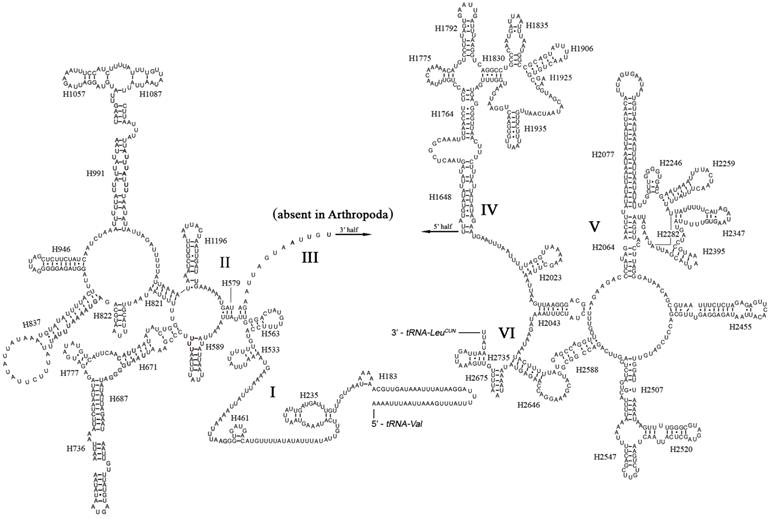
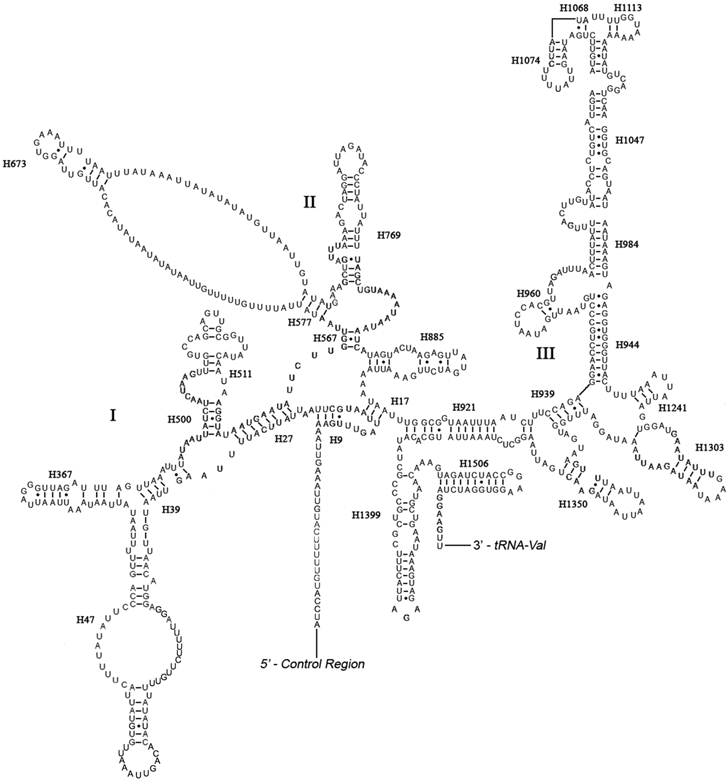
The boundaries of rRNA genes were determined by sequence alignment with that of Triatoma dimidiata [33] and Valentia hoffmanni [27]. As in most other insect mt genomes, the large and small ribosomal RNAs (rrnL and rrnS) genes in A. bakeri were located between tRNALeu(CUN) and tRNAVal and between tRNAVal and the control region, respectively (Fig. 1; Table 2). The length rrnL and rrnS were determined to be 1, 252 bp and 790 bp, respectively. The secondary structure of rrnL consisted of six structural domains (domain III is absent in arthropods) and 45 helices (Fig. 3), and the rrnS consisted of three structural domains and 27 helices (Fig. 4).
Predicted secondary structure of the rrnL gene in the A. bakeri mt genome. Roman numerals denote the conserved domain structure. The numbering system follows [20]. Dashed (-) indicate Watson-Crick base pairing and dot (•) indicate G-U base pairing.
Predicted secondary structure of the rrnS gene in the A. bakeri mt genome. Roman numerals denote the conserved domain structure. Dashed (-) indicate Watson-Crick base pairing and dot (•) indicate G-U base pairing. Structural annotations follow Fig. 3.
Protein-coding genes: Translation initiation and termination signals
All but one PCGs of A. bakeri initiated with ATN as the start codon (four with ATG, four with ATT and four with ATA) (Table 2). The only exception was the COI gene, which used TTG as a start codon.
The majority of the PCGs of A. bakeri had the complete termination codons TAA (ND2, ATP8, ATP6, ND3, ND4 and ND6) or TAG (ND4L and CytB), and the remaining five had incomplete termination codons, TA (COIII and ND5) or T (COI, COII and ND1) (Table 2).
Nucleotide composition and codon usage
The nucleotide composition of the A. bakeri mtDNA was significantly biased toward A and T. The A+T content was 73.5% (A = 40.1%, T = 33.4%, C = 16.3 %, G = 10.2%). The A+T content of isolated PCGs, tRNAs, rRNAs and the CR is 72.6%, 75.4%, 75.7% and 75.7%. The skew statistics of the total PCGs demonstrated that the J-strand PCGs were CG-skewed and consisted of nearly equal A and T while the N-strand PCGs were GC-skewed and much more TA-skewed, and the N-strand tRNAs had also higher GC-skewed than the J-strand tRNAs.
The nucleotide bias was also reflected in the codon usage. Analysis of base composition at each codon position of the concatenated 13 PCGs showed that the third codon position (81.2%) was higher in A+T content than the first (68.5%) and second (66.3%) codon positions (Table 3). There were different nucleotide frequencies in all codon position between the two strands in A. bakeri. If the J-strand alone was inspected, the third codon position sites showed a preponderance of A nucleotides, whereas for N-strand, the third codon position sites biased toward T (Table 3).
Nucleotide composition of the A. bakeri mt genome
| Proportion of nucleotides | ||||||||
|---|---|---|---|---|---|---|---|---|
| Feature | %T | %C | %A | %G | %A+T | AT Skew | GC Skew | No. of nucleotides |
| Whole genome | 33.4 | 16.3 | 40.1 | 10.2 | 73.5 | 0.09 | -0.23 | 15851 |
| Protein-coding genes | 40.6 | 14.0 | 32.0 | 13.3 | 72.6 | -0.12 | -0.03 | 11085 |
| First codon position | 33.7 | 13.1 | 34.8 | 18.4 | 68.5 | 0.02 | 0.17 | 3695 |
| Second codon position | 46.7 | 18.5 | 19.6 | 15.2 | 66.3 | -0.41 | -0.10 | 3695 |
| Third codon position | 41.5 | 10.4 | 39.7 | 8.3 | 81.2 | -0.02 | -0.11 | 3695 |
| Protein-coding genes-J | 35.9 | 17.0 | 35.5 | 11.6 | 71.4 | -0.01 | -0.19 | 6819 |
| First codon position | 28.6 | 15.8 | 38.0 | 17.6 | 66.6 | 0.14 | 0.05 | 2273 |
| Second codon position | 44.3 | 20.9 | 20.6 | 14.2 | 64.9 | -0.37 | -0.19 | 2273 |
| Third codon position | 34.6 | 14.4 | 47.9 | 3.0 | 82.5 | 0.16 | -0.66 | 2273 |
| Protein-coding genes-N | 48.3 | 9.2 | 26.4 | 16.1 | 74.7 | -0.29 | 0.27 | 4267 |
| First codon position | 41.9 | 8.8 | 29.7 | 19.5 | 71.6 | -0.17 | 0.38 | 1422 |
| Second codon position | 50.4 | 14.7 | 18.0 | 16.9 | 68.4 | -0.47 | 0.07 | 1422 |
| Third codon position | 52.5 | 4.1 | 31.6 | 11.9 | 84.1 | -0.25 | 0.49 | 1422 |
| tRNA genes | 36.7 | 10.6 | 38.7 | 14.1 | 75.4 | 0.03 | 0.14 | 1427 |
| tRNA genes-J | 35.2 | 12.3 | 39.9 | 12.6 | 75.1 | 0.06 | 0.01 | 908 |
| tRNA genes-N | 39.1 | 7.5 | 36.6 | 16.8 | 75.7 | -0.03 | 0.38 | 519 |
| rRNA genes | 41.2 | 8.8 | 34.5 | 15.6 | 75.7 | -0.09 | 0.28 | 2042 |
| Control region | 39.4 | 16.5 | 36.3 | 7.9 | 75.7 | -0.04 | -0.35 | 1312 |
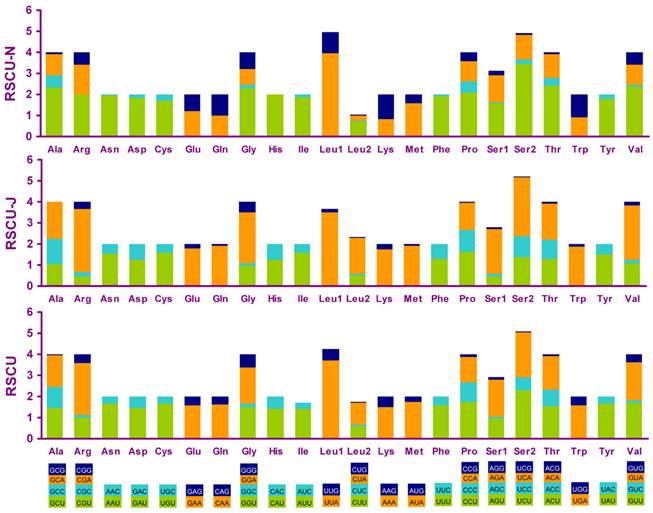
Four most frequently used codon, TTA (leucine), ATT (isoleucine), TTT (phenylalanine) and ATA(methionine), were all composed wholly of A and/or T, and NNA and NNC codons were more frequent than NNU and NNG in PCGs encoded on the J-strand, whereas the N-strand genes showed exactly the opposite trend (Fig. 5).
Relative synonymous codon usage (RSCU) in the A. bakeri mt genome. Codon families are provided on the x-axis.
The control region
The 1, 312 bp long control region of A. bakeri mt genome was located at the conserved position between rrnS and tRNAIle- tRNAGln- tRNAMet gene cluster (Fig. 1), and was composed of 75.7% A+T content, which was the most A+T-rich region (Table 3).
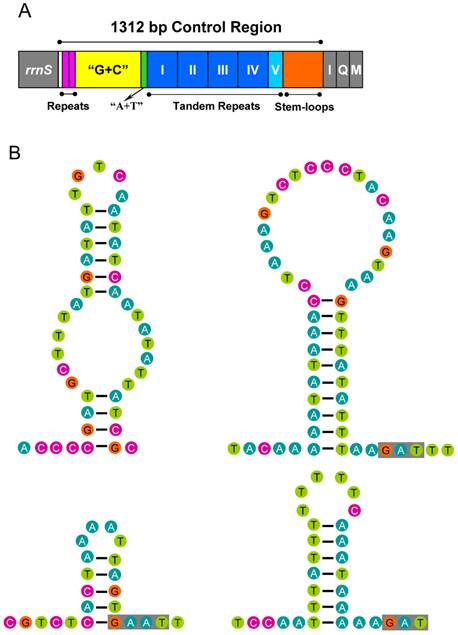
The control region of A. bakeri can be divided into four parts (Fig. 6A): (1) a 533 bp region that was bordered by rrnS, of which the G+C content (33.2%) is higher than the whole genome, and at the beginning of this region contained two 21 bp C-rich repetitive sequences (TCCCCCCTCCGGTGGTCGCTA); (2) a 39 bp region heavily biased toward A+T (89.7%); (3) a region composed of five tandem repeats; (4) a region at the end of control region containing 4 potential stem-loop structures, the largest one with a stem of 20 bp and 21 bp loop (Fig. 6B).
Control region of the A. bakeri mt genome. (A) Structure elements found in the control region of A. bakeri. The control region flanking genes rrnS, trnI (I), trnQ (Q), and trnM (M) are represented in grey boxes; the blue and azury boxes with roman numerals indicate the tandem repeat region; “G+C” (yellow) indicates high G+C content region; repeats (pink) indicate two repeat regions at the beginning of the high G+C content region; “A+T” (green) indicates high A+T content region. (B) The putative stem-loops structure found in the control region. The grey boxes indicate highly conserved flanking sequence.
Phylogenetic relationships
We performed phylogenetic analysis using nucleotide sequences of 13 mt PCGs from 32 heteropteran species and 4 outgroup hemipteran insect species [27, 34-37]. BI and ML analyses generated identical tree topologies (Fig. 7).
Phylogenetic tree inferred from the mt genomes of 32 heteropterans. Phylogenetic analysis was based on all 13 protein-coding genes. The tree was rooted with four outgroup taxa (P. venusta, A. pisum, S. damnosa and L. delicatula). Cycles indicate bootstrap support; percentages of Bayesian posterior probabilities (upper) and ML bootstrap support values (underside).
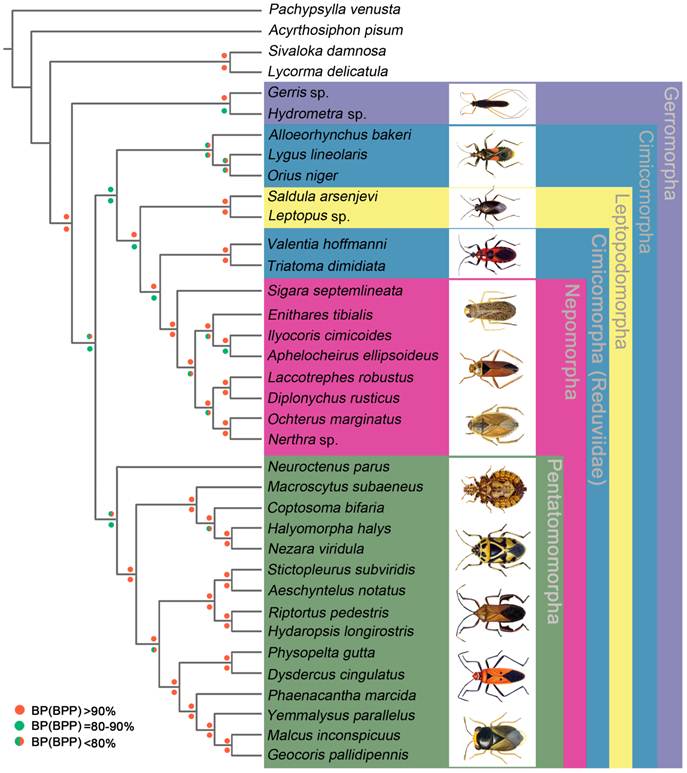
In the present study, the sister-relationship within the infraorders were supported for the Pentatomomorpha (14 taxa), Nepomorpha (8 taxa), Leptopodomorpha (2 taxa) and Gerromorpha (2 taxa) by BI and ML analysis. Two Gerromorpha superfamilies were monophyletic in the basal position of these five infraorders. Within Cimicomorpha, Reduviidae was paraphyletic with respect to the Nabidae, Anthocoridae and Miridae. The sister-relationship of Nabidae, Anthocoridae and Miridae was confirmed. The infraordinal relationships tended to be poorly resolved with low support.
Discussion
The mt genome of A. bakeri is a double-stranded circular molecule, with the same gene content (37 genes and 1 control region) and gene order as that in D. yakuba [38]. The overall organization of the A. bakeri mt genome is very compact, and the overlaps between ATP8/ATP6 (7 bp) and ND4/ND4L (7 bp) are often been found across the Metazoa [39, 40].
Dihydrouridine (DHU) arm of A. bakeri tRNASer (AGN) simply forms a loop. This phenomenon is common in sequenced true bug mt genomes, and has been considered as a typical feature of metazoan mtDNA [41]. Some A. bakeri tRNA genes possessed non-Watson-crick matches, aberrant loops, or even extremely short arms. It is not known whether the aberrant tRNAs lose their function in every case, but a post-transcriptional RNA editing mechanism has been proposed to maintain function of these tRNA genes [42, 43].
The secondary structures of the A. bakeri mt rrnL and rrnS are drawn following the previously published models for M. sexta [21]. In rrnL, H837 forms a long stem structure with a small loop in the terminal as observed in other insects [20, 21, 44, 45]. In many insect mtDNA, the helix H2077 is absent as the bases do not form complementary pairs [21, 46], whereas it includes a 23 paired bases stem and 12 bp loop in A. bakeri. The helix H2347 is also highly variable in insect, and in A. bakeri this region consisting of 5 paired bases is similar to that proposed for M. sexta [21]. H2735, the last stem-loop of rrnL, only forms a 4 bp stem and 6 bp loop in A. bakeri which is difference in size from M. sexta, 7 bp stem and 22 bp loop [21].
Domains I and II are alterable regions in terms of sequence and structure, whereas domain III is highly conserved part of the rrnS of A. bakeri. Helix 47 is variable among different insects, but the terminal portion of this stem is conserved [21, 45], and in A. bakeri two loops are formed similar to those in Evania appendigaster [45]. The sequence between H577 and H673 can't be folded, similar to that in M. sexta [21]. H1047 and associated stems H1068, H1074 and H1113 may yield multiple possible secondary structures due to its high AT bias and several non-canonical base pairs, as discussed in other insects [20, 21, 44, 47, 48].
An unconventional TTG start codon was detected only for the COI gene in A. bakeri, which is consistent with some other true bugs [27, 49, 50], and other insects (mainly in Diptera) [38, 51-54]. The presence of an incomplete stop codon is a common phenomenon found in mt genomes of insects and it has been proposed that the complete termination codon TAA could be generated by the posttranscriptional polyadenylation [55, 56].
The A+T content of A. bakeri corresponds well to the AT bias generally observed in hexapod mt genomes, which range from 64.8% in Japyx solifugus [57] to 87.4% in Diadegma semiclausum [44].
Metazoan mt genomes usually present a clear strand bias in nucleotide composition [58, 59], and the strand bias can be measured as AT- and GC-skews [26]. AT- and GC-skews of A. bakeri mt genomes is consistent compared to the usual strand biases of metazoan mtDNA (positive AT-skew and negative GC-skew for the J-strand, and whereas the reverse is observed in the N-strand). The underlying mechanism that leads to the strand bias has been generally related to replication, because this process has long been assumed to be asymmetric in the mtDNA and could therefore affect the occurrence of mutations between the two strands [58]. It is possible that the overall genome A-bias is driven by mutational pressure on the N-strand and the GC-skew may be correlated with the asymmetric replication process of the mtDNA [60].
The nucleotide bias is also reflected in the codon usage. As reported for other metazoan mtDNAs, the most commonly used codon in degenerate codon families often does not match the anticodon [6]. All codons are present in A. bakeri mtDNA PCGs, but GCG codon is not represented on the J-strand, and CAC and CGC codons for the N-strand, reflecting the influence of a strong biased codon usage [53]. Codon usage may be influenced by other molecular processes such as translational selection efficiency and accuracy, which apparently have a stronger influence in organisms with rapid growth rates [57, 61, 62].
The largest non-coding region (1, 312 bp) was flanked by rrnS and the tRNAIle gene in the A. bakeri mt genome. It was highly enriched in AT (75.7%) and could form stable stem-loop secondary structures. Repeated sequences are common in the control region for most insects, and length variations due to the various numbers of repeats are not without precedent [63]. In the case of A. bakeri, the control region includes four 133 bp tandem repeat units plus a partial copy of the repeat (28 bp of the beginning).
The stem-loop structure in the control region is suggested as the site of the initiation of secondary strand synthesis in Drosophila [64]. The flanking sequence of the structure is suggested to be highly conserved among some insects, possessing the consensus 'TATA' sequence at the 5' end and 'G(A)nT' at the 3' end [65, 66]. However, in the A. bakeri control region, no highly conserved flanking 'TATA' sequence existed at the 5' end, but we found 'G(A)nT' at the 3' end (Fig. 6B).
The poly-thymine stretch is relatively conserved across insects [63]. In A. bakeri this stretch locates in the beginning of the fourth part of control region and spans 12 thymine nucleotides with one adenine. It has been speculated that this poly-thymine stretch may be involved in transcriptional control or may be the site for initiation of replication [64].
The topology of infraordinal relationships of Heteroptera is similar to previous work [67], and future analyses should focus on phylogeny studies including Dipsocoromorpha and Enicocephalomorpha mt genome data and additional representatives for some poorly sampled clades. The sister-relationship of Nabidae, Anthocoridae and Miridae is confirmed in the present study. But the position of Reduviidae is not improved, although the mt genome of A. bakeri is added. Cimicomorpha comprise over 20, 000 species currently placed in 17 families [68], but only 4 families have the mt genome data, and it is too limited to resolve the phylogeny of Cimicomorpha, and increased taxon sampling may be the best way to resolve this problem.
Supplementary Material
Table S1: Primer sequences used in this study.
Acknowledgements
This research is supported by grants from the National Natural Science Foundation of China (Nos. 30825006, 30970394, 31061160186), Beijing Natural Science Foundation (No. 6112013), the Doctoral Program of Higher Education of China (No. 200800190015), the Key Laboratory of the Zoological Systematics and Evolution of the Chinese Academy of Sciences (No. O529YX5105) and the Innovation Program for Ph. D. Students of China Agricultural University (No. 15059211). We are very grateful to three anonymous reviewers' comments and suggestions. Specially thank goes to Dr. Thomas J. Henry of the Systematic Entomological Laboratory, USDA for his kind help in the bug identification.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Wilson K, Cahill V, Ballment E, Benzie J. The complete sequence of the mitochondrial genome of the crustacean Penaeus mondon: are malacostracan crustaceans more closely related to insects than to branchiopods? Mol Biol Evol. 2000;17:863-874
2. Salvato P, Simonato M, Battisti A, Negrisolo E. The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae). BMC Genomics. 2008;9:331
3. Cummings MP, Otto SP, Wakeley J. Sampling properties of DNA sequence data in phylogenetic analysis. Mol Biol Evol. 1995;12:814-822
4. Russo CA, Takezaki N, Nei M. Efficiencies of different genes and different tree-building methods in recovering a known vertebrate phylogeny. Mol Biol Evol. 1996;13:525-536
5. Zardoya R, Meyer A. Phylogenetic performance of mitochondrial protein-coding genes in resolving relationships among vertebrates. Mol Biol Evol. 1996;13:933-942
6. Wolstenholme DR. Genetic novelties in mitochondrial genomes of multicellular animals. Curr Opin Genet Dev. 1992;2:918-925
7. Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767-1780
8. Fauron CMR, Wolstenholme DR. Extensive diversity among Drosophila species with respect to nucleotide sequences within the adenine+thymine-rich region of mitochondrial DNA molecules. Nucleic Acids Res. 1980;8:2439-2452
9. Inohira K, Hara T, Matsuura ET. Nucleotide sequence divergence in the A+T-rich region of mitochondrial DNA in Drosophila simulans and Drosophila mauritiana. Mol Biol Evol. 1997;14:814-822
10. Kristensen NP. Phylogeny of insect orders. Annu Rev Entomol. 1981;26:135-157
11. Whiting MF, Carpenter JC, Wheeler Q, Wheeler WC. The Strepsiptera problem: phylogeny of the holometabolous insect orders inferred from 18S and 28S ribosomal DNA sequences and morphology. Syst Biol. 1997;46:1-68
12. Curole JP, Kocher TD. Mitogenomics: digging deeper with complete mitochondrial genomes. Trends Ecol Evol. 1999;14:394-398
13. Schuh RT, Slater JA. True bugs of the word (Hemiptera: Heteroptera): classification and natural history. New York: Cornell University Press. 1995
14. Nokkala C, Kuznetsova V, Grozeva S, Nokkala S. Direction of karyotype evolution in the bug family Nabidae (Heteroptera): New evidence from 18S rDNA analysis. Eur J Entomol. 2007;104:661-665
15. Aljanabi SM, Martinez I. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nucleic Acids Res. 1997;25:4692-4693
16. Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu Rev Ecol Evol Syst. 2006;37:545-579
17. Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95-98
18. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955-964
19. Cannone JJ, Subramainian S, Schnare MN, Collett JR, D'Souza LM, Du YS, Feng B, Lin N, Madabusi LV, Müller KM, Pande N, Shang ZD, Yu N, Gutell RR. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs: Correction. BMC Bioinformatics. 2002;3:15
20. Gillespie JJ, Johnston JS, Cannone JJ, Gutell RR. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): Structure, organization and retrotransposable elements. Insect Mol Biol. 2006;15:657-686
21. Cameron SL, Whiting MF. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene. 2008;408:112-123
22. Zhou Z, Huang Y, Shi F. The mitochondrial genome of Ruspolia dubia (Orthoptera: Conocephalidae) contains a short A+T-rich region of 70 bp in length. Genome. 2007;50:855-866
23. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876-4882
24. Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596-1599
25. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406-3415
26. Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995;41:353-358
27. Hua JM, Li M, Dong PZ, Cui Y, Xie Q, Bu WJ. Phylogenetic analysis of the true water bugs (Insecta: Hemiptera: Heteroptera: Nepomorpha): evidence from mitochondrial genomes. BMC Evol Bio. 2009;9:134
28. Nylander JAA. MrModeltest v2; Program distributed by the author. Evolutionary Biology Centre, Uppsala University. 2004
29. Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817-818
30. Huelsenbeck JP, Ronquist F. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754-755
31. Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696-704
32. Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783-791
33. Dotson EM, Beard CB. Sequence and organization of the mitochondrial genome of the Chagas disease vector, Triatoma dimidiata. Insect Mol Biol. 2001;10:205-215
34. Thao ML, Baumann L, Baumann P. Organization of the mitochondrial genomes of whiteflies, aphids, and psyllids (Hemiptera: Sternorrhyncha). BMC Evol Biol. 2004;4:25
35. Barrett RJ, Crease TJ, Hebert PD, Via S. Mitochondrial DNA diversity in the pea aphid Acyrthosiphon pisum. Genome. 1994;37:858-65
36. Song N, Liang AP, Ma C. The complete mitochondrial genome sequence of the planthopper, Sivaloka damnosus. J Insect Sci. 2010;10:76
37. Lee W, Kang J, Jung C, Hoelmer K, Lee SH, Lee S. Complete mitochondrial genome of brown marmorated stink bug Halyomorpha halys (Hemiptera: Pentatomidae), and phylogenetic relationships of hemipteran suborders. Mol Cells. 2009;28:155-165
38. Clary DO, Wolstenholme DR. The ribosomal RNA genes of Drosophila mitochondrial DNA. Nucleic Acids Res. 1985;13:4029-4045
39. Stewart JB, Beckenbach AT. Insect mitochondrial genomics: The complete mitogenome sequence of the meadow spittlebug Philaenus spumarius (Hemiptera: Auchenorrhyncha: Cercopoidae). Genome. 2005;48:46-54
40. Carapelli A, Vannini L, Nardi F, Boore JL, Beani L, Dallai R, Frati F. The mitochondrial genome of the entomophagous endoparasite Xenos vesparum (Insecta: Strepsiptera). Gene. 2006;376:248-259
41. Lavrov DV, Brown WM, Boore JL. Phylogenetic position of the Pentastomida and (pan) crustacean relationships. Proc Biol Sci. 2004;271:537-544
42. Tomita K, Yokobori S, Oshima T, Ueda T, Watanabe K. The cephalopod Loligo bleekeri mitochondrial genome: multiplied noncoding regions and transposition of tRNA genes. J Mol Evol. 2001;54:486-500
43. Zhang CY, Huang Y. Complete mitochondrial genome of Oxya chinensis (Orthoptera, Acridoidea). Acta Biochim Biophys Sin. 2008;40:7-18
44. Wei SJ, Shi M. He JH, Sharkey M, Chen XX. The complete mitochondrial genome of Diadegma semiclausum (Hymenoptera: Ichneumonidae) indicates extensive independent evolutionary events. Genome. 2009;52:308-319
45. Wei SJ, Tang P, Zheng LH, Shi M, Chen XX. The complete mitochondrial genome of Evania appendigaster (Hymenoptera: Evaniidae) has low A+T content and a long intergenic spacer between atp8 and atp6. Mol Biol Rep. 2010;37:1931-1942
46. Niehuis O, Naumann CM, Misof B. Identification of evolutionary conserved structural elements in the mt SSU rRNA of Zygaenoidea (Lepidoptera): a comparative sequence analysis. Org Divers Evol. 2006;6:17-32
47. Hickson RE, Simon C, Cooper A, Spicer GS, Sullivan J, Penny D. Conserved sequence motifs, alignment, and secondary structure for the third domain of animal 12S rRNA. Mol Biol Evol. 1996;13:150-169
48. Page RDM. Comparative analysis of secondary structure of insect mitochondrial small subunit ribosomal RNA using maximum weighted matching. Nucleic Acids Res. 2000;28:3839-3845
49. Hua JM, Dong PZ, Li M, Cui Y, Zhu WB, Xie Q, Bu WJ. The analysis of mitochondrial genome of Stictopleurus subviridis Hsiao (Insect, Hemiptera-Heteroptera, Rhopalidae). Acta Zootax Sin. 2009;34:1-9
50. Hua JM, Li M, Dong PZ, Cui Y, Xie Q, Bu WJ. Comparative and phylogenomic studies on the mitochondrial genomes of Pentatomomorpha (Insecta: Hemiptera: Heteroptera). BMC Genomics. 2008;9:610
51. Beard CB, Hamm DM, Collins FH. The mitochondrial genome of the mosquito Anopheles gambiae: DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects. Insect Mol Biol. 1993;22:103-124
52. Spanos L, Koutoumbas G, Kosyfakis M, Louis C. The mitochondrial genome of the Mediterranean fruitfly, Ceratitis capitata. Insect Mol Biol. 2000;9:139-144
53. Lessinger AC, Martins Junqueira AC, Lemos TA, Kemper EL, da Silva FR, Vettore AL, Arruda P, Azeredo-Espin AML. The mitochondrial genome of the primary screwworm fly Cochliomyia hominivorax (Diptera: Calliphoridae). Insect Mol Biol. 2000;9:521-529
54. Castro L, Ruberu K, Dowton M. Mitochondrial genomes of Vanhornia eucnemidarum (Apocrita: Vsnhorniidae) and Primeuchroeus spp. (Aculeata: Chrysididae): evidence of rearranged mitochondrial genomes within the Apocrita (Insecta: Hymenoptera). Genome. 2006;49:752-766
55. Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290:470-474
56. Cha SY, Yoon HJ, Lee EM, Yoon MH, Hwang JS, Jin BR, Han YS, Kim I. The complete nucleotide sequence and gene organization of the mitochondrial genome of the bumblebee, Bombus ignitus (Hymenoptera: Apidae). Gene. 2007;392:206-220
57. Carapelli A, Comandi S, Convey P, Nardi F, Frati F. The complete mitochondrial genome of the Antarctic springtail Cryptopygus antarcticus (Hexapoda: Collembola). BMC Genomics. 2008;9:315
58. Hassanin A, Leger N, Deutsch J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst Biol. 2005;54:277-298
59. Hassanin A. Phylogeny of Arthropoda inferred from mitochondrial sequences: Strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol Phylogenet Evol. 2006;38:100-116
60. Reyes A, Gissi C, Pesole G, Saccone C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol Biol Evol. 1998;15:957-966
61. Sharp PM, Bailes E, Grocock RJ, Peden JF, Sockett RE. Varition in the strength of selected codon usage bias among bacteria. Nucleic Acids Res. 2005;33:1141-1153
62. Stoletzki N, Eyre-Walker A. Synonymous codon usage in Escherichia coli: selection for translational accuracy. Mol Biol Evol. 2007;24:374-381
63. Zhang DX, Hewitt FM. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem Syst Ecol. 1997;25:99-120
64. Clary DO, Wolstenholme DR. Drosophila mitochondrial DNA: conserved sequences in the A+T-rich region and supporting evidence for a secondary structure model of the small ribosomal RNA. J Mol Evol. 1987;25:116-125
65. Zhang DX, Szymura JM, Hewitt GM. Evolution and structural conservation of the control region of insect mitochondrial DNA. J Mol Evol. 1995;40:382-391
66. Song N, Liang AP. Complete mitochondrial genome of the small brown planthopper, Laodelphax striatellus (Delphacidae: Hemiptera), with a novel gene order. Zool Sci. 2009;26:851-860
67. Li H, Gao JY, Liu HY, Liu H, Liang AP, Zhou XG, Cai W. The architecture and complete sequence of mitochondrial genome of an assassin bug Agriosphodrus dohrni (Hemiptera: Reduviidae). Int J Bio Sci. 2011;7:792-804
68. Weirauch C, Schuh RT. Systematics and evolution of Heteroptera: 25 years of progress. Annu Rev Entomol. 2011;56:487-510
Author contact
![]() Corresponding author: Dr. Wanzhi Cai, Department of Entomology, China Agricultural University, Yuanmingyuan West Road, Beijing 100193, China. Phone: 86-10-62732885; Fax: 86-10-62732885; Email: caiwzedu.cn
Corresponding author: Dr. Wanzhi Cai, Department of Entomology, China Agricultural University, Yuanmingyuan West Road, Beijing 100193, China. Phone: 86-10-62732885; Fax: 86-10-62732885; Email: caiwzedu.cn